
Abstract
During operation of gas turbine engines, different salts develop that lead to a hot-corrosion attack. While the hot corrosion associated with Na2SO4 and Na3VO4 has received considerable attention, MgSO4 is also a main salt component, but it has received limited examination. For operation at temperatures beyond the capabilities of Ni-base superalloys, refractory-metal alloys with a Mo–Si–B-based coating have demonstrated a robust environmental resistance including hot corrosion attack by Na2SO4 and Na3VO4. In order to evaluate the performance of Mo with a Mo–Si–B-based amorphous coating, samples were exposed to MgSO4 under thermal cycling at temperatures from 800 to 1300 °C. Mass change measurements revealed minimal attack from the MgSO4 exposure except in local regions where cristobalite and MgMoO4 were detected. Even with the local region reaction, the mass change after a cumulative 100 h of exposure was about 3.5 mg/cm2. Exposure to a composite salt mixture containing Na2SO4, NaVO3, MgSO4, CaCl, and KCl revealed signs of interactivity compared to the corrosion due to the separate effects of the individual salts, but again the mass change stabilized to a minimal level demonstrating that the Mo–Si–B-based coating is resilient to hot corrosion.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the continuing effort to improve aircraft engine efficiency, it has become evident that new material systems with high-temperature capability are necessary. Turbine engine efficiency is improved with higher operating temperatures as modeled by the Brayton Cycle [1]. This route requires new materials, such as refractory metals, that maintain their structural performance at high temperatures. Among these, Mo–Si–B alloys have been demonstrated to be a promising candidate [2]. However, molybdenum and other refractory metals have poor oxidation resistance with Mo oxidizing to form low melting point and volatile oxides [3, 4]. To prevent this, a Mo–Si–B-based coating with an aluminoborosilica glass outer layer has been developed as a means to protect the substrate from oxidation [5]. The Mo–Si–B-based coatings are typically produced via a pack cementation method [6]. This process produces a multilayer system on the molybdenum or other refractory metal-based substrate [7].
In addition to oxygen attack, a variety of salt deposits from fuel impurities and the environment have been found in gas turbine engines [8]. The degradation of materials by molten salt deposits at the operational temperatures of turbines is defined as hot corrosion [9]. The hot corrosion reaction occurs in two varieties: a lower temperature Type II and a higher temperature Type I [10]. Type II corrosion is typically active between 670 and 750 °C [11]. Type I corrosion is a two-phase process that begins with a low corrosion incubation period that is related to the formation of an oxide scale. This is followed by accelerated attack as the scale breaks down and is likely caused by salt fluxing [12]. For the most part, hot corrosion studies have focused on the attack by sodium sulfate (Na2SO4)/sodium metavanadate (NaVO3) on chromia or alumina forming alloys. There has also been some study on the attack on silica forming materials such as SiC [13]. At the same time, MgSO4 has been found to be as abundant as Na2SO4 in the gas turbine environment, but there has been little attention to the attack by MgSO4 or the combination of the magnesium and sodium sulfates.
A previous study that investigated the resistance of the Mo–Si–B coating to sodium sulfate (Na2SO4)/sodium metavanadate (NaVO3) reported good corrosion protection and a self-healing ability for the Mo–Si–B in this environment [7]. In the present study, the protective ability of the coating was investigated in the environments of magnesium sulfate (MgSO4) and a salt mixture analogous to real-world conditions. This mixture that is based upon analysis from Bornstein and Allen also contains vanadium, as another component present in fuel impurities [8, 14] (Table 1).
The coating in this composite system would be affected by both attack from the individual salts and the results of the interactivity of the salts. Previous experiments on silica forming materials showed that sodium and magnesium salt combinations increased magnesium mobility while decreased sodium mobility [15]. The presence of sodium, magnesium, vanadium, and sulfur enables the formation of a range of vanadates, oxides, and sulfates that can both promote and inhibit hot corrosion [14]. As a result, a composite salt mixture can behave differently than the contribution of each individual component.
