
Abstract
Narirutin (Nar) is a flavonoid that is abundantly present in citrus fruits and has attracted considerable attention because of its diverse pharmacological activities and low toxicity. Here, we evaluated the preventive effects of Nar in middle cerebral artery occlusion/reperfusion (MCAO/R)-injured mice and oxygen–glucose deprivation/reperfusion (OGD/R)-injured bEnd.3 cells. Pretreatment with Nar (150 mg/kg) for 7 days effectively reduced infarct volume, improved neurological deficits, and significantly inhibited neuronal death in the hippocampus and cortex in MCAO/R-injured mice. Moreover, anti-apoptotic effects of Nar (50 µM) were observed in OGD/R-injured bEnd.3 cells. In addition, Nar pre-administration regulated blood-brain barrier function by increasing tight junction-related protein expression after MCAO/R and OGD/R injury. Nar also inhibited NOD-like receptor protein 3 (NLRP3) inflammasome activation by reducing the expression of thioredoxin-interacting protein (TXNIP) in vivo and in vitro. Taken together, these results provide new evidence for the use of Nar in the prevention and treatment of ischemic stroke.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is considered a major fatal disease worldwide, with high incidence, mortality, disability, and recurrence rates [1]. Nearly 87% of stroke cases result from ischemic stroke. At present, there is a lack of effective treatments for ischemic stroke, mainly because of the complex molecular mechanisms underlying this disease [2]. Ischemic stroke occurs when a sudden lack of blood flow, oxygen, or nutrients to the brain leads to rapid cell death. Rapid reperfusion is the standard treatment for cerebral ischemia; however, it usually causes inflammation, oxidative stress, and damage to the blood-brain barrier (BBB), resulting in severe neuronal damage and ischemic neuronal cell death [3, 4]. Therefore, prevention may be an effective strategy to protect against cerebral ischemia-reperfusion (I/R) injury [5].
Previous studies have shown that inflammation plays a key role in stroke pathology and has a significant impact on stroke prognosis [6, 7]. The increase in inflammatory cell infiltration mediated by inflammatory factors leads to the release of inflammatory mediators and reactive oxygen species, which trigger an inflammatory cascade, causing neurovascular, BBB, and neuronal injury [8]. Therefore, inhibition of the inflammatory response may be effective in controlling ischemic stroke. The NOD-like receptor protein 3 (NLRP3) inflammasome, a multimolecular complex comprising the NLRP3 receptor, an apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1, is currently considered an important molecular player for regulating the release of pro-inflammatory cytokines and mediating inflammatory damage after cerebral ischemia [9,10,11]. Many drugs and natural substances that inhibit NLRP3 inflammasome activation have been studied for the treatment of ischemic stroke. For example, NLRP3 inflammasome inhibitors, such as MCC950, CY-09, and the herbal component oridonin, have been shown to reduce infarct volume and improve neurological deficits and BBB integrity by inhibiting inflammatory cytokines, pyroptosis, and oxidative stress in ischemic regions after cerebral I/R injury [11,12,13,14]. Therefore, the NLRP3 inflammasome has been identified as a potential therapeutic target for ischemic stroke. In addition, recent studies have reported that thioredoxin-interacting protein (TXNIP), a redox-regulated protein, dissociates from the complex under oxidative stress conditions, such as stroke, and rapidly binds to and activates the NLRP3 inflammasome [15, 16]. Furthermore, TXNIP binding to NLRP3 is crucial for inflammasome activation, and inhibition of TXNIP could directly reduce the activation of the NLRP3 inflammasome. Also, the expression of TXNIP and the NLRP3 inflammasome exhibit the same trend in cerebral I/R injury [15, 17, 18]. Therefore, inhibiting activation of the TXNIP/NLRP3 pathway may be an important strategy for the prevention and treatment of cerebral I/R injury.
