
Abstract
Cu-based chalcogenide materials have attracted a great deal of attention due to their promising optoelectronic properties. The density functional theory (DFT) framework is used in order to estimate the optical and electronic properties of XCu2GeQ4 (X = Ba, Sr and Q = S, Se). We report the optical and electronic properties of Cu-based chalcogenides in this study, which have narrow and direct bandgap materials. The calculated energy bandgap of quaternary chalcogenide materials decreases in the following sequence: SrCu2GeS4 (0.697 eV), BaCu2GeS4 (0.667 eV), BaCu2GeSe4 (0.378 eV), and SrCu2GeSe4 (0.195 eV). This reduction in energy bandgaps shows significant effect of changing dopants on electronic and consequently optical properties of XCu2GeQ4 (X = Ba, Sr and Q = S, Se). The optical characteristics of these materials are investigated in order to explore their potential for optoelectronic applications. However, other materials are emerging as contenders for solar cells, which operate from UV to infrared regions. Initially in infrared region, we can note a redshift in the maximum absorption of incident photons from \({\varepsilon }_{2}(\omega )\) plots in the following sequence: BaCu2GeS4 (1.52 eV), SrCu2GeS4 (1.50 eV), BaCu2GeSe4 (1.30 eV), and SrCu2GeSe4 (0.93 eV). The approximated values of reflectivity, R(ω) are plotted against incident photon energy from 0 to14 eV. Thus, the reflectivity is approximately below 50% before E ≈ 12.0 eV and then increased to 70% reflection at ~ 13.0 eV. Based on calculated thermoelectric properties, these chalcogenides are promising thermoelectric materials. The ZT values of XCu2GeQ4 (X = Ba, Sr and Q = S, Se) decreased in the following sequence: SrCu2GeSe4 (2.6), BaCu2GeSe4 (1.85), SrCu2GeS4 (1.01) and BaCu2GeS4 (0.94). Hence, we believe our findings propose promising materials for anti-reflecting coating layers in optoelectronic technology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Scientists are extremely encouraged by the insufficiency of the natural energy sources in order to develop novel renewable energy sources. Quaternary sulfides are a significant family of inorganic materials with exciting application potential for renewable energy devices (Zhang et al. 2018). Significant advancements have been made in the synthesis and characterizations of quaternary sulfides during the past few decades (Brant et al. 2014; Chen et al. 2018; Li et al. 2012; Zhang et al. 2016). The transition metal elements (Cu, Ag, Zn, Cd, and Hg) and main group elements (In, Ga, Sn, Ge, As, and Sb) typically make up the frameworks of these sulfides/selenides. Researchers are drawn to quaternary sulfides due to enhanced probability for discovering novel functional materials because of diversity in their composition and structure. These quaternary chalcogenide materials are promising materials for various device applications such as radiation detection, catalysts, non-linear optics, and solar energy conversions.
The global energy crisis can be addressed by using thermoelectric (TE) materials, because they are efficient alternatives for fossil fuels (He et al. 2013; Snyder and Toberer 2008). The performance of TE materials is evaluated by using the figure of merit \(ZT=\sigma {S}^{2}T/\kappa\), where κ, T, S, and σ represents thermal conductivity, temperature, the Seebeck coefficient, and electrical conductivity, respectively. Thermal conductivity includes both phonon (\({\kappa }_{l}\)) and electronic (\({\kappa }_{e}\)) components. Improving ZT by the subjective tuning of these parameters is a challenging task, which is due to the close relationship between \({\kappa }_{e}\), σ, and S. Thus, scientists have employed several strategies, which include nanostructures (Gao et al. 2020; Wang et al. 2020), power factor (\({S}^{2}\sigma\)) enhancement by band structure engineering (Huang et al. 2017; Li et al. 2016b; Mobaraki et al. 2018; Pei et al. 2011; Zhao et al. 2013), and the weakening of thermal conductivity by alloy defects (Pei and Morelli 2009; Poudel et al. 2008; Shen et al. 2017), in order to improve the ZT values. Furthermore, the ZT values can also be considerably improved by discovering novel TE materials with low thermal conductivity and high-power factors (Ding et al. 2018; Llanos et al. 2001; Wang et al. 2018).
Scientists have developed an enormous amount of interest in the quaternary metal chalcogenides AB2XY4 (A = Sr, Ba; B = Cu, Ag; X = Sn, Ge, Si; Y = Se, S) in recent decades, which is due to their prospective photovoltaic and optoelectronic applications (Assoud et al. 2005; Ding et al. 2019; Hong et al. 2016; Jiahong Kuo et al. 2019; Llanos et al. 2001; Nian et al. 2017; Shin et al. 2016; Teymur et al. 2018; Xiao et al. 2017; Zhu et al. 2017). BaAg2SnSe4, BaCu2GeSe4, SrCu2GeSe4, and BaCu2SnSe4 were anticipated to have high power factors among these compounds. Nian et al. (2017) explored the optical characteristics of BaCu2XY4 (X = Sn, Si, Ge; Y = S, Se). It was revealed that the orthorhombic BaCu2SnSe4 absorber shows exceptional photovoltaic characteristics, which is based on systematic theoretical and experimental investigations (Xiao et al. 2017). Four compounds according to Kuo et al., which include (BaAg2SnSe4, BaCu2SnSe4, SrCu2GeSe4 and BaCu2GeSe4) more significantly show low thermal conductivities (0.48–0.34 Wm−1 K−1 at 673 K) (Jiahong Kuo et al. 2019). The crystalline BaCu2SnSe4 and SrCu2GeSe4 have the same structures and belong to the same space group, which is Ama2. There is a very limited amount of literature that is available on the TE characteristics of these chalcogenide materials, but their ZT values and underlying electronic transport characteristics are still ambiguous. Metal chalcogenides have evolved as fascinating optoelectronic materials during the last few decades, which is due to their captivating physiochemical characteristics and structural diversities (Guo et al. 2017; Li et al. 2016a, 2017; Liang et al. 2017; Lin et al. 2017; Ma et al. 2014; Mei et al. 2014). Tetrahedral units, which have been verified as active units, in order to make a primary contribution for the nonlinear optical (NLO) effect, are present in most of them. Chalcogens and group 14 elements, which include Sn, Ge, and Si are usually coupled in order to generate the deformed [MIVQ4] tetrahedra (Brant et al. 2014; Morris et al. 2013; Rosmus et al. 2014; Wu et al. 2015a, b; Zhen et al. 2016). Numerous novel metal chalcogenides with appealing NLO performances and the [MIVQ4] tetrahedra have been reported in the literature to date, such as Na2BaSnS4 (Wu et al. 2016), β-K2Hg3Ge2S8 (Liao et al. 2003), Na2Hg3Ge2S8 (Kuei et al. 2016), and Li2CdGeS4 (Lekse et al. 2009). In literature, various studies are available that report thermoelectric, half-metallic, structural, and optical properties of different materials using DFT calculations (Boochani et al. 2010; Lahourpour et al. 2019; Yeganeh et al. 2020, 2022).
