
Abstract
Neuroinflammation plays crucial role in the development and progression of depression. Large conductance calcium- and voltage-dependent potassium (BK) channels mediate the activation of microglia. Herein, we investigated whether BK channels could serve as a target for the treatment of inflammation-associated depression. Lipopolysaccharide (LPS, 0.83 mg/kg) was injected intraperitoneally (i.p.) to induce neuroinflammation and depressive-like behavior in 6–8 week ICR mice. Adeno-associated virus (AAV) constructs (AAV9-Iba1p-BK shRNA-EGFP (BK shRNA-AAV) or AAV9-Iba1p-NC shRNA-EGFP (NC shRNA-AAV)) were unilaterally injected intracerebroventricularly to selectively knock down BK channels in microglia. The tail suspension test (TST) and forced-swim test (FST) were used to evaluate depressive-like behavior in mice 24 h after LPS challenge. The morphology of microglia, expression of BK channels, levels of cytokines, and expression and activity of indoleamine 2,3-dioxygenase (IDO) were measured by immunohistochemistry, western blot, quantitative real time PCR, and enzyme-linked immunosorbent assay (ELISA), respectively. Either paxilline (i.p.), a specific BK channel blocker, or BK shRNA-AAV effectively inhibited the activation of microglia, reduced the production of IL-1β in the hippocampus and suppressed the expression and activity of IDO in the hippocampus and prefrontal cortex, resulting in the amelioration of depressive-like behavior in mice. These data suggest for the first time that BK channels are involved in LPS-induced depressive-like behaviors. Thus, microglia BK channels may be a potential drug target for the depression treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is one of the most prevalent neuropsychiatric disorders. MDD affects more than 280 million people, and is one of the top leading causes of burden of disease worldwide [1, 2]. Growing evidence shows that central inflammation plays an important role in the development and progression of depression [3,4,5]. Pro-inflammatory cytokines such as interleukin 1β (IL-1β), IL-6 and tumor necrosis factor-α (TNF-α) are significantly elevated in the cerebrospinal fluid (CSF) of patients with depression [6, 7]. Up to 30% of patients receiving interferon-α (IFN-α) treatment developed depression within the first three months of therapy [8]. Signals detected by positron emission tomography (PET) from the translocator protein (TSPO), a protein located on outer mitochondrial membranes in microglia, become significantly increased in various brain regions including the hippocampus of depressed patients, which suggests an increase in activated microglia [9,10,11]. Post-mortem studies have also found evidence of activated microglia and enhanced neuroinflammation in the brains of patients suffering from depression [12,13,14,15]. Furthermore, antidepressants including classical selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and atypical antidepressants such as ketamine can reduce neuroinflammation [16,17,18,19]. Conversely, some drugs with anti-inflammatory effects such as minocycline, nonsteroidal anti-inflammatory drugs, and statins, can improve depressive symptoms in both preclinical and clinical studies [20,21,22,23].
Large conductance calcium- and voltage-dependent potassium (BK) channels are ubiquitously expressed in both excitable and non-excitable cells and are associated with various neurological diseases such as neuropathic pain and stroke [24,25,26]. In recent years, mounting evidence suggests that BK channels are involved in microglial activation and are therefore related to neuroinflammation [27]. In hippocampal slices from juvenile mice, BK currents were solely recorded in activated microglia [28]. In mice that had undergone nerve injury, BK channel blockade inhibited the production of mature IL-1β, proliferation of spinal microglia, and ultimately attenuated tactile allodynia while BK channel activation promoted the development of tactile allodynia in these mice [29]. Using spinal cord slices from Iba1+/GFP mice after a five-day treatment with morphine (10 mg/kg, i.p., twice per day), Hayashi and colleagues found that BK channels in microglia rather than neurons or astrocytes were activated via the phospholipase A2-arachidonic acid pathway, which led to microglia activation and morphine-induced hyperalgesia [30]. In addition, inhibition of BK channels suppressed the expression of the NOD-like receptor family, the pyrin domain-containing 3 (NLRP3) inflammasome, decreased the levels of IL-1β and IL-18 in the cerebral cortex of rats with blast-induced traumatic brain injury, and ultimately alleviated brain edema [31]. Our previous study found that BK channels are located on both the plasma membrane and nucleus of murine microglia. The membrane-associated BK channels participate in the early stages of lipopolysaccharide (LPS)-induced activation of microglia via regulating nuclear factor-κB (NF-κB). Whereas the nuclear BK channels function in the late stages of LPS-induced microglia activation [32].
In the present study, we aim to investigate the potential roles of microglia BK channels in LPS-induced depressive-like behavior in mice. The pharmacologic blockade or genetic knockdown of microglia BK channels repress the activation of microglia, reduce the level of IL-1β and the activity of indoleamine 2, 3-dioxygenase (IDO), and lead to amelioration of depressive-like behavior of mice induced by LPS.
Materials and Methods
Chemicals
LPS (IL2020, from Escherichia coli 055:B5) was purchased from Solarbio Life Sciences (Beijing, China). Paxilline (p2928) was purchased from Sigma-Aldrich (St. Louis, MO). All of the other reagents were obtained from Sigma-Aldrich, except where indicated.
Animals and Treatments
Six to eight weeks old male Crl:CD1 (ICR) mice were purchased from Zhejiang Vital River Laboratory Animal Technology Co., Ltd (Zhejiang, China). Mice were housed in an animal facility under constant environmental conditions (21 ± 1 °C, 40–70% relative humidity and a 12/12 h light/dark cycle) and had access to food and water. A total of seventy three mice were used in this study.
LPS and paxilline solutions were prepared on the day of injection. For LPS solution, the drug was dissolved in sterile phosphate buffer solution (PBS) to 0.083 mg/mL and intraperitoneally (i.p.) injected at a dose of 0.83 mg/kg (10 mL/kg) [33]. Paxilline was first dissolved in dimethyl sulfoxide (DMSO) to 10 mM and then diluted to 0.51 μM with sterile PBS, finally was administered i.p. at a dose of 2.2 μg/kg (10 mL/kg). Blank PBS or PBS containing 0.0051% DMSO was administered i.p. at 10 mL/kg as the vehicle control for LPS or paxilline, respectively.
