
Abstract
The importance of manufacturing route of Pt-aluminide bond coatings in determining the life of thermal barrier coatings on Ni-based superalloys is demonstrated. It is shown that bond coatings aluminized by chemical vapor deposition exhibit higher resistance to oxidation in comparison with the pack cementation route, which results in about twofold increase in the life of the thermal barrier coating as determined under cycling oxidation conditions. This difference in behavior is correlated with the initial microstructures of the bond coatings and their effect on thermal stability and oxidation resistance. However, thermal barrier coatings utilizing the two types of bond coatings are found to fail by the same mechanism involving spallation of the top coating due to loss of adhesion between the thermally grown oxide and underlying bond coating. It is concluded that manufacturing by the CVD route with low Al activity decelerates the kinetics of the processes leading to degradation of the coating system in comparison with the pack cementation route with high Al activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aluminizing is an integral part of synthesizing protective coatings for the Ni-based superalloys used to manufacture gas turbine blades. In this process, an Al-rich surface layer is built up to eventually allow the coating to develop a protective oxide layer based on Al2O3 scale. Platinum is added to improve the diffusional stability of the coating and therefore enhance selective oxidation of Al to maintain scale of higher purity, slower growth rate and better adherence to the substrate [1]. In the case of the more advanced thermal barrier coatings, the Al2O3 scale developed by the bond coating also enhances the adhesion between the top ceramic coating (usually yttria-stabilized zirconia) and the superalloy substrate [2].
It is well known that the oxidation resistance of the bond coating and thermal expansion mismatch at the bond coating–oxide interface play important roles in determining the useful life of thermal barrier coatings [3,4,5]. On the other hand, the oxidation behavior of the bond coatings at elevated temperatures is influenced by the composition of the superalloy substrate [6,7,8,9,10,11]. This is because interdiffusion can lead to contamination of the bond coating with superalloy substrate elements such as Ti and transition metals which degrade its oxidation resistance [12]. However, manipulation of the superalloy composition to enhance the oxidation resistance of the bond coating is limited by the stringent requirements of their high-temperature mechanical strength [13]. Another important parameter is the manufacturing route of the bond coating as discussed below.
In practice, Pt-modified aluminide bond coatings are manufactured by two main processes: (1) electrodeposition of a Pt layer about 6–8 μm in thickness on the surface of the turbine blade superalloy followed by a diffusion heat treatment and (2) an aluminizing treatment to form an Al-rich surface layer [14]. Deposition of Pt is usually carried out by wet electroplating techniques based upon dissociation of Pt-rich electrolytes, particularly chlorides [15].
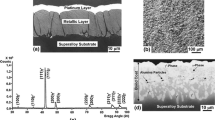
Aluminizing is carried out by two main versions of chemical vapor deposition (CVD). Both versions use vapors of aluminum chloride (AlCl3) as the source of Al. However, they differ in the method by which the vapor is created and how it is transported to the superalloy substrate. In the case of the earliest version of CVD, which is commonly known as the pack cementation process, the vapors are created by a pack enveloping the blades to be coated. Typically, the pack consists of a powder of either Al or an Al alloy, a halide activator usually NH4Cl or NaCl and a dilutent such as aluminum oxide [16,17,18]. As illustrated in the schematic of Fig. 1a [16, 19, 20], the blades are immersed in the pack mixture and contained in an airtight retort which is then heated to a temperature ranging from 800 to 1000 °C. The Al source is created by decomposition of the activator, reaction between the resulting Cl and Al to form AlCl3 vapor and dissociation of the vapor resulting in deposition of Al at the superalloy’s surface. In the other version of CVD, the AlCl3 vapors are created in an external reactor where a precursor gas, usually HCl, is passed over pure Al or an Al alloy and then transported into the reaction vessel as schematically illustrated in Fig. 1b [14, 19, 20]. Therefore, the pack and the superalloy are separated from each other unlike the process shown in Fig. 1a, which results in cleaner bond coatings [19]. Since both processes shown in Fig. 1 are by definition CVD, a clear distinction between them is sometimes made by referring to the process shown in Fig. 1a as closed-retort CVD and that shown in Fig. 1b as an open-retort CVD [21]. However, the standard terminology of pack cementation (Fig. 1a) and CVD (Fig. 1b) will be followed throughout the text.