In this article, GGA + U based DFT calculations have been employed to investigate structural, electronic, optical, and thermoelectric properties for chalcogenide materials XCu2GeQ4 (X = Ba, Sr and Q = S, Se). The significant effect of replacing Ba with Sr and S with Se is evident from the calculated results (structural, electronic, optical and thermoelectric). Band gap engineering is possible due to these replacements; hence, other properties can also be tuned. From optical properties, we can note that SrCu2GeSe4 is the most promising candidate for solar applications. However, BaCu2GeS4 is the most promising candidate for thermoelectric device applications. Our calculated results show that these compounds are promising candidates for photovoltaic and thermoelectric device applications. We believe that we got improved results in this study compared to previous studies.
2 Methodology
The ground state properties of metal chalcogenides are reported in this paper, which are based on the density functional theory (DFT) (Abbas et al. 2020b) calculations using the full potential linearized augmented plane wave (FP-LAPW) (Abbas et al. 2020a) procedure that was executed in the WIEN2K code (Abbas et al. 2022b, 2023a). The generalized gradient approximation (GGA) with addition of Hubbard correction (\(U=7.0\)) is used, because they seem to be crucial in regards to handling correlated electron systems in order to calculate exchange and correlation potentials (Azam et al. 2018). The unit cell space is generated in the FP-LAPW approach by combining muffin-tin spheres, which are atomic spheres, and the interstitial region (IR). The potential is assumed to be constant in the interstitial region, but it is assumed to be a spherically symmetric as a function of r ≤ RMT in the muffin–tin spheres. It is possible to prevent current leakage from the atoms’ core states as well as guarantee that there is no overlapping of atomic spheres during the convergence of total energy by selecting appropriate muffin-tin radii for various elements. The Schrodinger wave equation (SWE) is solved in the muffin-tin spheres and the interstitial region using the plane wave function and the radial wave function’s time spherical harmonics.
Here, Fermi wave vector (\(\overrightarrow{k}\)) and plane wave’s amplitude at \(\overrightarrow{k}\) (\({V}_{k}\)) is used to calculate spherical potential.
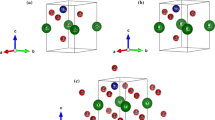
The wave functions are expanded by choosing the maximum angular momentum (\({l}_{max}\)) as 10 in the muffin-tin radii, and the cutoff energy is specified to be − 6.0 Ry. We choose the convergence value for the force to be 0.0001 Ry, and we choose total energy to be 0.001 Ry/au. The values of \({G}_{max}=12\) and \({R}_{MT}\times {K}_{max}=7.0\) are set to be cutoff values for plane wave in the first Brillouin zone, and all calculations are performed using a dense k-mesh of 500 points. The largest reciprocal lattice vector is illustrated by \({K}_{max},\) and the smallest muffin-tin radius is indicated. The relaxed geometry was achieved by reducing the forces on all the atoms to less than 1 mRy/au for all calculations. The optimized unit cell structures for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are presented in Fig. 1a–d.

Optimized unit cell structures of a BaCu2GeS4, b BaCu2GeSe4, c SrCu2GeS4, and d SrCu2GeSe4
3 Results and discussion
3.1 Structural properties
The structural stability of XCu2GeQ4 (X = Ba, Sr and Q = S, Se) is evaluated in this section by calculating their structural properties. The structural stability for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) is investigated by probing their essential structural parameters such as ground state energy \({E}_{0}\), Bulk modulus (B), pressure derivate of Bulk modulus (\({B}{\prime}\)) and optimized volume (V). The optimized lattice parameters (a & c) are also calculated as these structures crystallize in hexagonal symmetry. The ground state properties for studied chalcogenides has been evaluated using total energies corresponding to optimized volume. For XCu2GeQ4 (X = Ba, Sr and Q = S, Se), Birch-Murnaghan equation of state (EOS) has been employed to perform volume optimization (Abbas et al. 2023b).
Here, optimized volume (V), ground state energy (\({E}_{0}\)), Bulk modulus (B), and first pressure derivative of Bulk modulus (\({B}{\prime}\)) are used to calculate total energy (\({\mathrm{E}}_{\mathrm{tot}}\)) using above equation. The computed energy spectra as a function of unit cell volume for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are shown in Fig. 2. The compound’s ground state energy is represented by the minimum of the E-V curve and the lattice parameters corresponding to this point are equilibrium lattice parameters. The calculated values of optimized parameters such as lattice parameters (a & c), ground state energy (\({E}_{0}\)), Bulk modulus (B), pressure derivate of Bulk modulus (\({B}{\prime}\)) and optimized volume (V) are presented in Table 1. It is evident from the calculated ground state energies that BaCu2GeSe4 (due to lowest value of \({E}_{0}\)) is the most stable compound compared to other studied chalcogenide materials.

Optimized energy vs volume plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se)
3.2 Electronic structure
The energy band structure and density of states (DOS) provide deep insights in regards to the optoelectronic properties of crystalline materials. Energy band structures, partial density of states (PDOS), and total density of states (TDOS) are explained in this section of the manuscript in order to discuss the electronic properties of XCu2GeQ4 (X = Ba, Sr and Q = S, Se). The origin of sub energy bands and an explanation regarding the influence of various angular momentum components are provided below.
3.2.1 Band structure
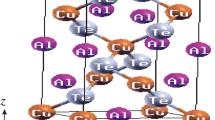
A comprehensive analysis of the energy band structures for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) shows that these Cu based chalcogenides are semiconductor compounds. A comparison between the effect of the cation (Ba, Sr) and anion (S, Se) replacements on the electronic behavior of Cu based chalcogenides was investigated. We demonstrated how the substitution of Sr for Ba and Se for S affected the band structures of XCu2GeQ4 (X = Ba, Sr and Q = S, Se) by analyzing TDOS and PDOS. The calculated energy band dispersions for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are presented in Fig. 3a–d from − 3.0 to 3.0 eV. Zero-energy also corresponds to the Fermi level (\({E}_{F}\)) in the energy band structures. The energy band structures for the aforesaid compounds have been investigated along the high symmetric axis of the irreducible Brillouin zone (IBZ). The conduction band minima (CBM) is located at a high symmetric point Γ for all compounds. Furthermore, the valence band maxima (VBM) can also be spotted at Γ from Fig. 3, which confirms direct energy bandgaps of 0.25, 0.38, 0.18, and 0.31 in BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4, and SrCu2GeSe4, respectively. The bandgaps of these materials are tuned by replacing Ba with Sr and S with Se because the atomic radius of Ba is larger than Sr and same is for Se and S. The presence of more electrons in the elements offer more electronic states (both in valence and conduction bands) that results in the tuning of energy bandgaps.

Calculated energy band structures for a BaCu2GeS4, b BaCu2GeSe4, c SrCu2GeS4 and d SrCu2GeSe4
3.2.2 Density of state
The density of state (DOS) calculations are used in order to investigate the genesis of the different electronic states in the energy band structures, because the electronic characteristics are mostly dependent on the conduction and valence bands of the material. The spectra for TDOS and PDOS for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) were also computed, which are shown in Figs. 4, 5, 6, and 7. Information regarding the number of energy states at a specific energy can be obtained from the calculated spectra of the TDOS for each compound.