Narirutin (Nar) is a flavonoid that is abundantly present in citrus fruits, such as grapefruits, mandarins, and oranges [19]. Nar has been reported to exert multiple biological activities, including anti-inflammatory, antioxidant, anti-obesity, anti-allergy, neuroprotective, anti-Alzheimer’s disease, and anti-tumor activities [20,21,22,23,24]. A recent study indicated that the Nar-rich fraction obtained from grapefruit peels can protect against cerebral I/R injury caused by transient bilateral common carotid artery occlusion (tBCCAO) in rats by reducing oxidative damage [25]. In addition, Ri et al. reported that the anti-inflammatory properties of Nar were mediated by the inhibition of NLRP3 inflammasome priming processes and NLRP3-ASC interactions in macrophages, thereby suppressing the NLRP3 inflammasome [26]. However, it is unclear whether Nar plays a neuroprotective role in cerebral I/R injury by suppressing NLRP3 inflammasome activation.
In the present study, an in vivo middle cerebral artery occlusion/reperfusion (MCAO/R) mouse model and an in vitro oxygen–glucose deprivation/reperfusion (OGD/R) cell model were established to determine the preventive effects of Nar on cerebral I/R injury and to explore the possible underlying molecular mechanisms, thus providing a basis for the development of new therapies to prevent cerebral I/R injury.
Materials and Methods
Drugs and Antibodies
Nar was purchased from Shanghai Pure One Biotechnology (purity > 98%, Shanghai, China). The following antibodies were used: anti-Bax (14,796, Cell Signaling Technology, 1:1000), anti-Caspase-3 (9664, Cell Signaling Technology, 1:1000), anti-Bcl-2 (3498, Cell Signaling Technology, 1:1000), anti-P-glycoprotein (ab170904, Abcam, 1:1000), anti-ZO-1 (21773-1-AP; Proteintech, 1:500), anti-occludin (13409-1-AP, Proteintech, 1:1000), anti-claudin3 (ab15102, Abcam, 1:1000), anti-TXNIP (ab188865, Abcam, 1:1000), anti-NLRP3 (15,101 S, Cell Signaling Technology, 1:1000), anti-ASC (NBP1-78977, Novus Biologicals, 1:1000), anti-Caspase-1 (24,232 S, Cell Signaling Technology, 1:1000), anti-IL-1β (16806-1-AP, Proteintech, 1:1000), anti-IL-18 (ab207323, Abcam, 1:1000), and anti-β-actin (A5441, Sigma, 1:10000).
Animals and Treatment
Male C57BL/6 mice (6–8 week) were used in the study. All mice were maintained at a temperature of 25 ± 1 °C, a relative humidity of 50–60%, and under a 12:12 h light–dark cycle. Food and water were freely available to the mice, which were kept in random groups of six per cage. Before the experiments, all mice were fed a commercial chow diet and acclimated to the laboratory environment for at least 1 week. All procedures were approved by the Ethics Committee for Animal Experimentation of the Fourth Military Medical University (approval no. KY20193145).
The mice were randomly divided into four groups: sham, sham + Nar (150 mg/kg), MCAO/R, and MCAO/R + Nar (150 mg/kg). Each group consisted of 16 mice. The dose of Nar was selected based on a previous study [25]. Nar was dissolved in 0.9% saline containing 1% dimethyl sulfoxide (DMSO) and administered daily via oral gavage for seven consecutive days before MCAO/R. The sham and MCAO/R groups were injected with an equal volume of 0.9% saline containing 1% DMSO at the same time.
In Vivo Experiments
MCAO/R Model
The MCAO model was established as described previously [27]. Briefly, anesthetized mice were exposed to right carotid bifurcation. A nylon monofilament (RWD Life Science, San Diego, CA, USA) was inserted into the common carotid artery and the anchored line was inserted into the internal carotid artery after the external carotid artery was cut open. Blood flow was monitored using a Laser Speckle Doppler flowmeter (RFLSI III; RWD Life Sciences). Cerebral blood flow was restored by removing the monofilament after 1.5 h of transient occlusion, which was monitored to guarantee blood reperfusion. A similar treatment was administered to the sham and sham + Nar groups but without a monofilament. Brain samples were collected 24 h after reperfusion.
Neurobehavioral Evaluation
Neurobehavioral tests were conducted 24 h after reperfusion using a modified neurological severity scoring (mNSS) system, according to a previous report [27]. The higher the mNSS score (0, normal; 18, maximal deficit), the more severe the neurological and behavioral disorders.