Adeno-Associated Virus (AAV) Construct Injection
AAV9-Iba1p-BK shRNA-EGFP (BK shRNA-AAV) and AAV9-Iba1p-NC shRNA-EGFP (NC shRNA-AAV) virus-based constructs were purchased from GENECHEM Company (Shanghai, China). The sequences of BK shRNA and NC shRNA are TTTGAACAGAGCCTCAAGCTC and ACGTGACACGTTCGGAGAA, respectively. Mice were anesthetized with 1% pentobarbital sodium (50 mg/kg) and placed in a stereotaxic apparatus (502300, WPI, Shanghai, China) with a mouse adaptor. The viral constructs (1 × 1011 viral particles/mL, 5 μL) were unilaterally injected intracerebroventricularly at a flow rate of 1 μL/min. The injections will allow the viruses to infect the entire brain. The coordinates for stereotaxic injection into the lateral ventricle were AP − 1.22 mm from bregma, ML 2.0 mm from the midline, and DV 2.25 mm from the brain surface. The injection needle remained in place for 10 min post injection to minimize retrograde flow along the needle track. Two weeks after the virus injection, mice were sacrificed and the brain slices were prepared to evaluate the efficacy of the BK shRNA-AAV infection. Three mice were used for this purpose.
Study Design
The mice were randomized into one of the following experimental protocols
Paxilline protocol: In this protocol, paxilline (PAX, 2.2 μg/kg, i.p.) was administered 30 min before LPS to pre-block the BK channels [30]. The tail suspension test (TST) was conducted 24 h after LPS (0.83 mg/kg, i.p.) injection, followed by the forced-swim test (FST) 4 h later. Brain tissues were collected after completing behavioral tests or at 29 h after LPS administration. The mice were randomly allocated to four experimental groups: CON (control group, 10 mL/kg vehicle, i.p.), PAX (2.2 μg/kg paxilline, i.p.), LPS (0.83 mg/kg LPS, i.p.), and PAX + LPS (2.2 μg/kg paxilline and 0.83 mg/kg LPS, i.p.). Thirty mice participated in the behavioral tests (8, 8, 7, 7 mice for CON, PAX, LPS or PAX + LPS group, respectively), and 12 of them were used for immunohistochemistry experiments (3 mice per group), while 18 of them were used for RT-PCR and ELISA analyses (5, 5, 4, 4 mice for CON, PAX, LPS or PAX + LPS group, respectively). Additionally, there are 12 more mice were used for RT-PCR and ELISA analyses (3 mice per group) without undergoing behavioral tests.
AAV protocol: The mice were randomly assigned to receive either BK shRNA-AAV or NC shRNA-AAV injections. Two weeks after the virus injection, mice were randomly divided into control (CON) and LPS groups. A single dose of LPS (0.83 mg/kg) or PBS (10 mL/kg, CON) was applied i.p. The tail suspension test (TST) was conducted 24 h after LPS administration, followed by the forced-swim test (FST) 4 h later. Brain tissues were collected after completing behavioral tests or at 29 h after LPS administration. Twenty mice participated in the behavioral tests (5 mice per group), and 4 of these mice were used for immunohistochemistry experiments (1 mouse per group), while the remaining 16 mice were used for RT-PCR and ELISA analyses (4 mice per group). Additionally, there are 8 more mice were used for immunohistochemistry experiments (2 mice per group) without undergoing behavioral tests.
Behavioral Experiments
Tail Suspension Test (TST)
Mice were suspended 45 cm above the ground by their tails with tapes. The adhesive tape was placed 1 cm from the tip of the tail. The video camera and a tripod had been set up 2 m in front of the mice. The behaviors of suspended mice were recorded for a period of 10 min by the video camera and the whole session was scored. The time of immobility was measured by a trained blinded observer. Small movements restricted to the front legs were scored as immobility if the hind legs weren’t involved. Oscillations and pendulum like swings resulting from the momentum gained during previous bouts of mobility were also scored as immobility [34].
Forced-Swim Test (FST)
Mice were placed individually in a cylinder (diameter: 16 cm; height: 25 cm) containing 20 cm of water (23 ± 1 °C) for 6 min and then returned to their home cage. During the test, the movements of each mouse were video recorded from alongside. The duration of immobility over the last 4 min was measured by a trained blinded observer. Small movements to balance the body and keep the head above the water were scored as immobility. Drifting in the water due to the momentum obtained after a single bout of mobility was also scored as immobility [35].
Immunohistochemistry
Mice were anesthetized with 1% pentobarbital sodium (50 mg/kg) and then underwent transcardial perfusion with ice-cold 0.01 M PBS followed by ice-cold fixative containing 4% paraformaldehyde (PFA) in 0.1 M PBS. The whole brains were quickly dissected and postfixed in the same fixative for 12 h at 4 °C. The brain tissues were then dehydrated in a 30% sucrose solution at 4 °C for at least one week and then cut into 20 μm sections via microtome (Leica CM1950) at − 20 °C. All tissues from AAV-injected mice were maintained and handled avoiding light. The slices were washed in PBS and blocked in PBS containing 5% bovine serum albumin (BSA) and 0.5% Triton X-100 for 2 h. Afterwards, the slices were incubated with Iba1 (ionized calcium-binding adapter molecule 1) (1:100, sc-32725, Santa Cruz Biotechnology, Shanghai, China) or BK (1:200, APC-021, Alomone Labs, Jerusalem, Israel) primary antibody in PBS containing 5% BSA and 0.5% Triton X-100 overnight at 4 °C. After several washes with PBST (0.01 M PBS + 0.2% Triton X-100), the slices were incubated with secondary antibody (Goat anti-Rabbit Alexa Fluor 594, 1:1000, R37117, Thermo Fisher Scientific, Shanghai, China) for 2 h. Finally, the slices were stained with Hoechst 33258 (0.5 μg/mL, Thermo Fisher Scientific, Shanghai, China) and mounted using Immu-Mount (Thermo Fisher Scientific). Specimens were viewed using a Zeiss LSM 710 microscope with ZEN software (Carl Zeiss Microscopy GmbH, Jena, Germany).