Calculated density of states (DOS) for BaCu2GeS4

Calculated density of states (DOS) for BaCu2GeSe4

Calculated density of states (DOS) for SrCu2GeS4

Calculated density of states (DOS) for SrCu2GeSe4
The calculated spectra of the TDOS and PDOS for BaCu2GeS4 are presented in Fig. 4. We can explain the valence band of BaCu2GeS4 in two portions, which include (i) − 3.0 to − 1.5 eV and (ii) − 1.5 to 0 eV. Significant contributions come from the Cu-atoms in portion (i), but small contributions from the Ge- and S-atoms are also present. It is evident from Fig. 4 that significant contributions in the valence band in the PDOS come from the Cu[\({3d}^{9}\)] and S[\({3p}^{4}\)] electrons. However, small contributions from the Ba[\({5p}^{6}\)], Ba[\({4d}^{10}\)] and Ge[\({4p}^{2}\)] electrons are also present. Significant contributions come from the Cu-atoms in portion (ii), but small contributions from the S-atoms are also present. From PDOS of Fig. 4, that significant contributions in the valence band come from the Cu[\({3d}^{9}\)] electrons. However, small contributions from the S[\({3p}^{4}\)] electrons are also present. Significant contributions come from the Ba-atoms in the conduction band, but small contributions from the Ge- and S-atoms are also present. From PDOS of Fig. 4, significant contributions in the conduction band come from the Ba[\({5d}^{6}\)] electrons. However, small contributions from the S[\({3p}^{4}\)] and Ge[\({4p}^{2}\)] electrons are also present. We can observe that near Fermi level region of valence band contains dense bands due to hybridization of S[\({3p}^{4}\)], Ge[\({4p}^{2}\)], Cu[\({3d}^{9}\)] and Ba[\({5d}^{6}\)] sub-energy levels.
The calculated spectra of TDOS and PDOS for BaCu2GeSe4 are presented in Fig. 5. Significant contributions come from the Cu-atoms in the valence band, but small contributions from the Ge- and Se-atoms are also present. From the PDOS of Fig. 5, the significant contributions in the valence band come from the Cu[\({3d}^{9}\)] electrons. However, small contributions from the Se[\({4p}^{4}\)] and Ge[\({4p}^{2}\)] electrons are also present. Significant contributions come from the Ba-atoms in the conduction band, but small contributions from the Ge- and Se-atoms are also present. From the PDOS of Fig. 5, the significant contributions in the conduction band come from the Ba[\({5d}^{6}\)] electrons. However, small contributions from the Se[\({4p}^{4}\)] and Ge[\({4p}^{2}\)] electrons are also present. We can observe that near Fermi level region of valence band contains dense bands due to hybridization of Se[\({4p}^{4}\)], Ge[\({4p}^{2}\)], Cu[\({3d}^{9}\)] and Ba[\({5d}^{6}\)] sub-energy levels.
The calculated spectra of the TDOS and PDOS for SrCu2GeS4 are presented in Fig. 6. Significant contributions come from the Cu-atoms in the valence band, but small contributions from the Ge- and S-atoms are also present. From the PDOS of Fig. 6, the significant contributions in the valence band come from the Cu[\({3d}^{9}\)] electrons. However, small contributions from the S[\({3p}^{4}\)] and Ge[\({4p}^{2}\)] electrons are also present. Significant contributions come from the Sr- and Ge-atoms in the conduction band, but small contributions from the S-atoms are also present. From the PDOS of Fig. 6, the significant contributions in the conduction band come from the Sr[\({3d}^{10}\)] and Ge[\({4p}^{2}\)] electrons. However, small contributions from the S[\({3p}^{4}\)] electrons are also present. We can observe that near Fermi level region of valence band contains dense bands due to hybridization of S[\({3p}^{4}\)], Ge[\({4p}^{2}\)], Cu[\({3d}^{9}\)] and Sr[\({3d}^{10}\)] sub-energy levels.
The calculated spectra of the TDOS and PDOS for SrCu2GeSe4 are presented in Fig. 7. Significant contributions come from the Cu-atoms in the valence band, but small contributions from the Ge- and Se-atoms are also present. From the PDOS of Fig. 7, the significant contributions in the valence band come from the Cu[\({3d}^{9}\)] electrons. However, small contributions from the Se[\({4p}^{4}\)] and Ge[\({4p}^{2}\)] electrons are also present. Significant contributions come from the Sr- and Ge-atoms in the conduction band, but small contributions from the Se-atoms are also present. From the PDOS of Fig. 7, the significant contributions in the conduction band come from the Sr[\({3d}^{10}\)] and Ge[\({4p}^{2}\)] electrons. However, small contributions from the Se[\({4p}^{4}\)] and electrons are also present. We can observe that near Fermi level region of valence band contains dense bands due to hybridization of Se[\({4p}^{4}\)], Ge[\({4p}^{2}\)], Cu[\({3d}^{9}\)] and Sr[\({3d}^{10}\)] sub-energy levels.
3.3 Optical properties
Optical properties play a significant role in regard to characterizing and significantly understanding the optical response of the present Cu-based chalcogenides. Materials undergo electronic transitions to unoccupied states in the CB from their occupied states in the VB when photons with a suitable frequency (\(E=h\upsilon\)) strike the material. Meaningful information that is related to the optical characteristics of crystalline materials is provided by these types of transitions (Eibler and Neckel 1980). Figure 8 displays the optical dispersion results, which illustrate how the dielectric function is impacted by the interband/intraband transitions. Two categories of interband transitions exist, which include direct and indirect electronic transitions, and these transitions are more favorable in semiconductor materials. The electronic transitions between sub-energy levels of valence band and unoccupied sub-energy bands of conduction band are responsible for various peaks in absorption spectra. When an electron undergoes transition from VBM to CBM at the same high symmetric point, it is known as a direct electronic transition. Also, if the high symmetric point is different, it is then called an indirect electronic transition. The following quantities are generally investigated, which include dielectric constant \(\varepsilon (\omega )\), optical loss \(L(\omega )\), refractive index \(n(\omega )\), absorption coefficient \(I(\omega )\), and reflectivity \(R(\omega )\) in order to comprehend the optical responses and transitions (Abbas et al. 2022b; Anisimov et al. 1991; Blaha et al. 2001). The dielectric function \(\varepsilon (\omega )\) explains the photon-electron interaction and the system’s reaction to electromagnetic radiation. The dielectric function \(\varepsilon (\omega )\) is the sum of the real \({\varepsilon }_{1}(\omega )\) and imaginary \({\varepsilon }_{2}(\omega )\) components (Abubakr et al. 2023). The dielectric function \(\varepsilon (\omega )\) can be formulated as the equation that is provided below.

The calculated a \({\varepsilon }_{1}(\omega )\), b \({\varepsilon }_{2}(\omega )\), c \(n(\omega )\) and d \(K(\omega )\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se)
The ability of the materials to absorb incident photons can be explained by using the investigated imaginary part \({\varepsilon }_{2}\left(\omega \right)\) of the complex dielectric function. However, optical reflections can be explained by using the investigated real part \({\varepsilon }_{1}\left(\omega \right)\). We can express \({\varepsilon }_{2}\left(\omega \right)\) with the equation that is provided below.
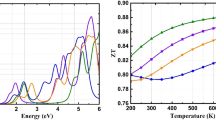
The calculated values of \({\varepsilon }_{2}\left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 8a. Various peaks of \({\varepsilon }_{2}\left(\omega \right)\) resulted from the electronic transitions between the conduction band, which is the unoccupied states, and valence band, which is the occupied states. Peaks for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) reach their highest points after a sharp increase at lower energy. It is evident that these compounds absorb the maximum number of incident photons in infrared and lower UV regions. Our investigated results show that BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 have the highest absorption power at approximately 5.42, 1.19, 5.77 and 0.72 eV, respectively. Their wide absorption spectrum make them ideal materials for tunable device applications.\({\varepsilon }_{1}\left(\omega \right)\) can be calculated using the following Kramer’s–Kronig transformation and the calculated values of \({\varepsilon }_{2}\left(\omega \right)\).