Experimental Procedures
The effects of the salt systems on Mo–Si–B coatings were explored by producing a series of coated molybdenum coins that were exposed to hot corrosion environments. Pure molybdenum (99.95%) rods were cut into coins approximately 1.25 cm in diameter and .3 cm thick. The edges of these coins were rounded and polished to a 5 micron finish.
A coating from a source of Si and B in the ratio of 35:1 silicon to boron was deposited via pack cementation. The coins were surrounded by a powder mixture and heated in an argon-filled tube furnace at 1000 °C for 50 h. The powder mixture contained 2.5 wt% NaF salt as an activator to promote the coating formation. In the mixture, 35 wt% was the 35:1 silicon to boron powder, with the remaining 62.5 wt% being inert 180 grit alumina filler. The void space between the alumina particles provided pathways for the active components to interact with the molybdenum substrate.
After the pack cementation, the coins were conditioned in laboratory air at 1300 °C for 10 h. This conditioning served two functions. Primarily, it oxidized the surface forming the vitreous outer layer [16]. Secondly, it verified the integrity and completeness of the coating. If the coating was incomplete, the molybdenum substrate would rapidly oxidize to molybdenum trioxide (MoO3) and vaporize. This failure of the coating was visually apparent and caused a significant mass loss allowing for easy detection of poorly coated samples.
For hot corrosion exposure, an aqueous slurry of magnesium sulfate or the composite salts was applied to the coated coins using an airbrush in sequential thin coatings in order to achieve a uniform deposit. The amount of salt applied was approximately 25 mg/cm2 for the magnesium sulfate and 10 mg/cm2 for the composite salt. This amount is several orders of magnitude beyond reported salt amounts known to promote corrosive effects [17]. The composite salt slurry was a mixture of salts found in existing jet engines, along with environmental salts, and elements related to fuel impurities [8, 18]. The final constituents are presented in Table 2. The coated specimens were dried in open air until the salt slurry densified. Vapor phase mass loss experiments using a salt reservoir in the furnace were not attempted as it has been shown to yield significantly lower mass losses [7].
The coated coins were loaded into a tube furnace. In the furnace, the coins were exposed to repeated thermal cycling of 5 h periods at either 900 °C or 1000 °C. During the cycles, the air was either left static in the furnace or laboratory air was flowed over the coins at a rate of approximately 10 l/min. The large quantities of salt and relatively slow air speeds make the conditions tested far more favorable for hot corrosion attack than turbine operation. After each cycle, the mass of the coin was measured, and X-ray diffraction was performed periodically. The salt coating was not reapplied during the cycles as Gray et al. [19] have shown that redeposition has little effect on corrosion for the time scales used here. Select coins were sectioned and polished for scanning electron microscopy. Before sectioning, the coins were coated with a sputtered gold and an electroplated nickel layer to preserve the glassy silica coating during the polishing according to a procedure outlined by Richter [20].
Results
Coating Structure
The pack cementation method produced a coating with several different phase layers covering the molybdenum substrate. The coating structure after exposure to MgSO4 for 20 thermal cycles at 1000 °C in flowing air is presented in the cross-section images in Fig. 1. The initial coating structure after conditioning was not altered by the exposure to MgSO4 at 1000 °C in flowing air. Moving away from the molybdenum substrate, a thin T2 layer of Mo5SiB2 develops, on top of that a T1 layer of boron saturated Mo5Si3, then a layer of MoSi2, and finally there is an outer layer of aluminoborosilica that develops during conditioning from the codeposition of Si and B with the incorporation of some alumina filler [7]. During cooling, a few cracks develop in the MoSi2 containing layer, but they are arrested by the T1 layer and do not reach the substrate. The vitreous outer layer is capable of flowing at elevated temperatures to fill the cracks and can be replenished by the lower layers in case of damage [21]. The T2 phase serves as a diffusion barrier and a source of silicon and boron [7]. This enables a self-healing mechanism that further mitigates corrosive attacks.