Quantitative Real-Time PCR
Mice were anesthetized with 1% pentobarbital sodium (50 mg/kg) and then transcardially perfused with ice-cold PBS. After that, the whole brain was quickly dissected. The hippocampus and prefrontal cortex (PFC) tissues were collected on ice rapidly and snap-frozen in liquid nitrogen. Total RNA was extracted from brain tissues (hippocampus and PFC) using RNAiso Plus (TaKaRa, Shiga, Japan) in accordance with the manufacturer’s instructions. The purity of nucleic acid in the samples was determined by measuring the A260/A230 ratio. One μg of total RNA was reverse-transcribed using the TaKaRa reverse transcription kit (PrimeScript™ 1st Strand cDNA Synthesis Kit) with Oligo (dT) primer for real-time PCR (RT-PCR) assays. Quantitative RT-PCR was set up using SYBR Green PCR Master Mix (TaKaRa) and conducted on the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). The selective primers for each gene are listed in Table 1. The amplification reactions were cycled 40 times at 95 °C for 30 s, 95 °C for 5 s, and 60 °C for 30 s. Finally the reactions were heated slowly from 60 °C to 95 °C. Relative quantification was calculated using the 2−ΔΔCT formula, where CT is the cycle threshold. The transcript abundance of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was measured as the control for expression of an endogenous house-keeping gene. The transcript abundance of each target gene was expressed as fold-change relative to the transcript abundance of the control.
Enzyme-Linked Immunosorbent Assay (ELISA)
Hippocampus and PFC tissues were harvested following the same procedure as described in the Quantitative Real-Time PCR section. The samples were homogenized in sterile PBS containing a protease inhibitor cocktail at 200 mg/mL on ice and then centrifuged at 12,500×g for 15 min at 4 °C. The supernatants were collected and assayed for IDO, tryptophan (Trp), and kynurenine (Kyn) using mouse-specific ELISA kits (Sanjia, China) according to the manufacturer’s instructions.
Cell Culture and Transfection
HEK293T cells were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The HEK293T cells were maintained under a humidified atmosphere of 5% CO2 at 37 °C in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS), 100 IU/mL penicillin G, and 100 μg/mL streptomycin (Thermo Fisher Scientific, Shanghai, China). For plasmid transfection, HEK293T cells were plated in 6-well plates with 2 mL/well of antibiotic-free growth medium at a density of 1 × 105 cells/well, grown for 24 h, and transfected with equal amounts of BK plasmid combining either BK shRNA or negative control shRNA (NC shRNA) using Lipofectamine 2000 (Thermo Fisher Scientific, Shanghai, China). Cells were collected for western blot analysis at 24 h after transfection. BK plasmid (Gene ID, NM_001253358), BK shRNA (GCTTGAGGCTCTGTTCAAA) and NC shRNA (CGCTGAGTACTTCGAAATGTC) were purchased from GENECHEM Company (Shanghai, China).
Western Blotting
HEK293T cells were washed with ice-cold PBS 24 h after transfection and lysed in chilled RIPA lysis buffer (Cell Signaling Technology, Boston, MA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF). The cell lysates were then centrifuged at 14,000×g for 10 min at 4 ℃ and the supernatants were used for assays. Equal quantities of protein were separated on 10% sodium dodecyl sulfate-PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes (0.2 μm, Merck, Darmstadt, Germany). The membranes were then incubated with BK (1:1000, 75-022, UC Davis/NIH NeuroMab Facility, Davis, CA) or β-actin (1:20,000, 66009-1-Ig, Proteintech Group, Rosemont, IL, USA) primary antibodies, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies. Immunoreactivity was measured using the Immobilon Western Chemiluminescent HRP Substrate (Merck). ImageJ software (National Institutes of Health, Bethesda, MD) was used to carry out the densitometric analysis.
Statistical Analysis
All experiments were conducted at least three times. Statistical analyses were performed by one-way or two-way ANOVA or Student’s t-test using GraphPad Prism 9.0.0 software (GraphPad Software, Inc., La Jolla, CA). The Shapiro–Wilk test was used to test for normal distribution of the data. The quantitative data obtained from the western blotting experiment exhibited normal distribution, and the two-group means were analyzed using an unpaired two-tailed Student’s t test. Before conducting one-way or two-way ANOVA tests, the normal distribution and variance equality of the data were examined using the Shapiro–Wilk test and the Brown-Forsythe test, respectively. All data from the Paxilline protocol were analyzed by one-way ANOVA. If the data showed normal distribution and equal variances, an ordinary one-way ANOVA with Tukey’s post hoc test was used. If the data showed normal distribution but unequal variances, then the Brown-Forsythe and Welch ANOVA with Dunnett’s T3 post hoc test was applied. For data obtained from the AAV protocol, two-way ANOVA with Tukey’s post hoc test was used. Data are presented as mean ± SEM. P-values of < 0.05 were considered significant.
Results
BK Channel Blockade Ameliorates LPS-Induced Depressive-Like Behavior in ICR Mice
Firstly, we evaluated the effects of a specific BK channel inhibitor, paxilline, on the depressive-like behavior induced by LPS in 6–8-week-old male ICR mice using the tail suspension test (TST) and the forced-swim test (FST), the two typical behavioral experiments for depression assessment in mice (Fig. 1a). At 24 h after LPS (0.83 mg/kg, i.p.) injection, the immobility time of mice in the TST was significantly prolonged (395.7 ± 20.3 s compared with 250.5 ± 17.4 s, LPS compared with CON, P = 0.002, n = 7 mice). Paxilline alone (2.2 μg/kg, i.p.) had no impact on TST immobility time, however pre-administration of paxilline reversed the effect of LPS (290.4 ± 17.5 s compared with 395.7 ± 20.3 s, PAX + LPS compared with LPS, P = 0.0385, n = 7 mice) (Fig. 1b). The FSTs were performed 4 h after the TSTs, and similar results were observed (Fig. 1c). This suggests that LPS successfully induced depressive-like behaviors in mice and that the inhibition of BK channels by paxilline could prevent the development of depression.