Assessment of Infarct Volume
Infarct volume was assessed using 2,3,5-triphenyltetrazolium chloride (TTC) staining as previously described [28]. Briefly, the mice were sacrificed, and the entire brain was rapidly removed and cooled for 30 min at − 20 °C. Starting from the frontal pole, coronal Sect. (2 mm) were dissected, immersed in 2% TTC for 30 min at 37 °C, and fixed with 4% paraformaldehyde for 24 h. After obtaining digital images of the brain slices, unstained areas (white areas) were defined as infarcts, and the sizes of the infarcts on both sides of each slice were measured using ImageJ software (NIH, Bethesda, MD, USA).
Hematoxylin and Eosin Staining
To evaluate pathological changes, hematoxylin and eosin (HE) staining was performed as previously reported [29]. The brains were perfused with 4% paraformaldehyde for 4 h at 4 °C, then dehydrated in 20% and 30% sucrose solutions for 24 h. Brain slices (5 μm) containing the hippocampus and cortex were cut using a cryostat (Leica CM1800) and then immediately mounted on slides. Sections were stained with hematoxylin solution for 15 min, counterstained with eosin solution for 5 min, subjected to gradient alcohol dehydration, made transparent, and sealed. The sections were observed under a high-power bright-field microscope (Nikon Ni). Cells with clearly visible nucleoli were considered intact and the proportion of intact cells was recorded.
Nissl Staining
Morphological changes in ischemic penumbra cells after MCAO/R were observed using Nissl staining [27]. Brain slices were prepared as previously described [22]. Frozen sections were stained with 0.1% toluidine blue for 5 min, rinsed with phosphate-buffered saline (PBS), dehydrated in a graded alcohol series, and mounted with a neutral sealant. The number of Nissl-positive cells in the penumbra of each section was counted under a light microscope (FV-1000; Olympus, Tokyo, Japan).
TUNEL Staining
A fluorescent terminal deoxynucleotidyl transferase nick-end labeling (TUNEL) kit was used to quantify cell death according to the manufacturer’s protocol (Roche Diagnostics Corporation, Indianapolis, IN, USA) [27]. The number of TUNEL-positive cells in the hippocampal CA1 region and cortex were counted using light microscopy at ×400 magnification (FV-1000; Olympus, Tokyo, Japan).
In Vitro Experiments
Cell Culture and Drug Treatment
Mouse brain microvascular vessel-derived endothelial (bEnd.3) cells were obtained from Sixin Biotechnology (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (AF29498406, HyClone) supplemented with 10% fetal bovine serum (2206993CP, Gibco) and 1% penicillin–streptomycin mixture (20191028, Solarbio). The medium was changed every 2 days.
The cells were divided into four groups: control, control + Nar, OGD/R, and OGD/R + Nar. Cells in the control and control + Nar groups were cultured under normal conditions. Nar was dissolved in DMSO and added to the cell culture medium at a final concentration of 0.1%. The effective concentration of Nar was determined by subjecting cells to a cell counting kit-8 (CCK-8) assay after exposure to various Nar concentrations (12.5, 25, 50, 100, or 200 µM). Nar was administered 4, 8, 12, and 24 h before OGD to determine the effective administration time.
OGD/R Model In Vitro
bEnd.3 cells were subjected to anoxia-glucose deprivation followed by glucose reoxygenation to simulate an in vitro ischemia/reperfusion environment [27]. Briefly, the cells were rinsed thrice with PBS, and glucose-free DMEM (2120596, GIBCO) was used to replace the normal medium. The cells were then transferred to a hypoxic chamber (95% N2/5% CO2, MIC-101, Billups-Rothenberg, USA) and incubated for 1.5 h at 37 °C. The cells were then cultured for an additional 24 h of reperfusion under normal conditions.
Cell Viability Analysis
Cell viability was determined using a CCK-8 assay according to the manufacturer’s instructions [27]. Briefly, 10 µl of CCK-8 reagent (BS350B, Biosharp, Hefei, China) was added to each well and cells were incubated at 37 °C for 4 h. Absorbance was measured at 490 nm using a microplate reader (Infinite M200 Pro, TECAN). Cell viability (%) = [OD (experiment) × OD (blank)]/[OD (control) × OD (blank)] ×100.