The calculated values of \({\varepsilon }_{1}\left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 8b. Information regarding the dispersion of incident photons can be obtained from the calculated \({\varepsilon }_{1}\left(\omega \right)\). We can note that the maximum dispersion of the incident photons occurs between 1.0 and 2.0 eV for XCu2GeQ4 (X = Ba, Sr and Q = S, Se). We can determine the static values of \({\varepsilon }_{1}\left(0\right)\) at zero frequency or a constant electric field for XCu2GeQ4 (X = Ba, Sr and Q = S, Se). The static values of \({\varepsilon }_{1}\left(0\right)\) are approximately 9.71, 13.87, 9.68 and 17.8 for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively. There is a sharp decrease in the spectra, and the values of \({\varepsilon }_{1}\left(\omega \right)\) go beyond zero at around 6.71, 5.93, 6.68 and 5.36 eV for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively, which corresponds to the plasmon frequency.
The dispersion of the incident photons and the light-matter interaction can be explained by calculating the ratio of the speed of light in a vacuum to the speed of light in a medium, which is known as the refractive index \(n\left(\omega \right)\). The calculated values of \({\varepsilon }_{1}\left(\omega \right)\) and \({\varepsilon }_{2}\left(\omega \right)\) can be used to calculate \(n\left(\omega \right)\).
The calculated values of \(n\left(\omega \right)\) are plotted against energy (0–14 eV), which are shown in Fig. 8c. Information regarding the dispersion of the incident photons can also be obtained from the calculated \(n\left(\omega \right),\text{ which is like }{\varepsilon }_{1}\left(\omega \right)\). We can see that the maximum dispersion of the incident photons occurs between 1.0 and 2.0 eV for XCu2GeQ4 (X = Ba, Sr and Q = S, Se). We can also see the static values of \(n\left(0\right)\) for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) at zero frequency or a constant electric field. The static values of \(n\left(0\right)\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 is approximately 3.01, 3.75, 3.02 and 4.31, respectively. There is a sharp decrease in the spectra, and the values of \(n\left(\omega \right)\) go beyond unity at around 8.5, 7.84, 8.63 and 7.09 eV for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively, which corresponds to plasmon frequency. The materials show a metallic behavior below this value.
Absorption of the incident photons can also be explained by using the extinction coefficient \(K\left(\omega \right)\), which is analogous to \({\varepsilon }_{2}\left(\omega \right)\). We can express \(K\left(\omega \right)\) as
The calculated values of \({\varepsilon }_{2}\left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 8d. Various peaks of \(K\left(\omega \right)\) resulted from the electronic transitions between the conduction band, which is the unoccupied states, and the valence band, which is the occupied states. The peaks for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) reach their highest points after a sharp increase at lower energy. It is evident that these compounds absorb the maximum number of incident photons in infrared and lower UV regions. The highest values of \(K\left(\omega \right)\) occur at approximately 7.14, 6.54, 7.2 and 0.99 eV for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively. It is evident from Our investigated results show that BaCu2GeS4/SrCu2GeS4 have the highest absorption power in the infrared region. However, BaCu2GeSe4/SrCu2GeSe4 has the highest absorption power in the lower UV region.
The reflection coefficient \(R(\omega )\) is the proportion of reflected photons to the incident photons. We can express \(R\left(\omega \right)\) with the equation below.
The calculated values of \(R\left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 9a. We can see that the maximum dispersion of the incident photons occurs around 13.0 eV for XCu2GeQ4 (X = Ba, Sr and Q = S, Se). We can also see the static values of \(R\left(0\right)\) for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) at zero frequency or a constant electric field. The static values of \(R\left(0\right)\) are approximately 0.26, 0.33, 0.28 and 0.39 for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively. And the highest reflectivity occur in upper UV region (~ 13 eV). These materials can be used as reflecting coatings in the upper UV region.

The calculated a \(R(\omega )\), b \(I(\omega )\), c real \(\sigma (\omega )\) and d \(L(\omega )\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se)
The maximum penetration length of the incident photon in the material before its complete absorption can be explained by using the absorption coefficient \(I\left(\omega \right)\). We can use the expression below to calculate \(I\left(\omega \right).\)
The calculated values of \(I\left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 9b. We can see that there are no peaks initially, and the peaks then start to appear from approximately 073, 0.42, 0.72 and 0.16 eV for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively, which is known as the threshold value of \(I\left(\omega \right)\). The peaks for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) reach their highest points by a showing gradual increase. It is evident that the penetration length of the incident photons increases by increasing the photon energy. The highest value of absorption coefficient for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 occur at approximately 13.56, 13.59, 13.61 and 6.19 eV, respectively. Our investigated results show that BaCu2GeSe4 has the highest values for the absorption coefficient.
We can use the expression below to calculate \(\sigma \left( \omega \right).\)
The calculated values of \(\sigma \left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 9c. We can see that there are no peaks initially, and the peaks for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 then start to originate from approximately 0.55, 0.30, 0.56 and 0.18 eV, which is known as the threshold value of \(\sigma \left(\omega \right)\). The peaks for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) reach their highest points after a sharp increase. It is evident that these materials show maximum optical conduction around 6.82, 5.01, 5.83 and 4.44 eV for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4, respectively. Our investigated results show that XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are potential candidates for solar cell applications in the infrared and UV regions.
The loss of rapidly moving electrons by the material can be explained by using the energy loss function \(L\left(\omega \right)\). We can use the expression below to calculate \(L\left(\omega \right).\)
The calculated values of \(L\left(\omega \right)\) are plotted against energy (0–14 eV), which is shown in Fig. 9d. There are notably no peaks initially, and the peaks for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 then start to originate from approximately 0.76, 0.39, 0.75 and 0.16 eV, respectively, which is known as the threshold value of \(L\left(\omega \right)\). The peaks for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) reach their highest points by showing a gradual increase. The point from where peaks start to fall is the point of plasmon resonance for the studied compounds. The energy loss function shows the exact opposite behavior to that of \({\varepsilon }_{2}\left(\omega \right)\). The tuning in optical properties comes from the tuning of energy bandgaps of these materials by replacing Ba with Sr and S with Se because the atomic radius of Ba is larger than Sr and same is for Se and S. The presence of more electrons in the elements offer more electronic states (both in valence and conduction bands) that results in the tuning of energy bandgaps. The optical properties are also affected significantly as they are connected with energy bandgap of the studied materials.