To meet the demands of continuing advances in gas turbine designs, there is always a need for coating systems with better temperature capabilities to be compatible with the requirement of higher engine efficiency and less fuel consumption. In this regard, yttria-stabilized zirconia top coating of TBC systems remains to represent state-of-the-art technology. Therefore, a great deal of attention is focused on enhancing the temperature capabilities of bond coatings. Toward that objective, the present investigation has been carried out to compare the oxidation resistance of platinum–aluminide bond coatings using the two aluminizing methods shown in Fig. 1, and the respective performance in TBC systems utilizing yttria-stabilized zirconia as top coating and Ni-based superalloy with commercial grade as substrate.
Experimental Procedures
Cylindrical samples (about 10 cm in length and 8 mm in diameter) of the directionally solidified alloy MAR M002DS were used as substrate in the present investigation. Table 1 lists the nominal chemical composition of the alloy as well as the composition of the samples used in the investigation as measured by inductively coupled plasma atomic emission spectroscopy except for the carbon content which was measured by combustion calorimetry. Disk-shaped specimens 8 mm in diameter and 6 mm in thickness were machined from the superalloy samples. All specimens were grit-blasted and then ultrasonically cleaned. This was followed by electroplating about 7-μm-thick layer of Pt and diffusing it into the substrate by 1-h annealing at 1100 °C under argon atmosphere according to industrial practice [22]. Some specimens were then aluminized by pack cementation (Fig. 1a) at 850 °C using a pack of high Al activity corresponding to Al content of 25 wt%. The remainder of the specimens was aluminized by CVD (Fig. 1b) at 1100 °C using low activity Al source with about 17 wt% Al. In the as-deposited condition, both coatings had a total thickness of about 60 μm. The pack cementation coating consisted of: (1) an outer 20-μm layer of β-NiAl + PtAl2, (2) an intermediate 20-μm layer of β-NiAl containing some Pt in solid solution as well as secondary precipitates including carbides and sigma phase and (3) an interdiffusion zone about 20 μm in thickness. In contrast, the CVD coating consisted of outer 30-μm layer of β-NiAl containing Pt in solid solution followed by an interdiffusion zone of about 30 μm in thickness.
Specimens reserved for investigation of the bare bond coatings were annealed for 3 h at 1150 °C under argon atmosphere to develop the respective microstructures. Other bond-coated specimens to be top coated were annealed for 3 h at 1150 °C in air to develop the bond coatings microstructures as well as thin layers of Al2O3 about 1–2 μm in thickness to enhance the adhesion of the top coating during processing. A layer of zirconia-7 wt% yttria about 250 μm in thickness was then deposited by electron-beam physical vapor deposition [2]. Some specimens were reserved for microstructural characterization in the as-deposited condition. Isothermal oxidation tests were carried out on bond-coated specimens in still air at 1000, 1050, 1100 and 1150 °C for up to 100 h at a temperature, and the respective kinetics were determined from weight change measurements. The thermal stability characteristics of the two bond coatings were determined from the effect of exposure time at the extreme temperature of 1150 °C on the microstructure (morphology, structure and composition).
To evaluate the performance of each bond coating in the respective TBC system, bond- and top-coated specimens were exposed at 1150 °C with 24-h cycling periods to room temperature. The average useful lives of the TBC systems were determined from the time required to cause macroscopic spallation of the top coating. In order to characterize the microstructure of the bond coatings near the surface on the finer scale of transmission electron microscopy (TEM), disk-shaped samples about 25 mm in diameter and 8 mm in thickness were bond-coated as described above. Thin-foil specimens transparent to electrons were prepared by the procedure described in Ref. [23] as detailed below.