Lactate Dehydrogenase Release Assay
Cytotoxicity was quantitatively detected by measuring the lactate dehydrogenase (LDH) activity released by injured cells into the medium [30]. After treating the cells with 0.5% Triton X-100, the medium containing detached cells was collected and centrifuged. The supernatant was used for an LDH activity assay using an assay kit (Beyotime Biotechnology) according to the manufacturer’s instructions.
Flow Cytometric Analysis
The annexin V/propidium iodide (PI) double staining procedure was used to assess apoptosis [27]. Briefly, 1 × 106 cells were seeded into each well of a six-well plate. Cells were harvested and washed with 1× Annexin V-FITC binding buffer, then stained with Annexin V-FITC (5 µl) and PI (5 µl) at 25 °C in the dark for 15 min. Cell fluorescence was immediately analyzed using flow cytometry. Annexin V-FITC-positive and PI-positive cells were used to calculate apoptotic rates.
Measurement of Transendothelial Electrical Resistance
Transendothelial electrical resistance (TEER) was measured using a Millicell ERS cell resistor (Millipore, USA) to assess the integrity of the monolayer cell barrier [27]. Briefly, 2 × 105 cells were seeded into the upper chamber of a Transwell insertion chamber (3460; Corning, NY, USA) in 0.5 ml DMEM (1.5 ml of the same medium was added to the lower chamber to inhibit pressure gradient formation). The TEER value (Ω cm2) was measured by subtracting the resistance of a blank Transwell chamber without cells and standardized by the area of the culture insert.
FITC-Dextran Assay
A FITC-dextran assay was used to investigate cell permeability, as previously reported [27]. After OGD and reoxygenation for 24 h, the medium was removed, and fresh medium containing 1% FITC-dextran (MW 1000, MCE) was added to the cells for 30 min. Then, 100 µl medium from the lower chamber was obtained, and the fluorescence intensity was measured using a microplate reader (Infinite M200 Pro, TECAN) at the excitation and emission wavelengths of 490 and 520 nm, respectively. The relative cell permeability was calculated by normalizing to the control group.
Western Blot Analysis
Western blot analysis was performed as previously described [31]. Ischemic penumbra tissue and cells were homogenized in ice-cold RIPA lysis buffer containing phosphatase and protease inhibitors. Protein concentration was determined using a bicinchoninic acid protein assay. Proteins were analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene fluoride membranes (Invitrogen, Carlsbad, CA, USA). The membranes were then incubated with primary antibodies overnight at 4 °C. Subsequently, the membranes were washed thrice with TBST and incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit or anti-mouse IgG, 1:10000). Protein bands were quantified using ImageJ (NIH, Bethesda, MD, USA). For data analysis, band intensities were normalized to actin and expressed as percentages of the relative intensities.
Statistical Analysis
Data analysis was performed using Prism 8 software (GraphPad Software, San Diego, CA, USA). Data are expressed as the mean ± SEM for at least three independent experiments. Multigroup analysis were performed using one-way analysis of variance (ANOVA). p < 0.05 was considered statistically significant.
Results
Nar Decreases Infarct Volume and Improves Neurological Deficits in MCAO/R Injured Mice
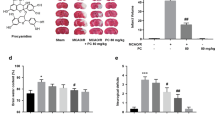
To determine the protective effects of Nar against MCAO/R injury, we performed TTC staining and neurological evaluations. The results indicated that, compared to the sham group, MCAO/R injury caused a larger infarct volume (Fig. 1B, C) and a significant increase in neurological deficits (Fig. 1D). Pretreatment with Nar (150 mg/kg) before MCAO/R injury significantly reduced infarct size (MCAO/R group vs. MCAO/R + Nar group: 36.25 ± 1.98% vs. 21.50 ± 1.54%) and improved neurological impairment (MCAO/R group vs. MCAO/R + Nar group: 9.75 ± 0.53 vs. 5.25 ± 0.37). No obvious differences were observed between the Sham and Sham + Nar groups in the above tests. These results suggest that pretreatment with Nar protected MCAO/R mice from injury.