4 Thermoelectric properties
In the recent past, scientists’ interest in thermoelectric (TE) materials has increased significantly due to the worldwide energy crisis. The thermoelectric effect is a phenomenon that describes the direct conversion of temperature gradient into electrical energy. In this section, the calculated thermoelectric (TE) properties for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are discussed to get an insight of their potential for TE device applications. The thermoelectric characteristics of the chalcogenide materials XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are estimated using the rigid band approximation and the conventional Boltzmann kinetic transport theory (Abbas et al. 2022a, c). To characterize the TE behavior of aforementioned compounds, the basic TE features like power factor (\(PF=\sigma {S}^{2}\)), figure of merit\(\left(ZT=\frac{{S}^{2}T}{\rho \kappa }\right)\), thermal conductivity\(\left(q=-k\frac{dT}{dx}\right)\), Seebeck coefficient\(\left(S=\frac{\Delta V}{\Delta T}\right)\), and electrical conductivity must be employed. Assuming the relaxation time constant, the Boltztrap package (based on semi-classical theory) is used to calculate the temperature-dependent TE parameters. Low thermal conductivity and resistivity values are essential for good thermoelectric materials, paired with high Seebeck coefficient (S) values. The calculated TE parameters plotted against temperature (0–1000 K) are shown in Fig. 10a–e.

The calculated a electrical conductivity \((\sigma )\), b thermal conductivity (\(\kappa\)), c Seebeck coefficient (S), power factor (PF) and figure of merit (ZT) for Xcu2GeQ4 (X = Ba, Sr and Q = S, Se)
Electrical conductivity (\(\sigma\)) can be used to evaluate the materials ability to conduct electricity. The calculated \(\sigma\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are shown in Fig. 10a. We can note that there are no peaks initially (i.e., \(\sigma =0\)) and then peaks for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 starts to originate from approximately 600, 300, 600 and 200 K, respectively, known as the threshold value of \(\sigma\). After threshold temperature, an exponential increase in \(\sigma\) plots are evident with increasing temperatures. The increasing nature of \(\sigma\) plots confirm semiconductor behavior of Cu-based chalcogenide materials XCu2GeQ4 (X = Ba, Sr and Q = S, Se). The values of \(\sigma\) for SrCu2GeSe4 > BaCu2GeSe4 > SrCu2GeS4≈BaCu2GeS4 in the entire temperature range. The highest values of \(\sigma\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are \(2.45\times {10}^{17}\), \(1.43\times {10}^{18}\), \(2.47\times {10}^{17}\) and \(2.27\times {10}^{18}\) \({\left(\Omega .m.s\right)}^{-1}\), respectively.
Lattice vibrations and electrons contribute in thermal conductivity (κ) of semiconductors. Fourier law can be employed to formulate thermal conductivity κ. The calculated \(\kappa\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are shown in Fig. 10b. We can note that there are no peaks initially (i.e., \(\kappa =0\)) and then peaks for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 starts to originate from approximately 500, 250, 500 and 150 K, respectively, known as the threshold value of \(\kappa\). After threshold temperature, an exponential increase in \(\kappa\) plots are evident with increasing temperatures. At room temperature, the increasing behavior of κ is because the crystal boundary is smaller than the phonon wavelength. All phonon contributions to κ are equal or less than the crystal boundary. There exists an inverse relation between phonon wavelengths and temperature. Furthermore, the values of \(\sigma\) for SrCu2GeSe4 > BaCu2GeSe4 > SrCu2GeS4 ≈ BaCu2GeS4 in the entire temperature range. The highest values of \(\kappa\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are \(5.02\times {10}^{13}\), \(1.37\times {10}^{14}\), \(5.09\times {10}^{13}\) and \(1.33\times {10}^{14}\) \(\left(\frac{W}{m \cdot K \cdot s}\right)\), respectively.
A potential difference (\(\Delta V\)) is established due to migration of electrons from higher temperature zone towards lower temperature zone as a result of temperature gradient created by connecting two distinct materials (Fatima et al. 2022). The efficiency of a thermocouple can be established using a ratio of \(\Delta V\) to \(\Delta T\) known as Seebeck coefficient. The calculated \(S\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are shown in Fig. 10c. It can be noted from S plots that BaCu2GeS4/SrCu2GeS4 are p-type semiconductor materials as their S values are positive whereas BaCu2GeSe4/SrCu2GeSe4 are n-type semiconductor materials. An exponential decrease and increase in the S values of p-type and n-type materials, respectively, with increasing temperature is a typical behavior for semiconductor materials. The highest values of \(S\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are \(2.7\times {10}^{-3}\), \(-4.10\times {10}^{-5}\), \(2.5\times {10}^{-3}\) and \(5.39\times {10}^{-5}\) \(\left(\frac{V}{K}\right)\), respectively. Materials are deemed to be good thermoelectric materials if their S values are more than 200 \(\left(\frac{\mu V}{K}\right)\). XCu2GeQ4 (X = Ba, Sr and Q = S, Se) have S values that are close to the specified standard, making them potentially useful thermoelectric materials for device applications.
The calculated \(PF\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are shown in Fig. 10d. We can note that there are no peaks initially (i.e., \(PF=0\)) and then peaks for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 starts to originate from approximately 300, 150, 300 and 100 K, respectively, known as the threshold value of \(PF\). After threshold temperature, an exponential increase in \(PF\) plots are evident with increasing temperatures. The highest values of \(PF\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are \(1.50\times {10}^{10}\), \(1.66\times {10}^{10}\), \(2.28\times {10}^{10}\) and \(1.17\times {10}^{10}\) \(\left(\frac{W}{m \cdot {K}^{2} \cdot s}\right)\), respectively.
Finally, the calculated \(ZT\) plots for XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are shown in Fig. 10e. The figure of merit (\(ZT\)) plots can be used to determine thermoelectric performance of the studied materials. For all compounds, initially \(ZT\) values are high and then we can gradual/abrupt decrease in \(ZT\) values with increasing temperature. The highest values of \(ZT\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are 0.94, 1.85, 1.01 and 2.6, respectively. The \(ZT\) value must be equal or greater than 1 for any thermoelectric material to be used in thermoelectric devices. BaCu2GeS4 and SrCu2GeS4 are potential candidates for TE device applications as their \(ZT\) values are ≈1 in the entire temperature range. It can be concluded that SrCu2GeS4 is the most efficient thermoelectric material for TE device applications based on the \(ZT\) values of XCu2GeQ4 (X = Ba, Sr and Q = S, Se).
5 Conclusions
In conclusion, we used GGA + U approximation in order to calculate the optoelectronic properties of Cu-based chalcogenides for solar cell and thermoelectric device applications by employing the FP-LAPW technique. It can be concluded that XCu2GeQ4 (X = Ba, Sr and Q = S, Se) are direct and narrow bandgap semiconductors, which is based on the energy band structures. The values of energy band gaps for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are approximately 0.667, 0.378, 0.697 and 0.195 eV, respectively. A blueshift is evident from the spectra of \({\varepsilon }_{2}(\omega )\) in following sequence: SrCu2GeSe4, BaCu2GeSe4, SrCu2GeS4 and BaCu2GeS4. Their calculated refractive index \(n(\omega )\) shows that these materials are active optical materials, which can be used in photovoltaic devices. These compounds show significantly less reflectivity on low energies and high reflectivity of ~ 13.0 eV. These materials can be used as reflecting coatings in the upper UV region. The highest values of \(ZT\) for BaCu2GeS4, BaCu2GeSe4, SrCu2GeS4 and SrCu2GeSe4 are 0.94, 1.85, 1.01 and 2.6, respectively. But BaCu2GeS4 is the most promising TE candidate for thermoelectric devices applications in the entire temperature range.
Data availability
All data is available in the article.