Various techniques used in detailed microstructural characterization included: (1) scanning electron microscopy combined with energy- and wavelength-dispersive X-ray spectroscopy, (2) X-ray diffraction using Cu–Kα radiation and (3) TEM. To prepare thin-foils for TEM, disks about 3 mm in thickness were sliced from the coated disks and ground on one side down to about 1 mm in thickness and then 3-mm-diameter specimens were punched from the disks. The specimens were electropolished on the superalloy side in a solution consisting of 30% nitric acid in methanol by volume until the specimen was perforated. Final thinning was carried out in an ion beam mill. All foils were examined at an accelerating voltage of 200 keV.
Results and Discussion
Kinetics of Oxidation
The functional dependence of the specific weight gain on time during isothermal oxidation in air for up to 100 h at 1150 °C is shown in Fig. 2 for each bond coating. An initial stage of rapid kinetics indicative of accelerated diffusion of oxygen into the bond coating is observed. Subsequently, a steady state is reached where the reaction becomes controlled by diffusion within the oxide film established in the initial stage. However, it is evident that the pack cementation bond coating is oxidized more rapidly in comparison with the OR-CVD bond coating. A plot of the specific weight gain vs. the square root of exposure time is shown in Fig. 3. Although for each bond coating an initial transient stage is observed, the CVD bond coating are observed to more closely approach parabolic rate behavior (x = k t1/2 where x is the mass gain per unit area, k is the reaction rate constant at a given temperature and t is the exposure time).

Comparative mass gain vs. time of the pack cementation and CVD bond coatings during isothermal oxidation at 1150 °C in still air

A plot of the mass gain shown in Fig. 4 versus the square root of time illustrating the extent of parabolic rate behavior followed by the pack cementation and CVD bond coatings
A plot of the reaction rate constant k versus the reciprocal of temperature T is shown in Fig. 4 as determined from the above parabolic rate relationship and Arrhenius rate equation k = ko exp−Q/RT where ko is a constant, Q is the activation energy of the reaction, R is the universal gas constant and T is the temperature in degrees K. The activation energy of the oxidation reaction in the pack cementation bond coating is estimated to be about 388 kJ/mol in comparison with 407 kJ/mol for the CVD bond coating. It is noted here that both activation energies are comparable with those reported for other Al2O3 formers such as Haynes alloy 214 (400 kJ/mol) [24], Ni–Cr–Al and Ni–Cr–Al–Y alloys (396 kJ/mol) [25], Fe–Cr–Al and Fe–Cr–Al–Y alloys (392 kJ/mol) [26]. The observed higher oxidation resistance of the bond coating processed by CVD is reflected on its performance in TBC systems as demonstrated below.

Functional dependence of the reaction rate constant k on temperature for the pack cementation and CVD bond coatings
Performance of TBC Systems
Consistent with the results of mass gain measurements on bare bond coatings, the thermally grown oxide (TGO) developed by the CVD bond coating in the respective TBC system is found to thicken at a slower rate in comparison with the pack cementation bond coating as demonstrated in Fig. 5. The insets are backscattered SEM images along the cross section of each TBC system illustrating the microstructure of the TGO after 120 h of exposure at 1150 °C. It is observed that the oxide layer of each bond coating consists of a dispersion of Hf-rich oxide pegs expected to be HfO2 in a matrix of Al2O3-based oxide as illustrated in the representative EDS spectra of Fig. 6. However, as shown in Fig. 5, the oxide of the pack cementation bond coating contains higher density of HfO2 with larger particle size enhancing internal oxidation which contributes to its higher oxidation rate and lower thermal stability as further demonstrated later. It is noted here that earlier studies have shown that higher density and excessive growth of oxide pegs of reactive elements such as Hf can be detrimental to oxidation resistance because they enhance internal oxidation causing higher oxide growth rate, deplete the surface in Al and can create high level of localized stress concentrations near the oxide-bond coating interface [6, 27,28,29,30].

Comparative thickening rates of the thermally grown oxide developed by the pack cementation and CVD bond coatings in the respective TBC systems during oxidation at 1150 °C in still air with 24-h cycling periods to room temperature; the insets are backscattered SEM images along TBC cross sections illustrating the microstructure of the oxide developed by each bond coating after 120 h of exposure (5 cycles)

Energy-dispersive X-ray spectra illustrating the elemental compositions of the regions marked Al2O3 and HfO2 in Fig. 10. a Al2O3. b HfO2
A common feature of the TBC systems utilizing the two bond coatings is that failure is observed to occur by spallation of the top coating due to loss of adhesion between the TGO and underlying bond coating. However, the kinetics of the processes leading to failure is found to be accelerated in the case of the TBC system utilizing the pack cementation bond coating as shown later.