BK channel blockade ameliorates lipopolysaccharide (LPS)-induced depressive-like behavior. a Experimental timeline of behavioral studies in ICR mice. b The duration of immobility during the tail suspension test (TST) was recorded 24 h following administration of LPS or PBS. c The duration of immobility during the forced-swim test (FST) was recorded 28 h post-LPS or PBS (n = 7–8 mice per group). Group information are as below: CON; control group, mice were injected intraperitoneally (i.p.) with PBS (10 mL/kg) 30 min after vehicle (PBS containing 0.0051% DMSO, 10 mL/kg) application. PAX; mice were injected i.p. with PBS (10 mL/kg) 30 min after paxilline (PAX, 2.2 μg/kg, i.p.) application. LPS; mice were injected i.p. with LPS (0.83 mg/kg) 30 min after vehicle (PBS containing 0.0051% DMSO, 10 mL/kg) application. PAX + LPS; mice were injected i.p. with LPS (0.83 mg/kg) 30 min after paxilline (PAX, 2.2 μg/kg) application. *P < 0.05; **P < 0.01. Data are presented as mean ± SEM. Statistical significance was determined using an ordinary one-way ANOVA with Tukey’s post hoc test
BK Channel Blockade Inhibits LPS-Induced Microglia Activation and IL-1β Production in the Brains of ICR Mice
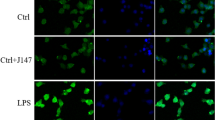
Because microglia activation plays key role in LPS-induced depressive-like behavior [36] and BK channels are reported to be involved in microglia activation [32], we then explored the effect of paxilline on LPS-induced microglia activation in the brain of mice (Fig. 2). As indicated by ionized calcium-binding adapter molecule 1 (Iba1, specific microglia marker) staining in brain slices, microglia displayed amoeboid morphology and the number of microglia increased in LPS group (Fig. 2a and b). There were no differences in either microglia morphology or the number of microglia between the control (CON) and paxilline alone (PAX) group while paxilline suppressed LPS-induced morphology change and the increase in the number of microglia (Fig. 2a and b). Furthermore, in hippocampus tissue, the transcript abundances of both the IL-1β- and TNF-α-encoding genes were significantly increased after LPS treatment from 1.2 ± 0.1 and 1.2 ± 0.2 to 25.1 ± 3.5 (P = 0.0025) and 6.0 ± 1.9 (P = 0.0147) (n = 7 mice), respectively. Pre-administration of paxilline attenuated the increased transcript abundance of IL-1β (from 25.1 ± 3.5 to 15.2 ± 2.3, P = 0.1943, n = 7 mice, Fig. 2c) and that of TNF-α (from 6.0 ± 1.9 to 3.7 ± 0.5, P = 0.3968, n = 7 mice, Fig. 2d) without statistical significance. Interestingly, there was not much difference in the transcript abundance of IL-6 in each group (Fig. 2e). However, in PFC tissue, only the transcript abundance of IL-1β was significantly increased after LPS treatment from 1.0 ± 0.1 to 5.2 ± 1.6 (P = 0.0410, n = 5 mice, Fig. S1a). These results imply that the cytokine production stimulated by systemic administration of LPS is confined to specific regions of the brain. Pre-administration of paxilline slightly lowered the transcript abundance of IL-1β without significance (Fig. S1a). Paxilline alone had no effects on the transcript abundances of all three of these pro-inflammatory cytokines in either hippocampus or PFC (Fig. 2c–e and Fig. S1). This suggests that paxilline did not affect microglia under basal conditions but prevented LPS-induced microglia activation.

BK channel blockade inhibits LPS-induced microglia activation and IL-1β production in the brain of mice. a Representative confocal images of Iba1 immunostaining (red, scale bar = 100 μm). Nucleus was stained with Hoechst 33258 (blue). These experiments were conducted on three mice per group with similar results. b Quantitative analysis of the number of Iba1 positive cells per field (n = 3 different slides per group). c–e The mRNA abundances of genes encoding interleukin-1β (IL-1β, c), TNF-α (d), and interleukin 6 (IL-6, e) in the hippocampus of ICR mice treated with vehicle (CON), PAX, LPS alone, or PAX/LPS in combination (PAX + LPS) (n = 7 mice per group). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant. Data are presented as mean ± SEM. Statistical significance was determined using an ordinary one-way ANOVA with Tukey’s post hoc test (b, d) or using the Brown-Forsythe and Welch ANOVA with Dunnett’s T3 post hoc test (c, e)
BK Channel Blockade Reduces the IDO Protein Level and Activity Induced by LPS in the Brains of ICR Mice
IDO is a specific enzyme catalyzing the degradation of Trp to Kyn. IDO has been reported to be a critical molecular mediator of LPS-induced depressive-like behavior [33]. We wonder whether the prevention of LPS-induced depression by paxilline is correlated with IDO. Next, we measured the levels of IDO, Kyn and Trp in both hippocampus and PFC tissues. The ratio of Kyn to Trp (Kyn/Trp) was used to quantify IDO activity. As shown in Fig. 3, both IDO and Kyn levels were significantly increased from 16.3 ± 0.6 ng/L and 4.2 ± 0.1 μmol/L to 33.8 ± 2.2 ng/L (P < 0.0001, n = 5 mice, Fig. 3a) and 6.1 ± 0.1 μmol/L (P < 0.0001, n = 5 mice, Fig. 3c) with Trp levels unchanged (41.8 ± 0.9 μmol/L compared with 40.0 ± 1.3 μmol/L, LPS compared with CON, P = 0.6750, n = 5 mice, Fig. 3d) by LPS. Therefore, the ratio of Kyn/Trp increased prominently (0.1470 ± 0.0059 compared with 0.1049 ± 0.0038, LPS compared with CON, P < 0.0001, n = 5 mice), indicating elevated IDO activity in the LPS group (Fig. 3b). Paxilline alone had no effect on the levels of IDO, Kyn, or Trp (Fig. 3a-d). However, paxilline could weaken the effect of LPS and decrease both IDO expression level and activity (Fig. 3a–d). Similar trends were observed in the PFC tissue (Fig. S2). These indicate that the inhibition of IDO can be associated with the suppression by paxilline of LPS-induced depressive-like behavior.