References
Abbas, Z., Iftikhar, I., Azam, S.: Band gap tailoring of Cu2Se by chlorine: a first principles study. EasyChair (2020a)
Abbas, Z., Munaf, S., Azam, S., Abubakar, M., Irfan, M.: First-principles calculations of optoelectronic properties of Sn1−xInxA (A= S and Se) for solar cell applications. EasyChair (2020b)
Abbas, Z., Fatima, K., Gorczyca, I., Irfan, M., Alotaibi, N., Alshahrani, T., Raza, H.H., Muhammad, S.: Proposition of new stable rare-earth ternary semiconductor sulfides of type LaTlS2 (La= Er, Eu, Tb): ab-initio study and prospects for optoelectronic, spintronic and thermoelectric applications. Mater. Sci. Semicond. Process. 146, 106662 (2022a). https://doi.org/10.1016/j.mssp.2022.106662
Abbas, Z., Fatima, K., Jaffery, S.H.A., Ali, A., Raza, H.H., Muhammad, S., Algarni, H., Hussain, S., Jung, J.: Ab-initio study of Nb-based complex materials: a new class of materials for optoelectronic applications. J. Comput. Sci. 63, 101791 (2022b). https://doi.org/10.1016/j.jocs.2022.101791
Abbas, Z., Hussain, S., Muhammad, S., Siddeeg, S.M., Jung, J.: A first-principles investigation on the structural, optoelectronic, and thermoelectric properties of pyrochlore oxides (La2Tm2O7 (Tm = Hf, Zr)) for energy applications. Int. J. Mol. Sci. 23, 15266 (2022c). https://doi.org/10.3390/ijms232315266
Abbas, Z., Fatima, K., Gorczyca, I., Abbas Jaffery, S.H., Ali, A., Irfan, M., Raza, H.H., Algarni, H., Muhammad, S., Teisseyre, H., Hussain, S., Siddeeg, S.M., Jung, J.: First-principles calculations to investigate electronic, optical, and thermoelectric properties of Na2GeX3 (X = S, Se, Te) for energy applications. Mater. Sci. Semicond. Process. 154, 107206 (2023a). https://doi.org/10.1016/j.mssp.2022.107206
Abbas, Z., Naz, A., Hussain, S., Muhammad, S., Algarni, H., Ali, A., Jung, J.: First-principles calculations to investigate structural, electronic, optical and magnetic properties of pyrochlore oxides Eu2Tm2O7 (Tm = Hf, Sn, Zr) for energy applications. Inorganics 11, 193 (2023b). https://doi.org/10.3390/inorganics11050193
Abubakr, M., Abbas, Z., Rehman, S., Ul Hassan, N., Ifseisi, A.A., Khan, M.A., Kim, H., Khan, K., Kim, D., Khan, M.F.: Systematic study on the optoelectronic and elastic properties of Cu-based ternary chalcogenides: using ab-initio approach. Mater. Sci. Semicond. Process. 162, 107512 (2023). https://doi.org/10.1016/j.mssp.2023.107512
Anisimov, V.I., Zaanen, J., Andersen, O.K.: Band theory and mott insulators: hubbard u instead of stoner I. Phys. Rev. B 44, 943–954 (1991). https://doi.org/10.1103/PhysRevB.44.943
Azam, S., Abbas, Z., Gul, B., Khan, M.S., Irfan, M., Sohail, M., Khan, S.A., Naseer, F., Irfan, A., Khan, G., Rai, D.P., Khenata, R.: First-principles calculations of optoelectronic properties of CaO: Eu+2 (SrO: Eu+2) for energy applications. Int. J. Mod. Phys. B 32, 1850333 (2018). https://doi.org/10.1142/S0217979218503332
Blaha, P., Schwarz, K., Medsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2k an augmented plane wave plus local orbitals program for calculating crystal properties. Vienna University Technology (2001)
Boochani, A., Abolhasani, M.R., Ghoranneviss, M., Elahi, M.: First principles study of half metallic properties of VSb surface and VSb/GaSb (001) interface. Commun. Theor. Phys 54(1), 148–158 (2010). https://doi.org/10.1088/0253-6102/54/1/28
Brant, J.A., Clark, D.J., Kim, Y.S., Jang, J.I., Zhang, J.-H., Aitken, J.A.: Li2CdGeS4, a diamond-like semiconductor with strong second-order optical nonlinearity in the infrared and exceptional laser damage threshold. Chem. Mater. 26, 3045–3048 (2014). https://doi.org/10.1021/cm501029s
Chen, R., Wu, X., Su, Z.: Structural insights into T2-cluster-containing chalcogenides with vertex-, edge- and face-sharing connection modes of NaQ6 ligands: Na3ZnMIIIQ4 (MIII = In, Ga; Q = S, Se). Dalton Trans. 47, 15538–15544 (2018). https://doi.org/10.1039/C8DT03281E
Ding, G., He, J., Cheng, Z., Wang, X., Li, S.: Low lattice thermal conductivity and promising thermoelectric figure of merit of Zintl type TlInTe2. J. Mater. Chem. C 6, 13269–13274 (2018). https://doi.org/10.1039/C8TC03492C
Ding, G., Li, S., He, J.: Origins of promising thermoelectric performance in quaternary selenide BaAg2SnSe4. Appl. Phys. Express 12, 071006 (2019). https://doi.org/10.7567/1882-0786/ab29e1
Eibler, R., Neckel, A.: The density-of-states functions of the ordered alloys FeAl, CoAl and NiAl and their relation to experimental data; calculations of the imaginary part of the complex dielectric function. J. Phys. F Met. Phys. 10, 2179–2195 (1980). https://doi.org/10.1088/0305-4608/10/10/015
Fatima, K., Abbas, Z., Naz, A., Alshahrani, T., Chaib, Y., Jaffery, S.H.A., Muhammad, S., Hussain, S., Jung, J., Algarni, H.: Shedding light on the structural, optoelectronic, and thermoelectric properties of pyrochlore oxides (La2Q2O7 (Q = Ge, Sn)) for energy applications: a first-principles investigation. J. Solid State Chem. 313, 123305 (2022). https://doi.org/10.1016/j.jssc.2022.123305
Gan, M., Chong, X., Yu, W., Xiao, B., Feng, J.: Understanding the ultralow lattice thermal conductivity of monoclinic RETaO4 from acoustic-optical phonon anti-crossing property and a comparison with ZrO2. J. Am. Ceram. Soc. 106, 3103–3115 (2023). https://doi.org/10.1111/jace.18988
Gao, B., Zhou, T., Hong, A.-J., Liang, F., Song, G., Xu, Q.-Q., Zhang, J., Li, G.-N., Wang, Y., Dang, C.: Study on thermoelectric properties of CrI3 monolayer. Appl. Phys. Express 13, 045001 (2020). https://doi.org/10.35848/1882-0786/ab7a44
Guo, S.-P., Chi, Y., Guo, G.-C.: Recent achievements on middle and far-infrared second-order nonlinear optical materials. Coord. Chem. Rev. 335, 44–57 (2017). https://doi.org/10.1016/j.ccr.2016.12.013