Figure 7 shows an example obtained from the TBC system utilizing the pack cementation bond coating to illustrate the failure mode after 8 cycles between 1150 °C and room temperature (192 h of exposure). A photograph illustrating the exposed bond coating surface of the failed specimen is shown in Fig. 7a. Photographs of the bottom and top surface of spalled-off piece of the top coating are shown in Fig. 7b. Backscattered SEM images showing the morphologies of exposed bond coating surface and bottom surface of the spalled-off top coating are shown in Fig. 7c and d, respectively. The bond coating surface is observed to contain islands of Al2O3 containing particles of HfO2 as shown in Fig. 7c. Reference to the inset showing cross section of the pack cementation bond coating in Fig. 5 indicates that those islands correspond to the surface depressions created by internal oxidation. As shown in Fig. 7d, the bottom surface of the top coating is covered with Al2O3 layer containing particles of the Hf-rich oxide. The elemental compositions of the regions marked Al2O3 and HfO2 in Fig. 7c and d are similar to those shown in Fig. 6. Evidently, fracture of the HfO2 pegs near the TGO-bond coating interface has been involved in the failure as indicated by the observation of HfO2 particles at both surfaces shown in Fig. 7c and d.

An example obtained from the TBC system utilizing the pack cementation bond coating to illustrate the failure characteristics of specimen exposed 192 h at 1150 °C (8 cycles). a A photograph of the exposed surface of the bond coating. b Photographs illustrating the bottom and top surfaces of spalled-off piece of the top ceramic coating. c Backscattered SEM image illustrating the microstructure of the exposed bond coating surface. d SEM image illustrating the microstructure of the exposed bottom surface of the top coating
To demonstrate the qualitative similarity between the failure modes of the two TBC systems, Fig. 8 shows backscattered SEM image and corresponding X-ray mapping images illustrating the distribution of various elements at the CVD bond coating surface exposed by failure of the respective TBC system after 17 cycles (408 h of exposure at 1150 °C). Similar to the case of the pack cementation bond coating shown in Fig. 8, the bond coating surface is observed to contain islands of Al2O3 containing particles of HfO2. This is indicated by the observation that Al, O and Hf make up the elemental composition of the islands with very little or no concentrations of other elements. The other elements observed in Fig. 8 make up the elemental composition of the bond coating, which appears to correspond to Ni-rich solid solution of γ-phase as elucidated later.

Backscattered SEM image (BEI) and corresponding X-ray mapping images of various elements at the exposed surface of the CVD bond coating due to failure of the respective TBC system after 408 h of exposure at 1150 °C (17 cycles)
Figure 9 illustrates the useful lives of the two TBC systems included in the investigation as determined from the results of cyclic oxidation tests on 12 specimens each (exposure at 1150 °C in still air with 24-h cycling periods to room temperature). Consistent with the higher oxidation resistance of the CVD bond coating, the life of the respective TBC system is determined to be 420 ± 48 h as compared to 215 ± 42 h for the TBC system utilizing the pack cementation bond coating under cycling oxidation conditions (24-h at 1150 °C). The observed higher oxidation resistance and TBC performance of the CVD bond coating in comparison with the pack cementation bond coating are correlated with differences in their initial microstructures which are reflected on their thermal stability characteristics as illustrated below.

Comparative lives of the TBC systems utilizing the pack cementation and CVD bond coatings as determined from oxidation tests at 1150 °C with 24-h cycling periods to room temperature
Initial Microstructures
It is well known that the major component of Pt-aluminide bond coatings is β-phase with NiAl base composition and B2-type superlattice [1, 14, 16, 17, 19, 31]. However, the exact morphology, structure and composition may differ from one coating to another depending upon the manufacturing route as demonstrated below.