BK channel blockade reduces both protein abundance and activity of indoleamine 2,3-dioxygenase (IDO) induced by LPS in the hippocampus of mice. a Protein level of IDO, b Ratio of kynurenine to tryptophan (Kyn/Trp), c Level of kynurenine (Kyn), d Level of tryptophan (Trp) in the hippocampus tissues of ICR mice treated with vehicle (CON), PAX, LPS alone, or PAX/LPS in combination (PAX + LPS) (n = 5 mice per group). ***P < 0.001; ****P < 0.0001; ns, not significant. Data are presented as mean ± SEM. Statistical significance was determined using an ordinary one-way ANOVA with Tukey’s post hoc test
Microglial BK Channel Knockdown Ameliorates LPS-Induced Depressive-Like Behavior in ICR Mice
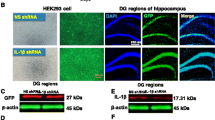
BK channels are expressed in different types of cells in the brain, including microglia and neurons. Therefore, systemic administration of paxilline would broadly block BK channels in various cell types. To further differentiate the role of microglia BK channels in LPS-induced depressive-like behavior, we generated an adeno-associated virus construct (BK shRNA-AAV) containing sequences for the microglia-specific promotor (Iba1), BK shRNA, and EGFP for selective knockdown of BK channels in microglia in vivo. The efficacy of BK shRNA was first tested in vitro. BK plasmid plus either BK shRNA or negative control shRNA (NC shRNA) were transfected into HEK293 cells in the same amounts. BK shRNA knocked down BK channel expression by 73.5%, as shown in Fig. 4a and b, providing evidence for the effectiveness of this shRNA. The BK shRNA-AAV was then injected into the left lateral ventricle, and the brain slices were prepared after 2 weeks. As shown in Fig. 4c, those GFP positive cells were all stained by Iba1 antibody, meaning that these viral constructs were specifically expressed in microglia. Furthermore, these GFP-positive cells show negative or weak BK staining (Fig. 4d), suggesting prominent knockdown of BK channel protein in the AAV-positive cells. All of these results indicate that BK shRNA-AAV selectively knocked down microglia BK channels in mice.

Microglial BK channel knockdown ameliorates LPS-induced depressive-like behavior. a Representative western blot image of BK channels in HEK cells co-transfected with BK shRNA and BK plasmid or negative control (NC) shRNA and BK plasmid. b Quantification of BK protein bands was normalized to β-actin levels (n = 3 biological replicates). **P < 0.01. Data are presented as mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t test. c, d Mice were unilaterally injected intracerebroventricularly with AAV carrying the sequences for the Iba1 promotor, BK shRNA, and GFP (AP -1.22 mm, ML 2.0 mm, DV 2.25 mm). Two weeks after virus injection, the mice were euthanized and whole brains were collected and sliced. The brain slices were then fixed and stained with anti-Iba1 antibody (red, Iba1) and Hoechst 33258 (blue, nucleus) (c) or anti-BK antibody (red, BK) and Hoechst 33258 (blue, nucleus) (d), and samples were analyzed by confocal fluorescence microscopy. Scale bar, 10 μm. These experiments were conducted on three mice with similar results. e Experimental timeline of behavioral studies. Mice were unilaterally injected intracerebroventricularly with BK shRNA-AAV or NC shRNA-AAV. Two weeks after virus injection, mice were injected intraperitoneally (i.p.) with PBS (10 mL/kg, CON) or LPS (0.83 mg/kg). f The duration of immobility during the TST was recorded 24 h following administration of PBS or LPS. g The duration of immobility during the FST was recorded 28 h post-PBS or LPS (n = 5 mice per group). *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SEM. Statistical significance was determined using a two-way ANOVA with Tukey’s post hoc test
After knockdown of microglia BK channels, the depressive-like behavior of mice was measured (Fig. 4e). For mice injected with NC shRNA-AAV, LPS (0.83 mg/kg, i.p.) extended the duration of immobility in both the TST (406.2 ± 11.1 s compared with 334.9 ± 7.9 s, LPS compared with CON, P = 0.0042, n = 5 mice) and the FST (181.4 ± 7.3 s compared with 114.0 ± 9.0 s, LPS compared with CON, P = 0.0003, n = 5 mice) (Fig. 4f and g). In contrast, in mice injected with BK shRNA-AAV, LPS failed to induce such a change in the duration of immobility in either the TST (327.6 ± 15.4 s compared with 319.9 ± 13.5 s, LPS compared with CON, P = 0.9702, n = 5 mice) or the FST (136.4 ± 5.1 s compared with 113.4 ± 12.6 s, LPS compared with CON, P = 0.3007, n = 5 mice). Further, the duration of immobility in the TST and the FST was similar in the control groups of both NC shRNA-AAV and BK shRNA-AAV-injected mice (P > 0.05, n = 5 mice) (Fig. 4f and g). These results suggest that knockdown of microglia BK channels did not affect the basal behavior of these mice but alleviated LPS-induced depressive-like behavior.
Microglial BK Channel Knockdown Suppresses LPS-Induced Microglia Activation and IL-1β Production in the Brains of ICR Mice
Next, mouse brain tissues were collected and examined. We observed that approximately 30% of microglia were GFP positive (Fig. 5a and Fig. S3), evidencing the efficacy of expression of these viral constructs. For mice injected with NC shRNA-AAV, microglia were ramified in the control (CON) group but developed amoebic morphology under LPS administration, indicating the activation of microglia. However, in mice injected with BK shRNA-AAV, microglia cells were ramified in both CON and LPS groups, suggesting that microglia BK channel knockdown inhibited microglia activation by LPS (Fig. 5a). In addition to the morphological changes in microglia, the alteration of microglia cell numbers showed a similar trend. LPS only stimulated microglia proliferation in the NC shRNA-AAV group but not in the BK shRNA-AAV group (Fig. 5b). Furthermore, LPS mainly stimulated the production of IL-1β (141.5 ± 24.7 compared with 1.3 ± 0.5, LPS compared with CON, P < 0.0001, n = 4 mice) (Fig. 5c) rather than TNF-α (4.2 ± 1.4 compared with 1.2 ± 0.4, LPS compared with CON, P = 0.0548, n = 4 mice) (Fig. 5d) or IL-6 (1.7 ± 0.5 compared with 1.1 ± 0.3, LPS vs CON, P = 0.5749, n = 4 mice) (Fig. 5e) in NC shRNA-AAV-injected mouse hippocampal tissue, which was significantly suppressed by BK shRNA-AAV (29.1 ± 5.5 compared with 141.5 ± 24.7, BK shRNA-AAV compared with NC shRNA-AAV, P = 0.0002, n = 4 mice) (Fig. 5c). And no matter which AAV construct was applied, there were no differences in the transcript abundances of IL-1β, TNF-α, or IL-6 under PBS administration (the control groups), indicating that microglia BK channel knockdown had no effect on production of these pro-inflammatory cytokines under basal conditions (Fig. 5c-e). Moreover, in the PFC tissue, there were no notable alterations in the transcript abundances of IL-1β, TNF-α, or IL-6 across all groups (Fig. S4).