a Backscatter electron image and b secondary electron image of Mo–Si–B coating after 20 thermal cycles at 1000 °C in flowing air
Magnesium Sulfate
Magnesium sulfate comprises about 22% of the salt deposits found in turbines [8]. Both MgSO4 and Na2SO4 have similar decomposition routes producing SO3 and an oxide [22, 23]. It has been proposed that sodium oxide (Na2O) diffuses into the outer vitreous layer of the Mo–Si–B-based coating promoting the nucleation of cristobalite [7]. The resulting crystallization front advances into the silica and causes some of the coating to break off via spalling during cooling. In contrast, magnesium ions have virtually no mobility in silica [15]. Consequently, magnesium is restricted to forming cristobalite only on the surface and maintains a larger amount of the vitreous surface which enhances the preservation of the Mo–Si–B coating.
The mass change of the sample in the MgSO4 environment after each cycle is presented in Fig. 2. There is an initial gain in mass from the deposited salt coating. After this gain, the sample stabilizes and the total mass change plateaus to near the initial mass before the salt coating application. The coated coins that were in a flowing air environment showed a stable mass with little mass change. The 1000 °C cycles in the static environment showed some mass change, but the amount appears to level off at about 3.5 mg/cm2 which is still minimal. The 900 °C static salt environment did not have a net mass change as some initial amount of salt remained on the surface throughout the experiment. The presence of some amount of cristobalite on these coins, as determined by XRD, indicates that some corrosion has occurred but the mass of the salt remaining overcompensates for this mass loss (Fig. 3).

Mass change of the coated Mo coins with a Mo–Si–B-based coating in different MgSO4 environments

Mass change data for coated Mo coins with a Mo–Si–B-based coating in both MgSO4 and Na2SO4/NaVO3 environments. The Na2SO4/NaVO3 data are reproduced from [7]
These results show a clear difference with the previous work on attack by Na2SO4/NaVO3 salts where the mass change is shown in Fig. 4 [7]. In most cases, there is significantly less mass change for the exposed MgSO4-coated samples than those coated with Na2SO4/NaVO3. The mass change for both MgSO4- and Na2SO4-/NaVO3-coated samples was similar only for the 900 °C flowing air environment, which was relatively inactive for both. The consistent reduction in mass change can be attributed to the lack of magnesium mobility in the amorphous surface layer. In static environments, the mass change during MgSO4 exposure is consistently 0.1 times that of the Na2SO4-based mass change. The mass changes in flowing air environments were more temperature-dependent. At 900 °C, the MgSO4 corrosion was 0.2 times that due to Na2SO4/NaVO3, whereas at 1000 °C the corrosion was 0.05 times that due to Na2SO4/NaVO3, as a result of increased reactivity in the Na2SO4/NaVO3 system at higher temperatures. Since the magnesium does not move through the amorphous surface layer advancing a crystallization front, large cristobalite regions do not form and spall off, as described in the Na2SO4/NaVO3 environments [7, 15]. Additionally, a sample was tested at 1300 °C in a static environment. The mass change of this sample stabilized at about − 3 mg/cm2, comparable to the mass change in the 1000 °C static environment. This is in contrast to coated samples without salt deposits at 1000 °C which show no mass change during thermal cycling. This mass change could be a result of the time the sample was at lower temperatures, while the furnace was heating up and/or more aggressive corrosion above 1000 °C.