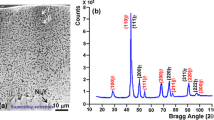
Figure 10 summarizes the microstructural characteristics of the pack cementation bond coating in the as-deposited condition. Typical of coatings processed using high-activity Al pack at low temperatures where the microstructure is developed by inward diffusion [17], the coating consists of three layers of β-phase marked I, II and III in the backscattered SEM image of Fig. 10a with total thickness of about 60 μm. The corresponding X-ray diffraction pattern in Fig. 10b shows that the outermost layer consists of a mixture of β-phase with a ≈ 0.29 nm and PtAl2 (cubic with CaF2-type superlattice and a ≈ 0.59 nm). Figure 10c shows the concentration profiles of Pt and Al along the coating cross section. As can be seen, most of the Pt is concentrated in the outermost layer (layer I in Fig. 10a) with thickness of about 20 μm. It is well known that Pt can substitute for Ni in the β-phase [32], and therefore excess Pt over the solubility limit is precipitated in the form of PtAl2. The second layer marked II in Fig. 10a consists of Pt-depleted β-phase with fine dispersion of secondary precipitates including carbides and intermetallics, particularly MC-type carbides (M stands for metal such as Ta, Ti, Hf and W) and sigma phase. The inner layer marked III is the interdiffusion zone, which consists of coarse aggregate of sigma phase with lamellar morphology and (N–Cr–W)-type composition, and blocky particles of MC-type carbides in a matrix of β-phase. Figure 10d is a dark-field TEM image showing a fine dispersion of PtAl2 particles near the surface of the bond coating. The corresponding convergent beam electron diffraction pattern is consistent with the cubic structure of PtAl2 in [001] orientation, which further confirms the results of X-ray diffraction in Fig. 10b.

Microstructure of the pack cementation bond coating in the as-deposited condition. a Backscattered SEM image along cross section. b Corresponding X-ray diffraction pattern derived from the surface. c Concentration profiles of Pt and Al along the cross section shown in a. d Dark-field TEM image illustrating fine dispersion of PtAl2 particles near the surface. e Corresponding convergent beam electron diffraction pattern in [001] orientation of PtAl2
In contrast with the pack cementation bond coating, the CVD bond coating is observed to consist of two layers marked I and II with total thickness of 60 μm as illustrated in backscattered SEM image of Fig. 11a, which typifies an outward-type coating produced from low activity source at high temperatures [19]. As shown in the X-ray diffraction pattern of Fig. 11b, the outermost coating layer solely consists of β-phase. As can be seen, this layer with a thickness of about 30 μm is relatively free of secondary precipitates. Figure 11c shows the concentration profiles of Pt and Al along the coating cross section where Pt is observed to be uniformly distributed across the entire layer marked I in Fig. 11a. Evidently in this case, the entire Pt content is accommodated in solid solution within the outer layer of β-phase with about the same Al content. Although the interdiffusion zone (layer II in Fig. 11a) is thicker than that of the pack cementation bond coating, it also consists of β-phase containing coarse aggregate of sigma phase and MC-type carbides. The structure of the outermost coating layer is further confirmed by the electron diffraction patterns of Fig. 11d and e, which are consistent with the B2 superlattice of β-phase in [111] and [211] orientations, respectively. As shown below, the differences in microstructures of the two bond coatings are reflected on their thermal stability during exposure at elevated temperatures.