Microglial BK channel knockdown suppresses LPS-induced microglia activation and IL-1β production in the brain of mice. a Representative confocal images of Iba1 immunostaining (red, scale bar = 50 μm). GFP (green) indicates the expression of the AAV virus sequences. Nuclei were stained with Hoechst 33258 (blue). These experiments were conducted on three mice per group with similar results. b Quantitative analysis of the number of Iba1-positive cells per field (n = 3 different slides per group). c-e The mRNA abundances of genes encoding interleukin-1β (IL-1β, c), TNF-α (d), and interleukin 6 (IL-6, e) in the hippocampus of BK shRNA-AAV or NC shRNA-AAV-injected ICR mice treated with PBS (CON) or LPS (n = 4 mice per group). **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant. Data are presented as mean ± SEM. Statistical significance was determined using a two-way ANOVA with Tukey’s post hoc test
Microglial BK Channel Knockdown Reduces the IDO Protein Level and Activity Induced by LPS in the Brains of ICR Mice
Finally, the protein abundance and activity of IDO were examined in both hippocampus and PFC tissues of AAV-injected mice (Fig. 6). For mice injected with NC shRNA-AAV, the contents of IDO and Kyn were elevated after LPS treatment from 25.6 ± 0.4 ng/L and 2.2 ± 0.1 μmol/L to 29.2 ± 0.1 ng/L (P = 0.0005, n = 3 mice) and 3.0 ± 0.1 μmol/L (P = 0.0011, n = 3 mice), respectively (Fig. 6a and c). Trp level did not change much (54.6 ± 1.6 μmol/L compared with 52.6 ± 1.4 μmol/L, LPS compared with CON, P = 0.7672, n = 3 mice) (Fig. 6d), so the ratio of Kyn/Trp increased significantly (0.0558 ± 0.0032 compared with 0.0422 ± 0.0002, LPS compared with CON, P = 0.0023, n = 3 mice) (Fig. 6b). However, for mice injected with BK shRNA-AAV, there were no differences in levels of IDO, Kyn, Trp, or the ratio of Kyn/Trp between CON and LPS groups (Fig. 6a–d), suggesting that microglia BK channel knockdown effectively prevented the effect of LPS on IDO expression and activity. Moreover, for mice administered PBS (the control groups), the contents of IDO, Kyn, Trp, and the ratio of Kyn/Trp were similar in both NC shRNA-AAV and BK shRNA-AAV-injected mice (Fig. 6a–d), indicating that microglia BK channel knockdown itself did not affect the expression and activity of IDO. Similar results were obtained with the PFC tissues (Fig. S5).

Microglial BK channel knockdown reduces both protein abundance and activity of IDO induced by LPS in the hippocampus of mice. a Protein level of IDO, b Ratio of kynurenine to tryptophan (Kyn/Trp), c Level of kynurenine (Kyn), d Level of tryptophan (Trp) in the hippocampus tissues of BK shRNA-AAV or NC shRNA-AAV-injected ICR mice treated with PBS (CON) or LPS (n = 3 mice per group). **P < 0.01; ***P < 0.001; ns, not significant. Data are presented as mean ± SEM. Statistical significance was determined using a two-way ANOVA with Tukey’s post hoc test
Discussion
In this study, the blockade of BK channels by specific inhibitor or the selective genetic deletion of BK channels by AAV in microglia prevented LPS-induced prolongation of immobility duration in TSTs and FSTs, the activation of microglia, and the production of IL-1β in hippocampus and the protein level and activity of the IDO in both PFC and hippocampus.
A low dose of paxilline (2.2 μg/kg) was injected i.p. to inhibit BK channels in the brains of mice. This dose of paxilline has been proven sufficient to block BK channels in both neurons [37, 38] and microglia [30] in vivo without affecting motor function. We also observed indistinguishable behavior in mice injected with paxilline alone or vehicle. Although it is difficult to exclude the contribution of BK channel blockade in other cell types on the improvement of depression by paxilline, we obtained similar results by selective knockdown of microglial BK channels with AAV. This implies that BK channels in microglia play a key role in LPS-induced depressive-like behavior.
Studies have shown that the hippocampus and PFC strongly respond to intraperitoneal LPS challenge in rodents [39,40,41]. These two regions are also the most extensively studied brain regions in the LPS-induced depressive mouse model. Therefore, our study focuses on exploring these two brain regions. We examined the transcript abundances of genes encoding three different cytokines including IL-1β, TNF-α, and IL-6, and found that the response to peripheral stimulation by LPS in the hippocampus is stronger than that in the PFC. Increased transcript abundances of both IL-1β and TNF-α were significantly induced by LPS in the hippocampus. However, only IL-1β transcripts were elevated in the PFC, and were 1/5 of their abundance in the hippocampus under LPS stimulation. Additionally, AAV infection itself also affected the production of cytokines, especially IL-1β, following LPS challenge to a certain degree. In AAV- injected mice, the level of IL-1β following LPS administration in both the hippocampus and PFC were almost fivefold that in mice without AAV injection. Similarly, there are differences in the expression and activity of IDO between hippocampus and PFC, although relatively small. The expression of IDO in the hippocampus is higher than that in the PFC, whereas the activity of IDO in the hippocampus is lower than that in the PFC. These spatial differences in the profiles of expression of proinflammatory cytokines and IDO in the murine brain in response to i.p. LPS injection has been reported previously by others as well [42, 43]. However, the underlying mechanism related to the regulation of IL-1β production and IDO activity by microglia BK channels needs further exploration. Moreover, some other brain regions such as the amygdala and hypothalamus are also associated with depression [44], the effect of BK channels on cytokines and IDO in these regions needs to be investigated in the future.
In summary, this study identifies a potential new drug target, BK channels in microglia, for the treatment of inflammation-associated depression. By blockade or knockdown of this channel, the depressive-like behavior induced by LPS in mice can be effectively improved. Thus, BK channel blockers might serve as antidepressants. An important limitation of the present study is the use of only an acute depression mouse model. Whether microglia BK channels broadly mediate depressive conditions associated with other inflammatory states, such as chronic stress, needs further investigation. In addition, drugs specifically targeting BK channels in microglia need to be developed in the future. To achieve this, targeting the microglia-specific auxiliary β3 subunit [30] or developing a nano-drug delivery system might serve as key breakthroughs.