X-ray diffraction data for coated samples after 20 5 h cycles at 1000 °C under a static condition a a typical region of Mo–Si–B coin surface after magnesium sulfate exposure and b regions containing MgMoO4
X-ray diffraction was used to identify the composition and structure of the Mo–Si–B coatings after the conditioning and following the thermal cycling exposure. The majority of the coating is the expected vitreous layer that appears as a broad amorphous maximum in the X-ray diffraction scan. In addition, multiple series of peaks associated with the molybdenum silicide structures of T1 and MoSi2 are detected clearly. Some areas of the coins showed peaks consistent with magnesium molybdate (MgMoO4) as shown in Fig. 4. The formation of MgMoO4 from reaction of MgSO4 with MoO3 was evaluated by free energy calculations [24, 25]. The result of this calculation is presented in Fig. 5 and shows that the reaction between MgSO4 and MoO3 to yield MgMoO4 and SO2 and O2 is favorable thermodynamically over the entire temperature range investigated.

Temperature dependence of the free energy for the reaction of MgSO4 and MoO3 to yield MgMoO4 + SO2 + 1/2O2
The relative scarcity of MgMoO4 and the lack of significant mass change implies that the presence of MoO3 as a reactant with MgSO4 occurs in localized regions of the coating. The SEM image in Fig. 6 shows localized cracks in the silicide layers that likely formed upon cooling and were subsequently filled by flow of the amorphous surface layer during heating. These gaps are a probable source of the molybdenum that forms MoO3 during heating before the glass has sufficient fluidity to cover the gap. This MoO3 can then act as a reactant to form MgMoO4 locally.

SE-SEM image of cracks in the surface coating that have been filled in by glass during thermal cycling at 1000 °C under a static condition
In order to assess the self-healing capability of the coating system, it is necessary to consider the flow velocity of the amorphous surface layer. The works of Papko and Levitskii and Saiz et al. [26, 27] provide the foundation to approximate the self-healing rate. Papko and Levitskii found the viscosity, η of a 70% silica glass at 900 °C, with other oxides including B2O3 accounting for no more than 10% each, to be 105.2 Pas s [26]. Saiz et al. found the spreading velocity, v of a silica glass on molybdenum at 1200 °C to be approximately 90 µm s−1 and related to the capillary number, Ca = ηv/γlv. Using their capillary number of 9 × 10−2 and an interfacial tension, γlv of ~ 1 J m−1 an approximate viscosity of 105 Pas s can be calculated [27]. Given the proximity of these two viscosities, an overestimated spreading velocity of 90 µm s−1 can be used to generalize the flow speed of the glass. Using this, cracks such as A and B in Fig. 6 could be filled in about 10 s. At the same time, upon reheating the formation of MoO3 becomes appreciable above 700 °C where the glass flow rate is slower. As a result, the interval between 700 and 1000 °C which corresponds to about 20 min during heating is available for MoO3 formation. However, due to the highly localized crack regions the impact on the mass change is small.
XRD comparisons between the coated samples exposed in the Na2SO4/NaVO3 environment and MgSO4 environment are presented in Fig. 7 [7]. Both coatings showed the expected peaks corresponding to molybdenum silicide phases such as MoSi2 and Mo5Si3. However, the cristobalite peaks are more pronounced in the coating with Na2SO4/NaVO3 deposit. This coupled with a reduced relative amorphous signal indicates some crystallization of the amorphous coating. These peak differences are in agreement with the expected scarcity of cristobalite nucleation due to the immobility of magnesium in the amorphous surface layer.

X-ray diffraction information of typical surfaces in a magnesium sulfate environment and b Na2SO4/NaVO3 environment [7]
In Fig. 8 X-ray diffraction results are presented for samples exposed in a MgSO4 environment at 1000 °C under a static condition for 20 5 h cycles. For a typical surface region, Fig. 8a reveals small peaks for cristobalite and the expected molybdenum silicides. Figure 8b from isolated regions shows peaks corresponding to cristobalite, molybdenum silicides, and magnesium molybdate. These regions are accompanied by a negligible amorphous signal associated with glass surface. This is likely due to the immobile molten salt causing crystallization on the surface. The structural change and spallation from this transformation are likely the cause of the mass loss in this situation via the same mechanism described earlier [7]. However, a more detailed examination into spallation and cristobalite formation mechanisms is beyond the scope of this paper.