Microstructure of the CVD bond coating in the as-deposited condition. a Backscattered SEM image along cross section. b Corresponding X-ray diffraction pattern derived from the surface. c Concentration profiles of Pt and Al along the cross section shown in a. d and e are electron diffraction patterns derived from the β-phase near the surface in [111] and [211] orientations, respectively
Thermal Stability Characteristics
It is well known that Pt is a key element in improving the oxidation resistance of aluminide coatings due to its recognized role in enhancing selective oxidation of Al, which results in purer scale of slower growth rate [1, 7, 22, 32,33,34,35,36,37,38,39,40,41,42]. Therefore, the concentration profile of Pt as a function of distance from the bond coating surface can serve as a useful index of thermal stability. Figure 13 summarizes the effect of exposure time up to 120 h at the extreme temperature of 1150 °C on the concentration of Pt along cross sections of the two bond coatings included in the investigation. Due to the initial concentration gradient of Pt along the cross section of the pack cementation bond coating (Fig. 10c), Pt is observed to diffuse inward upon thermal exposure as shown in Fig. 12a in order to eliminate the concentration gradient. In contrast, the initial uniform distribution of Pt along the cross section of the CVD bond coating (Fig. 11c) provides higher stability toward inward diffusion of Pt, which maintains its uniform distribution for longer times as illustrated in Fig. 11b. This difference in behavior is also reflected on the effect of exposure time on the microstructure of each bond coating as exemplified below.

Effect of exposure time up to 120 h at 1150 °C on the concentration profiles of Pt along cross sections of the bond coatings. a Pack cementation bond coating. b CVD bond coating
Figure 13 illustrates the effect of 120 h of exposure at 1150 °C on the microstructure of the pack cementation bond coating. A backscattered SEM image along the cross section is shown in Fig. 13a and illustrates the presence of secondary precipitates including ternary sigma phase based upon the Ni–Cr–W composition and MC-type as well as partial transformation of the β-phase into the Ni-rich and Al-depleted γ′-phase with Ni3Al base composition and cubic L12 superlattice. The EDS spectrum of Fig. 13b shows the elemental composition of the sigma phase with Ni, Cr and W as the major elemental constituents. As shown in the EDS spectrum of Fig. 13c, the metallic MC-forming elements include Ta and Ti with smaller concentration of Hf. The bright-field TEM image of Fig. 13d shows a mixture of β- and γ′-phases near the surface as confirmed by the corresponding diffraction patterns of β-phase in [111] orientation (Fig. 13e) and γ′-phase in [111] orientation (Fig. 13f). It is noted here that in such orientation, the β-phase exhibits only body-centered cubic fundamental reflections, while the γ′-phase is distinguished by the presence of superlattice reflections at 1/2-positions of all the fundamental face-centered cubic reflections.

Effect of 120 h of exposure at 1150 °C on the microstructure of the pack cementation bond coating. a Backscattered SEM image along cross section. b Energy-dispersive X-ray spectrum showing the elemental composition of the sigma phase particle marked in a. c Energy-dispersive X-ray spectrum illustrating the elemental composition of the carbide particle marked in a. d Bright-field TEM image illustrating crystals of β- and γ′-phases near the surface. e [111] diffraction pattern of the β-phase in d. f [111] diffraction pattern of the γ′-phase in d
Evidently, the above changes in the microstructure of the pack cementation bond coating are indicative of significant interdiffusion involving inward diffusion of Pt and outward diffusion of substrate elements which can lead to rapid degradation of oxidation resistance and in turn shortening the useful life of the respective TBC system. On the other hand, the microstructure of the CVD bonding coating is found to have higher thermal stability consistent with the data shown in Fig. 12b. This is demonstrated in Fig. 14 which shows its microstructure after 360 h of exposure at 1150 °C. Figure 14a shows a backscattered SEM image along the cross section and also concentration profiles of Ni and Al. As can be seen, the microstructure remains to be cleaner than that of the pack cementation bond coating (Fig. 13a) even after three times the exposure time. The only change observed near the surface is the partial transformation of β-phase into the γ′-phase as confirmed by the concentration profiles of Ni and Al in Fig. 14a and the corresponding X-ray diffraction pattern shown in Fig. 14b. This indicates that the CVD bond coating is more resistant to interdiffusion than the pack cementation bond coating consistent with the observed higher oxidation resistance and performance in TBC systems as demonstrated earlier. However, it is anticipated that with extended exposure and continued outward diffusion of Ni and depletion of Al due to oxidation, the coating layer will ultimately transform into Ni-rich solid solution (γ-phase) as noted earlier in Fig. 8.