Data Availability
All the data supporting the findings of this study are available on request from the corresponding author.
Abbreviations
- AAV:
-
Adeno-associated virus
- BK:
-
Large conductance calcium- and voltage-dependent potassium channel
- BSA:
-
Bovine serum albumin
- CSF:
-
Cerebrospinal fluid
- DMEM:
-
Dulbecco’s modified eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- ELISA:
-
Enzyme-linked immunosorbent assay
- FBS:
-
Fetal bovine serum
- FST:
-
Forced-swim test
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN-α:
-
Interferon-α
- IL-1β:
-
Interleukin 1β
- IL-6:
-
Interleukin 6
- i.p.:
-
Intraperitoneally
- Kyn:
-
Kynurenine
- LPS:
-
Lipopolysaccharide
- MDD:
-
Major depressive disorder
- NF-κB:
-
Nuclear factor-κB
- PAX:
-
Paxilline
- PBS:
-
Phosphate buffer solution
- PET:
-
Positron emission tomography
- PFA:
-
Paraformaldehyde
- PFC:
-
Prefrontal cortex
- RT-PCR:
-
Real-time PCR
- SNRIs:
-
Serotonin-norepinephrine reuptake inhibitors
- SSRIs:
-
Selective serotonin reuptake inhibitors
- TNF-α:
-
Tumor necrosis factor-α
- Trp:
-
Tryptophan
- TSPO:
-
Translocator protein
- TST:
-
Tail suspension test
References
Collaborators GMD (2022) Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet Psych 9:137–150. https://doi.org/10.1016/s2215-0366(21)00395-3
Malhi GS, Mann JJ (2018) Depression. Lancet 392:2299–2312. https://doi.org/10.1016/s0140-6736(18)31948-2
Hodes GE, Kana V, Menard C, Merad M, Russo SJ (2015) Neuroimmune mechanisms of depression. Nat Neurosci 18:1386–1393. https://doi.org/10.1038/nn.4113
Enache D, Pariante CM, Mondelli V (2019) Markers of central inflammation in major depressive disorder: a systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav Immun 81:24–40. https://doi.org/10.1016/j.bbi.2019.06.015
Afridi R, Suk K (2021) Neuroinflammatory basis of depression: learning from experimental models. Front Cell Neurosci 15:691067. https://doi.org/10.3389/fncel.2021.691067
Wang AK, Miller BJ (2018) Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder, and depression. Schizophr Bull 44:75–83. https://doi.org/10.1093/schbul/sbx035
Levine J, Barak Y, Chengappa KN, Rapoport A, Rebey M, Barak V (1999) Cerebrospinal cytokine levels in patients with acute depression. Neuropsychobiology 40:171–176. https://doi.org/10.1159/000026615
Schaefer M, Capuron L, Friebe A, Diez-Quevedo C, Robaeys G, Neri S et al (2012) Hepatitis C infection, antiviral treatment and mental health: a European expert consensus statement. J Hepatol 57:1379–1390. https://doi.org/10.1016/j.jhep.2012.07.037
Yao R, Pan R, Shang C, Li X, Cheng J, Xu J et al (2020) Translocator protein 18 kDa (TSPO) deficiency inhibits microglial activation and impairs mitochondrial function. Front Pharmacol 11:986. https://doi.org/10.3389/fphar.2020.00986
Li H, Sagar AP, Kéri S (2018) Translocator protein (18kDa TSPO) binding, a marker of microglia, is reduced in major depression during cognitive-behavioral therapy. Prog Neuropsychopharmacol Biol Psychiatry 83:1–7. https://doi.org/10.1016/j.pnpbp.2017.12.011
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G et al (2015) Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiat 72:268–275. https://doi.org/10.1001/jamapsychiatry.2014.2427
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z et al (2011) Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflam 8:94. https://doi.org/10.1186/1742-2094-8-94
Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N (2014) Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun 42:50–59. https://doi.org/10.1016/j.bbi.2014.05.007
Rahimian R, Wakid M, O’Leary LA, Mechawar N (2021) The emerging tale of microglia in psychiatric disorders. Neurosci Biobehav Rev 131:1–29. https://doi.org/10.1016/j.neubiorev.2021.09.023
Clark SM, Pocivavsek A, Nicholson JD, Notarangelo FM, Langenberg P, McMahon RP et al (2016) Reduced kynurenine pathway metabolism and cytokine expression in the prefrontal cortex of depressed individuals. J Psychiatry Neurosci 41:386–394. https://doi.org/10.1503/jpn.150226
Dionisie V, Filip GA, Manea MC, Manea M, Riga S (2021) The anti-inflammatory role of SSRI and SNRI in the treatment of depression: a review of human and rodent research studies. Inflammopharmacology 29:75–90. https://doi.org/10.1007/s10787-020-00777-5
Lyu D, Wang F, Zhang M, Yang W, Huang H, Huang Q et al (2022) Ketamine induces rapid antidepressant effects via the autophagy-NLRP3 inflammasome pathway. Psychopharmacology 239:3201–3212. https://doi.org/10.1007/s00213-022-06201-w
Lu Y, Ding X, Wu X, Huang S (2020) Ketamine inhibits LPS-mediated BV2 microglial inflammation via NMDA receptor blockage. Fundam Clin Pharmacol 34:229–237. https://doi.org/10.1111/fcp.12508
Mariani N, Everson J, Pariante CM, Borsini A (2022) Modulation of microglial activation by antidepressants. J Psychopharmacol 36:131–150. https://doi.org/10.1177/02698811211069110
Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT et al (2008) Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation 5:15. https://doi.org/10.1186/1742-2094-5-15
Nettis MA, Lombardo G, Hastings C, Zajkowska Z, Mariani N, Nikkheslat N et al (2021) Augmentation therapy with minocycline in treatment-resistant depression patients with low-grade peripheral inflammation: results from a double-blind randomised clinical trial. Neuropsychopharmacology 46:939–948. https://doi.org/10.1038/s41386-020-00948-6
Cao ZY, Liu YZ, Li JM, Ruan YM, Yan WJ, Zhong SY et al (2020) Glycyrrhizic acid as an adjunctive treatment for depression through anti-inflammation: a randomized placebo-controlled clinical trial. J Affect Disord 265:247–254. https://doi.org/10.1016/j.jad.2020.01.048