He, J., Kanatzidis, M.G., Dravid, V.P.: High performance bulk thermoelectrics via a panoscopic approach. Mater. Today 16, 166–176 (2013). https://doi.org/10.1016/j.mattod.2013.05.004
Huang, Z., Lü, T.-Y., Wang, H.-Q., Yang, S.-W., Zheng, J.-C.: Electronic and thermoelectric properties of the group-III nitrides (BN, AlN and GaN) atomic sheets under biaxial strains. Comput. Mater. Sci. 130, 232–241 (2017). https://doi.org/10.1016/j.commatsci.2017.01.013
Jiahong Kuo, J., Aydemir, U., Pöhls, J.-H., Zhou, F., Yu, G., Faghaninia, A., Ricci, F., Anne White, M., Rignanese, G.-M., Hautier, G., Jain, A., Jeffrey Snyder, G.: Origins of ultralow thermal conductivity in 1-2-1-4 quaternary selenides. J. Mater. Chem. A 7, 2589–2596 (2019). https://doi.org/10.1039/C8TA09660K
Kuei, C.-Y., Tsai, W.-L., Tong, B., Jiao, M., Lee, W.-K., Chi, Y., Wu, C.-C., Liu, S.-H., Lee, G.-H., Chou, P.-T.: Bis-tridentate Ir(III) complexes with nearly unitary RGB phosphorescence and organic light-emitting diodes with external quantum efficiency exceeding 31%. Adv. Mater. 28, 2795–2800 (2016). https://doi.org/10.1002/adma.201505790
Lahourpour, F., Boochani, A., Parhizgar, S.S., Elahi, S.M.: Structural, electronic and optical properties of graphene-like nano-layers MoX2(X:S, Se, Te): DFT study. J. Theor. Appl. Phys. 13, 191–201 (2019). https://doi.org/10.1007/s40094-019-0333-4
Lekse, J.W., Moreau, M.A., McNerny, K.L., Yeon, J., Halasyamani, P.S., Aitken, J.A.: Second-harmonic generation and crystal structure of the diamond-like semiconductors Li2CdGeS4 and Li2CdSnS4. Inorg. Chem. 48, 7516–7518 (2009). https://doi.org/10.1021/ic9010339
Li, H., Malliakas, C.D., Liu, Z., Peters, J.A., Jin, H., Morris, C.D., Zhao, L., Wessels, B.W., Freeman, A.J., Kanatzidis, M.G.: CsHgInS3: a new quaternary semiconductor for γ-ray detection. Chem. Mater. 24, 4434–4441 (2012). https://doi.org/10.1021/cm302838v
Li, G., Wu, K., Liu, Q., Yang, Z., Pan, S.: Na2ZnGe2S6: a new infrared nonlinear optical material with good balance between large second-harmonic generation response and high laser damage threshold. J. Am. Chem. Soc. 138, 7422–7428 (2016a). https://doi.org/10.1021/jacs.6b03734
Li, W., Lin, S., Ge, B., Yang, J., Zhang, W., Pei, Y.: Low sound velocity contributing to the high thermoelectric performance of Ag8SnSe6. Adv. Sci. 3, 1600196 (2016b). https://doi.org/10.1002/advs.201600196
Li, G., Liu, Q., Wu, K., Yang, Z., Pan, S.: Na2CdGe2Q6 (Q = S, Se): two metal-mixed chalcogenides with phase-matching abilities and large second-harmonic generation responses. Dalton Trans. 46, 2778–2784 (2017). https://doi.org/10.1039/C7DT00087A
Liang, F., Kang, L., Lin, Z., Wu, Y.: Mid-infrared nonlinear optical materials based on metal chalcogenides: structure-property relationship. Cryst. Growth Des. 17, 2254–2289 (2017). https://doi.org/10.1021/acs.cgd.7b00214
Liao, J.-H., Marking, G.M., Hsu, K.F., Matsushita, Y., Ewbank, M.D., Borwick, R., Cunningham, P., Rosker, M.J., Kanatzidis, M.G.: α- and β-A2Hg3M2S8 (A = K, Rb; M = Ge, Sn): polar quaternary chalcogenides with strong nonlinear optical response. J. Am. Chem. Soc. 125, 9484–9493 (2003). https://doi.org/10.1021/ja034121l
Lin, H., Chen, L., Yu, J.-S., Chen, H., Wu, L.-M.: Infrared SHG materials CsM3Se6 (M = Ga/Sn, In/Sn): phase matchability controlled by dipole moment of the asymmetric building unit. Chem. Mater. 29, 499–503 (2017). https://doi.org/10.1021/acs.chemmater.6b05026
Ma, Z., Weng, F., Wang, Q., Tang, Q., Zhang, G., Zheng, C., Han, R.P.S., Huang, F.: Low temperature synthesis and structures of alkaline earth metal chalcogenides Ba3Cu4SbS6OH, BaCuSbS3 and BaCu2S2. RSC Adv. 4, 28937–28940 (2014). https://doi.org/10.1039/C3RA46878J
Mei, D., Gong, P., Lin, Z., Feng, K., Yao, J., Huang, F., Wu, Y.: Ag3Ga3SiSe8: a new infrared nonlinear optical material with a chalcopyrite structure. CrystEngComm 16, 6836–6840 (2014). https://doi.org/10.1039/C4CE00817K
Mobaraki, A., Kandemir, A., Yapicioglu, H., Gülseren, O., Sevik, C.: Validation of inter-atomic potential for WS2 and WSe2 crystals through assessment of thermal transport properties. Comput. Mater. Sci. 144, 92–98 (2018). https://doi.org/10.1016/j.commatsci.2017.12.005
Morris, C.D., Li, H., Jin, H., Malliakas, C.D., Peters, J.A., Trikalitis, P.N., Freeman, A.J., Wessels, B.W., Kanatzidis, M.G.: Cs2MIIMIV3Q8 (Q = S, Se, Te): an extensive family of layered semiconductors with diverse band gaps. Chem. Mater. 25, 3344–3356 (2013). https://doi.org/10.1021/cm401817r
Nian, L., Huang, J., Wu, K., Su, Z., Yang, Z., Pan, S.: BaCu2MIVQ4 (MIV = Si, Ge, and Sn; Q = S, Se): synthesis, crystal structures, optical performances and theoretical calculations. RSC Adv. 7, 29378–29385 (2017). https://doi.org/10.1039/C7RA05022D
Pei, Y., Morelli, D.T.: Vacancy phonon scattering in thermoelectric In2Te3–InSb solid solutions. Appl. Phys. Lett. 94, 122112 (2009). https://doi.org/10.1063/1.3109788
Pei, Y., Shi, X., LaLonde, A., Wang, H., Chen, L., Snyder, G.J.: Convergence of electronic bands for high performance bulk thermoelectrics. Nature 473, 66–69 (2011). https://doi.org/10.1038/nature09996
Poudel, B., Hao, Q., Ma, Y., Lan, Y., Minnich, A., Yu, B., Yan, X., Wang, D., Muto, A., Vashaee, D., Chen, X., Liu, J., Dresselhaus, M.S., Chen, G., Ren, Z.: High-thermoelectric performance of nanostructured bismuth antimony telluride bulk alloys. Science 320, 634–638 (2008). https://doi.org/10.1126/science.1156446
Rosmus, K.A., Brant, J.A., Wisneski, S.D., Clark, D.J., Kim, Y.S., Jang, J.I., Brunetta, C.D., Zhang, J.-H., Srnec, M.N., Aitken, J.A.: Optical nonlinearity in Cu2CdSnS4 and α/β-Cu2ZnSiS4: diamond-like semiconductors with high laser-damage thresholds. Inorg. Chem. 53, 7809–7811 (2014). https://doi.org/10.1021/ic501310d