Effect of 360 h of exposure at 1150 °C on the microstructure of the CVD bond coating. a Backscattered SEM image along cross section and concentration profiles of Ni and Al. b Corresponding X-ray diffraction pattern derived from the surface
The above observations indicate that aluminizing by the CVD route decelerates the kinetics of the processes leading to degradation of the bond coating, which include oxidation and interdiffusion. On the other hand, bond coatings composed of PtAl2 and beta phase processed by pack cementation such as the present coating have lower thermal stability due to the rapid decomposition of the PtAl2 phase (Fig. 13) in agreement with the results reported earlier for other similar systems [43, 44]. Another important difference between the two bond coatings included in the present investigation is the solubility of Hf in Al2O3. This can have important effect on oxidation resistance [33, 41], which is reflected on the size and density of Hf-rich oxide pegs observed in Fig. 6.
To summarize, the difference in behavior between the two bond coatings can be related to the differences in their starting microstructures particularly the cleanliness of CVD bond coatings and the uniform distribution of Pt, which produces a single beta phase containing Pt in solid solution. Also, it is noted here that earlier studies have shown that a direct correlation exists between the concentration of refractory transition metals in the interdiffusion zone and resistance to interdiffusion between aluminide coatings and superalloy substrates [17, 31]. Therefore, it is possible that the thicker interdiffusion zone of the CVD bond coating contributes to its better performance.
Conclusions
It is concluded from this investigation that the manufacturing route of Pt-modified aluminide bond coatings plays a key role in determining their microstructure, oxidation resistance, thermal stability and performance in TBC systems. Aluminizing by the CVD process provides a microstructure which leads to higher oxidation resistance, thermal stability and performance in TBC systems as compared to aluminizing by the pack cementation process. The advantage of the CVD process is realized in about twofold increase in the useful TBC life as determined from cycling oxidation tests at 1150 °C. Although the adhesion of the thermally grown oxide to both bond coatings represents the weakest link in the two TBC systems, aluminizing by the CVD process decelerates the kinetics of oxidation and interdiffusion which eventually leads to spallation of the top coating.
References
D. K. Das, Progress in Materials Science 58, 2013 (151).
H. Lammermann and G. Kienel, Advanced Materials and Processes 140, 1991 (18).
H. E. Evans, Surface and Coatings Technology 206, 2011 (1512).
N. P. Padture, M. Gell and E. H. Jordan, Science 296, 2002 (280).
A. G. Evans, D. R. Mumm, J. W. Hutchinson, G. H. Meier and F. S. Pettit, Progress in Materials Science 46, 2001 (505).
H. M. Tawancy, Metallography, Microstructure and Analysis 7, 2018 (65).
J. A. Haynes, K. A. Unocic, M. J. Lance and B. A. Pint, Oxidation of Metals 86, 2016 (453).
M. Chieux, C. Duhamel, R. Molins, L. Remy and J.-Y. Guedou, Oxidation of Metals 81, 2014 (57).
H. M. Tawancy, A. I. Mohamed, N. M. Abbas, R. E. Jones and D. S. Rickerby, Journal of Materials Science 38, 2003 (3797).
B. A. Pint, I. G. Wright, W. Y. Lee, Y. Zhang, K. Prubner and K. B. Alexander, Materials Science and Engineering A A245, 1998 (201).
H. M. Tawancy, N. M. Abbas and T. N. Rhys-Jones, Surface and Coatings Technology 54, 1992 (1).
H. M. Tawancy, L. M. Al-Hadhrami, Journal of Engineering for Gas Turbines and Power-Transactions of the ASME, 133, Article No. 042101 (2011).
C. T. Sims, Advanced Materials and Processes 139, 1999 (32).
J. S. Smith, D. H. Boone, Platinum modified aluminides-present status, 1990 International Gas Turbine and Aeroengine Congress and Exposition, ASME Paper No. 90-GT-319, The American Society of Mechanical Engineers, New York, 1990.