X-ray diffraction patterns of coating surface after exposure to MgSO4 at 1000 °C for 20 5 h cycles under a static condition, a a typical surface region and b a surface region containing magnesium molybdate and cristobalite
Composite Salt
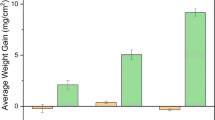
The furnace cycles of the composite salt mixture were only performed in flowing laboratory air. The mass change stabilized after 6–8 iterations of 5 h cycles. The composite salt samples showed little variation in mass change at 900 °C and 1000 °C as indicated in Fig. 9. The samples cycled at 800 °C exhibited lower mass change (less than 1 mg/cm2) compared to the higher temperature exposures. The extent of the corrosion at 900 °C is greater than the separate MgSO4 and Na2SO4/NaVO3 cycles indicating some increased activity from the interaction of the salts and the coating. At 900 °C, the salt composite exhibited twice the mass change as in the Na2SO4/NaVO3 environment. At 1000 °C, the composite only had 0.25 the mass change as the Na2SO4/NaVO3 mixture. The cause of this increased corrosion at a lower temperature will be addressed in future study. A series of extended duration cycles of 20 h each were performed on the 900 °C sample as it showed the most corrosion. After a total of 150 h, no further mass change occurred beyond the 2 mg/cm2 that resulted from 10 five-hour cycles. The lack of significant change in activity at 1000 °C is in agreement with previous research that the presence of magnesium in corrosive systems containing sodium and vanadium suppresses the corrosive activity [14]. This is contrasted by the results of the Na2SO4/NaVO3 system that had a clear increase in corrosion at higher temperatures.

Mass loss of composite salt mixture compared to Na2SO4/NaVO3 and MgSO4 salt environments
X-ray diffraction of the composite salt surface showed the same regions of molybdenum silicides and a distinct cristobalite peak as illustrated in Fig. 10. This indicates that the salt mixture, likely due to the large Na2SO4 portion, causes crystallization on the surface. However, the lower amount of mass change than that with the 1000 °C Na2SO4 salt indicates that less spalling is occurring in this environment than in the environment with only Na2SO4. Some of the peaks potentially correspond to vanadium compounds such as vanadium sulfide (VS) and vanadium silicides (V3Si/V5Si3), which suggests that there may be an intermixing between molybdenum and vanadium. Additionally, some peaks were observed corresponding to MgMoO4. Clear identification of components through X-ray diffraction was complicated by the number of phases present in the system, each with a variety of possible products.

Typical XRD pattern of the coating surface after exposure to the composite salt system after repeated thermal cycling at 900 °C
Conclusions
Magnesium sulfate in isolation has little detrimental effect on Mo–Si–B coatings for refractory metals in normal operating conditions. Unlike sodium sulfate, there is little crystallization of the outer vitreous silica layer. Examinations of the mass changes during repeated thermal cycles and changes in the X-ray diffraction of the surface support this. It is noted that magnesium molybdate does form on the surface of some coatings in localized regions as a result of the reaction of the magnesium sulfate and molybdenum oxide.
Repeated thermal cycling of samples with the composite salt mixture of Na2SO4, NaVO3, MgSO4, CaCl, KCl demonstrated a consistent level of impact on the coating. Following an initial period of corrosion and mass change during exposure to MgSO4, the amorphous coating stabilizes. This mass change remained at about or less than 3.5 mg/cm2 for all tested environments. After investigating the composite salt mixture, isolated sodium salts remain the most deleterious to the coating, but did not induce failures exposing the molybdenum core to oxygen. Against the range of salts and impurities that occur in turbine engines under normal operations, the amorphous aluminoborosilica coating derived from the Mo–Si–B system demonstrates significant and reliable resilience.