Kang HJ, Bae KY, Kim SW, Kim JT, Park MS, Cho KH et al (2016) Effects of interleukin-6, interleukin-18, and statin use, evaluated at acute stroke, on post-stroke depression during 1-year follow-up. Psychoneuroendocrinology 72:156–160. https://doi.org/10.1016/j.psyneuen.2016.07.001
Sun X, Zaydman MA, Cui J (2012) Regulation of voltage-activated K+ channel gating by transmembrane beta subunits. Front Pharmacol 3:63. https://doi.org/10.3389/fphar.2012.00063
Chen SR, Cai YQ, Pan HL (2009) Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J Neurochem 110:352–362. https://doi.org/10.1111/j.1471-4159.2009.06138.x
Huang S, Chen T, Suo Q, Shi R, Khan H, Ma Y et al (2021) BK channel-mediated microglial phagocytosis alleviates neurological deficit after ischemic stroke. Front Cell Neurosci 15:683769. https://doi.org/10.3389/fncel.2021.683769
Sun X (2023) BK channels in microglia. Brain Sci Adv 9:15–23. https://doi.org/10.26599/BSA.2023.9050001
Schilling T, Eder C (2007) Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res 1186:21–28. https://doi.org/10.1016/j.brainres.2007.10.027
Hayashi Y, Kawaji K, Sun L, Zhang X, Koyano K, Yokoyama T et al (2011) Microglial Ca2+-activated K+ channels are possible molecular targets for the analgesic effects of S-ketamine on neuropathic pain. J Neurosci 31:17370–17382. https://doi.org/10.1523/jneurosci.4152-11.2011
Hayashi Y, Morinaga S, Zhang J, Satoh Y, Meredith AL, Nakata T et al (2016) BK channels in microglia are required for morphine-induced hyperalgesia. Nat Commun 7:11697. https://doi.org/10.1038/ncomms11697
Ma J, Wang J, Deng K, Gao Y, Xiao W, Hou J et al (2022) The effect of MaxiK channel on regulating the activation of NLRP3 inflammasome in rats of blast-induced traumatic brain injury. Neuroscience 482:132–142. https://doi.org/10.1016/j.neuroscience.2021.12.019
Yang X, Wang G, Cao T, Zhang L, Ma Y, Jiang S et al (2019) Large-conductance calcium-activated potassium channels mediate lipopolysaccharide-induced activation of murine microglia. J Biol Chem 294:12921–12932. https://doi.org/10.1074/jbc.RA118.006425
O’Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N et al (2009) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14:511–522. https://doi.org/10.1038/sj.mp.4002148
Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, Gould TD (2012) The tail suspension test. J Vis Exp 59:e3769. https://doi.org/10.3791/3769
Can A, Dao DT, Arad M, Terrillion CE, Piantadosi SC, Gould TD (2012) The mouse forced swim test. J Vis Exp 59:e3638. https://doi.org/10.3791/3638
Li W, Ali T, Zheng C, Liu Z, He K, Shah FA et al (2021) Fluoxetine regulates eEF2 activity (phosphorylation) via HDAC1 inhibitory mechanism in an LPS-induced mouse model of depression. J Neuroinflam 18:38. https://doi.org/10.1186/s12974-021-02091-5
Sheehan JJ, Benedetti BL, Barth AL (2009) Anticonvulsant effects of the BK-channel antagonist paxilline. Epilepsia 50:711–720. https://doi.org/10.1111/j.1528-1167.2008.01888.x
Liu J, Ye J, Zou X, Xu Z, Feng Y, Zou X et al (2014) CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat Commun 5:3924. https://doi.org/10.1038/ncomms4924
Yu X, Yao H, Zhang X, Liu L, Liu S, Dong Y (2022) Comparison of LPS and MS-induced depressive mouse model: behavior, inflammation and biochemical changes. BMC Psychiatry 22:590. https://doi.org/10.1186/s12888-022-04233-2
Alzarea S, Abbas M, Ronan PJ, Lutfy K, Rahman S (2022) The effect of an α-7 nicotinic allosteric modulator PNU120596 and NMDA receptor antagonist memantine on depressive-like behavior induced by LPS in mice: the involvement of brain microglia. Brain Sci 12:1493. https://doi.org/10.3390/brainsci12111493
Sales AJ, Joca SRL, Del Bel E, Guimarães FS (2024) The antidepressant-like effect of doxycycline is associated with decreased nitric oxide metabolite levels in the prefrontal cortex. Behav Brain Res 458:114764. https://doi.org/10.1016/j.bbr.2023.114764
André C, O’Connor JC, Kelley KW, Lestage J, Dantzer R, Castanon N (2008) Spatio-temporal differences in the profile of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration. J Neuroimmunol 200:90–99. https://doi.org/10.1016/j.jneuroim.2008.06.011
Bay-Richter C, Janelidze S, Hallberg L, Brundin L (2011) Changes in behaviour and cytokine expression upon a peripheral immune challenge. Behav Brain Res 222:193–199. https://doi.org/10.1016/j.bbr.2011.03.060
Corrigan M, O’Rourke AM, Moran B, Fletcher JM, Harkin A (2023) Inflammation in the pathogenesis of depression: a disorder of neuroimmune origin. Neuro Signal. https://doi.org/10.1042/ns20220054
Funding
This work was supported by the National Natural Science Foundation of China (31400924), the National Innovation of Science and Technology-2030 (Program of Brain Science and Brain-Inspired Intelligence Technology) Grant (2021ZD0204004), National Key Research and Development Program of China (2021YFE0206000) and Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD).
Author information
Authors and Affiliations
Contributions
XS and XZ designed the research. GL and LH performed the experiments. XG and WZ helped to perform part of biological assays. GL prepared the original draft. XS and XZ revised the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical Approval
This study was approved by the Animal Care and Use Committee of Soochow University (Approval number 202201A0459), and all experiments were performed in accordance with all relevant ethical regulations approved by Soochow University.
Additional information
Publisher's Note
Springer nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, G., Hu, L., Gu, X. et al. Targeting Large-Conductance Calcium-Activated Potassium Channels to Ameliorate Lipopolysaccharide-Induced Depressive-Like Behavior in Mice. Neurochem Res 49, 1239–1253 (2024). https://doi.org/10.1007/s11064-024-04111-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-024-04111-1