C. P. K. Rao and D. C. Trivedi, Coordination Chemical Reviews 249, 2005 (613).
G. W. Goward and L. W. Cannon, Journal of Engineering for Gas Turbines and Power-Transactions of ASME 110, 1988 (150).
P. C. Patnaik, Materials and Manufacturing Processes 4, 1989 (133).
S. R. Levin and R. M. Caves, Journal of the Electrochemical Society 121, 1974 (121).
B. M. Warnes and D. C. Punola, Surface and Coatings Technology 94–95, 1997 (1).
B. M. Warnes, Surface and Coatings Technology 146–147, 2001 (7).
H. O. Pierson, Handbook of Chemical Vapor Deposition: Principles, Technology and Applications, 2nd ed, (Noyes Publications, Norwich, 1999), p. 110.
H. M. Tawancy, L. M. Al-Hadhrami, Journal of Engineering for Gas Turbines and Power-Transactions of ASME, 132, Article No. 022103 (2010).
P. J. Goodhew, J. Hymphreys and R. Beanland, Electron Microscopy and Analysis, 3rd ed, (Taylor and Francis, New York, 2001), p. 21.
H. M. Tawancy, Oxidation of Metals 37, 1992 (143).
H. M. Tawancy, Oxidation of Metals 86, 2016 (371).
T. A. Ramanarayanan, R. Ayer, R. Petkovic-Luton and D. P. Leta, Oxidation of Metals 29, 1988 (445).
B. A. Pint, J. A. Haynes and T. M. Besmann, Surface Coatings and Technology 204, 2010 (3287).
J. A. Haynes, B. A. Pint, K. L. More, Y. Zhang and I. G. Wright, Oxidation of Metals 58, 2002 (513).
B. A. Pint, K. L. More and I. G. Wright, Oxidation of Metals 59, 2003 (257).
R. Pendse and J. Stringer, Oxidation of Metals 23, 1985 (1).
P. Tomaszewicz and G. R. Wallwork, Reviews of High temperature Materials 5, 1982 (49).
B. Gleeson, N. Mu and S. Hayashi, Journal of Materials Science 44, 2009 (1704).
R. Streiff and D. H. Boone, Journal of Materials Engineering and Performance 22, 2013 (2801).
B. A. Pint, J. A. Haynes, K. L. More, J. H. Scheibel, Y. Zhang and I. G. Wright, in Superalloys 2008, eds. R. C. Reed, K. A. Green, T. P. Gabb, M. G. Fahrmann, E. S. Huron and S. A. Woodard (The Minerals, Metals and Materials Society, Warrendale, 2008), p. 641.
H. M. Tawancy, N. M. Abbas and M. O. Aboelfotoh, Journal of Materials Science 43, 2008 (2978).
J. A. Haynes, B. A. Pint, K. L. More, Y. Zhang and I. G. Wright, Oxidation of Metals 58, 2002 (513).
H. M. Tawancy, N. Sridhar, N. M. Abbas and D. S. Rickerby, Scripta Metallurgica et Materialia 33, 1995 (1431).
H. M. Tawancy, N. M. Abbas and T. N. Rhys-Jones, Surface Coatings and Technology 49, 1991 (1).
J. Schaeffer, G. M. Kim, G. H. Meier and F. S. Pettit, in The Role of Active Elements in the Oxidation Behavior of High Temperature Metals and Alloys, ed. E. Lang (Elsevier, Amsterdam, 1989), p. 231.
M. S. Farrell and D. H. Boone, Surface Coatings and Technology 32, 1987 (69).
R. Streiff, O. Cerclier and D. H. Boone, Surface and Coatings Technology 32, 1987 (111).
M. R. Jackson and J. R. Rairden, Metallurgical Transactions A 8A, 1977 (1697).
M. Gobel, A. Rahmel, M. Schutze, M. Schorr and W. T. Wu, Materials at High Temperatures 12, 1994 (301).
J. Angenete, K. Stiller and E. Bakchinova, Surface and Coatings Technology 176, 2004 (272).
Acknowledgements
The continued support of King Fahd University of Petroleum and Minerals is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tawancy, H.M. Influence of Manufacturing Route on the Oxidation Resistance of Platinum-Modified Aluminide Bond Coatings and Their Performance in Thermal Barrier Coatings Deposited on a Ni-Based Superalloy. Oxid Met 90, 435–452 (2018). https://doi.org/10.1007/s11085-018-9846-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-018-9846-5