References
J. H. Perepezko, Science326, 2009 (1068).
J. H. Perepezko and R. Sakidja, JOM62, 2010 (13).
S. Burk, B. Gorr, M. Krüger, M. Heilmaier and H.-J. Christ, JOM63, 2011 (32).
M. S. Samant, A. S. Kerkar, S. R. Bharadwaj and S. R. Dharwadkar, J. Alloys Compd.187, 1992 (373).
L. Su, O. Lu-Steffes, H. Zhang and J. H. Perepezko, Appl. Surf. Sci.337, 2015 (38).
R. Bianco, R. A. Rapp and K. H. Stern, in Metallurgical and Ceramic Protective Coatings, ed. K. Stern (Springer, Dordrecht, 1996), p. 236.
M. Taylor and J. H. Perepezko, Oxid. Met.87, 2017 (705).
N. S. Bornstein and W. P. Allen, Mater. Sci. Forum251–254, 1997 (127).
N. Eliaz, G. Shemesh and R. M. Latanision, Eng. Fail. Anal.9, 2002 (31).
G. Y. Lai, High Temperature Corrosion and Materials Application, (ASM International, Materials Park, OH, 2007), p. 249.
J. Stringer, Surf. Coat. Technol.108–109, 1998 (1).
R. A. Rapp, Mater. Sci. Eng.87, 1987 (319).
N. S. Jacobson and J. L. Smialek, J. Am. Ceram. Soc.68, 1995 (432).
E. Rocca, L. Aranda, M. Moliere and P. Steinmetz, J. Mater. Chem.12, 2002 (3766).
P. Mukundhan, H. H. Du and S. P. Withrow, J. Am. Ceram. Soc.85, 2002 (1613).
F. A. Rioult, S. D. Imhoff, R. Sakidja and J. H. Perepezko, Acta Mater.57, 2009 (4600).
R. L. Ashbrook, A survey of salt deposits in compressors of flight gas turbine engines, p. NASA TN D-4999 (1969).
N. Bornstein, H. Roth, and R. Pike, Vanadium Corrosion Studies, ONR Contract N00014-89-C-0053 (1993).
J. Sumner, Q. Aksoul, J. Delgado, A. Potter and S. Gray, Oxid. Met.87, 2017 (767).
S. Richter and J. Mayer, Sample Preparation for EPMA - EMAS 2012—10th Regional Workshop on Electron Probe Microanalysis Today—Practical Aspects, Padua, Italy (2012).
J. F. Stebbins and S. Sen, J. Non-Cryst. Solids224, 1998 (80).
M. N. Scheidema and P. Taskinen, Ind. Eng. Chem. Res.50, 2011 (9550).
M. G. Lawson, H. R. Kim, F. S. Pettit and J. R. Blachere, J. Am. Ceram. Soc.73, 1990 (989).
David R. Lide, in CRC Handbook, 84th ed. (CRC Press, Boca Raton, 2003), pp. 5:5–5:60, 5:85–5:86.
O. Kubaschewski, C. B. Alcock and P. J. Spencer, Materials Thermochemistry, (Pergamon Press, Oxford, 1993).
I. A. Levitskii and L. F. Papko, Glass Ceram.67, 2011 (336).
E. Saiz, A. P. Tomsia, N. Rauch, C. Scheu, M. Ruehle, M. Benhassine, D. Seveno, J. de Coninck and S. Lopez-Esteban, Phys. Rev. E76, 2007 (041602).
Acknowledgements
The support of the ONR (N00014-17-1-2575) is gratefully acknowledged. The constructive comments from the reviewer are appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Auchter, E., Taylor, M. & Perepezko, J.H. Resistance of a Mo–Si–B-Based Coating to Environmental Salt-Based Hot Corrosion. Oxid Met 93, 387–399 (2020). https://doi.org/10.1007/s11085-020-09961-y
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-020-09961-y