Shen, J., Zhang, X., Chen, Z., Lin, S., Li, J., Li, W., Li, S., Chen, Y., Pei, Y.: Substitutional defects enhancing thermoelectric CuGaTe2. J. Mater. Chem. A 5, 5314–5320 (2017). https://doi.org/10.1039/C6TA10770B
Shin, D., Saparov, B., Zhu, T., Huhn, W.P., Blum, V., Mitzi, D.B.: BaCu2Sn(S, Se)4: earth-abundant chalcogenides for thin-film photovoltaics. Chem. Mater. 28, 4771–4780 (2016). https://doi.org/10.1021/acs.chemmater.6b01832
Snyder, G.J., Toberer, E.S.: Complex thermoelectric materials. Nat. Mater. 7, 105–114 (2008). https://doi.org/10.1038/nmat2090
Teymur, B., Zhou, Y., Ngaboyamahina, E., Glass, J.T., Mitzi, D.B.: Solution-processed earth-abundant Cu2BaSn(S, Se)4 solar absorber using a low-toxicity solvent. Chem. Mater. 30, 6116–6123 (2018). https://doi.org/10.1021/acs.chemmater.8b02556
Wang, C., Ding, G., Wu, X., Wei, S., Gao, G.: Electron and phonon transport properties of layered Bi2O2Se and Bi2O2Te from first-principles calculations. New J. Phys. 20, 123014 (2018). https://doi.org/10.1088/1367-2630/aaf664
Wang, C., Zheng, C., Gao, G.: Bulk and monolayer ZrS3 as promising anisotropic thermoelectric materials: a comparative study. J. Phys. Chem. C 124, 6536–6543 (2020). https://doi.org/10.1021/acs.jpcc.0c00298
Wu, K., Pan, S., Yang, Z.: Ba2GeS4 and Mg2SnS4: synthesis, structures, optical properties and electronic structures. RSC Adv. 5, 33646–33652 (2015a). https://doi.org/10.1039/C5RA00264H
Wu, K., Su, X., Yang, Z., Pan, S.: An investigation of new infrared nonlinear optical material: BaCdSnSe4, and three new related centrosymmetric compounds: Ba2SnSe4, Mg2GeSe4, and Ba2Ge2S6. Dalton Trans. 44, 19856–19864 (2015b). https://doi.org/10.1039/C5DT03215F
Wu, K., Yang, Z., Pan, S.: Na2BaMQ4 (M = Ge, Sn; Q = S, Se): infrared nonlinear optical materials with excellent performances and that undergo structural transformations. Angew. Chem. Int. Ed. 55, 6713–6715 (2016). https://doi.org/10.1002/anie.201602317
Xiao, Z., Meng, W., Li, J.V., Yan, Y.: Distant-atom mutation for better earth-abundant light absorbers: a case study of Cu2BaSnSe4. ACS Energy Lett. 2, 29–35 (2017). https://doi.org/10.1021/acsenergylett.6b00577
Yeganeh, M., Kafi, F., Boochani, A.: Thermoelectric properties of InN graphene-like nanosheet: a first principle study. Superlattices Microstruct. 138, 106367 (2020). https://doi.org/10.1016/j.spmi.2019.106367
Yeganeh, M., Boochani, A., Yari, A., Amiri, M., Shahraki, A.T.: The WS2/AlN heterostructure band alignment by semi-local functional and its optical properties by the Bethe–Salpeter approximation on top of GW calculations. Mater. Sci. Semicond. Process. 148, 106772 (2022). https://doi.org/10.1016/j.mssp.2022.106772
Yu, F., Yu, S., Li, C., Li, Z., Song, F., Xu, Z., Zhu, Y., Dai, C., Cao, X., Zhang, Z., Liu, Y., Qiu, J.: Molecular engineering of biomimetic donor-acceptor conjugated microporous polymers with full-spectrum response and an unusual electronic shuttle for enhanced uranium(VI) photoreduction. Chem. Eng. J. 466, 143285–143297 (2023). https://doi.org/10.1016/j.cej.2023.143285
Zhang, X., Yi, N., Hoffmann, R., Zheng, C., Lin, J., Huang, F.: Semiconductive K2MSbS3(SH) (M = Zn, Cd) featuring one-dimensional ∞1[M2Sb2S6(SH2)]4− Chains. Inorg. Chem. 55, 9742–9747 (2016). https://doi.org/10.1021/acs.inorgchem.6b01529
Zhang, R.-C., Zhang, J.-C., Cao, Z., Wang, J.-J., Liang, S.-S., Cong, H.-J., Wang, H.-J., Zhang, D.-J., An, Y.-L.: Unusual flexibility of microporous sulfides during ion exchange. Inorg. Chem. 57, 13128–13136 (2018). https://doi.org/10.1021/acs.inorgchem.8b01238
Zhao, L.D., Wu, H.J., Hao, S.Q., Wu, C.I., Zhou, X.Y., Biswas, K., He, J.Q., Hogan, T.P., Uher, C., Wolverton, C., Dravid, V.P., Kanatzidis, M.G.: All-scale hierarchical thermoelectrics: MgTe in PbTe facilitates valence band convergence and suppresses bipolar thermal transport for high performance. Energy Environ. Sci. 6, 3346–3355 (2013). https://doi.org/10.1039/C3EE42187B
Zhao, Q., Yang, H., Liu, H., Zeng, G., Huang, B.: High birefringence D-shaped germanium-doped photonic crystal fiber sensor. Micromachines. 13, 826–10 (2022). https://doi.org/10.3390/mi13060826
Zhen, N., Wu, K., Wang, Y., Li, Q., Gao, W., Hou, D., Yang, Z., Jiang, H., Dong, Y., Pan, S.: BaCdSnS4 and Ba3 CdSn2S8: syntheses, structures, and non-linear optical and photoluminescence properties. Dalton Trans. 45, 10681–10688 (2016). https://doi.org/10.1039/C6DT01537A
Zhu, T., Huhn, W.P., Wessler, G.C., Shin, D., Saparov, B., Mitzi, D.B., Blum, V.: I2–II–IV–VI4 (I = Cu, Ag; II = Sr, Ba; IV = Ge, Sn; VI = S, Se): chalcogenides for thin-film photovoltaics. Chem. Mater. 29, 7868–7879 (2017). https://doi.org/10.1021/acs.chemmater.7b02638
Funding
This research was supported by the National Research Foundation of Korea (NRF), ICT (Grant no: 2022R1F1A1075229). This work was also funded by the Researchers Supporting Project Number (RSPD2023R664) King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
MA: Conceptualization, methodology, writing—original draft. ZA: Data curation, software. AN: Investigation, writing—original draft. MAK: Methodology, supervision. SR: Software, methodology: HK: Data curation, investigation. MO: Investigation, editing. KK: Analysis, review and editing. DK: Writing-reviewing and editing. MFK: Supervision, funding, writing-reviewing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abubakr, M., Abbas, Z., Naz, A. et al. First principles quantum analysis of structural, electronic, optical and thermoelectric properties of XCu2GeQ4 (X = Ba, Sr and Q = S, Se) for energy applications. Opt Quant Electron 55, 849 (2023). https://doi.org/10.1007/s11082-023-05136-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05136-6