Pretreatment with narirutin reduces MCAO-induced brain infarction and neurological deficits. A The chemical structure of narirutin (Nar). B Representative photographs of TTC staining 24 h after MCAO/R injury. C Statistical analysis of infarct volume. D Neurological deficit scores 24 h after MCAO/R injury. n = 8 mice per group; ∗∗p < 0.01 versus Sham group; ##p < 0.01 versus MCAO/R group, one-way ANOVA with Tukey’s multiple comparisons test
Nar Reduces Neuronal Death After MCAO/R Injury
To examine the effect of Nar on neuronal death, HE and Nissl staining were performed to assess morphological changes in the hippocampus and cortex. In the Sham and Sham + Nar groups, the cell morphology and density were normal, and no cell death was observed in the hippocampal CA1 region (Fig. 2A–D). Similarly, there were no obvious changes in cell morphology or density in the cortex between the Sham and Sham + Nar groups (Fig. S1A–D), indicating that administration of Nar had no effect on normal cells. However, most MCAO/R-injured mice displayed a disordered cell arrangement and neuronal death, and pretreatment with Nar significantly ameliorated these pathological abnormalities in the hippocampal CA1 region (Fig. 2A–D) and cortex (Fig. S1A–D). Consistent with these results, TUNEL staining revealed that the percentage of apoptotic cells in the hippocampal CA1 region was significantly higher in the MCAO/R group compared to the Sham group, but was obviously reduced after Nar pretreatment (Fig. 3A, B). Meanwhile, the same results were observed in the cortex, where the number of TUNEL-positive cells decreased remarkably in the MCAO/R + Nar group compared to the Sham group (Fig. S1E, F). In addition, compared with the Sham and Sham + Nar groups, the expression of the apoptosis-related proteins Caspase-3 and Bax in the infarct area of MCAO/R mice was elevated and that of Bcl-2 was decreased, while pretreatment with Nar effectively reversed these alterations (Fig. 3C–F). These findings further indicate that pre-administration of Nar can inhibit neuronal death after MCAO/R in mice.

Pretreatment with narirutin improves morphological changes in the hippocampus of MCAO/R-injured mice. A, B HE staining showing morphological neuronal changes (A) and the number of intact cells (B) in the hippocampal CA1 region after MCAO/R injury. Scale bar = 50 μm. C, D Nissl staining showing morphological neuronal changes (C) and the number of intact cells (D) in the hippocampal CA1 region after MCAO/R injury. Scale bar = 50 μm. n = 6 slices from two mice per group; ∗∗p < 0.01 versus Sham group; ##p < 0.01 versus MCAO/R group; ns not significant, one-way ANOVA with Tukey’s multiple comparisons test

Narirutin inhibits neuronal death in the hippocampus of MCAO/R-injured mice. A, B TUNEL staining showing the survival of neurons (A) and the number of TUNEL-positive cells (B) in the hippocampal CA1 region after MCAO/R injury. Scale bar = 50 μm. n = 6 slices from two mice per group. C–F Western blotting analysis of apoptosis-related proteins Caspase-3 (D), Bax (E), and Bcl-2 (F) in the infarct brain tissue. n = 6 mice per group; ∗∗p < 0.01 versus Sham group; ##p < 0.01 versus MCAO/R group; ns not significant, one-way ANOVA with Tukey’s multiple comparisons test
Nar Inhibits OGD/R-Induced Apoptosis of bEnd.3 Cells
We further evaluated the protective effects of Nar against apoptosis in bEnd.3 cells. First, CCK-8 and LDH release assays were performed to determine the appropriate Nar concentration and time of administration. Normal cultured bEnd.3 cells were treated with different concentrations of Nar (12.5, 25, 50, 100, or 200 µM), and toxicity was evaluated using the CCK-8 assay. The results showed no significant changes in cell viability (Fig. 4A), indicating that Nar did not have a cytotoxic effect on bEnd.3 cells. Next, we explored the effective concentration of Nar and found that cell viability was significantly increased after co-incubation with 25, 50, 100, or 200 µM Nar compared to the untreated OGD/R group, while there was no significant change in cell viability after exposure to 12.5 µM Nar (Fig. 4B). Therefore, we chose 50 µM as the effective concentration for subsequent experiments. Next, the cells were treated with Nar (50 µM) for 4, 8, 12, or 24 h before exposure to OGD/R to determine the appropriate administration time. Cell viability was dramatically reduced (Fig. 4C) and the release of LDH was significantly elevated (Fig. 4D) in the OGD/R group compared to the control group, and pre-administration of Nar reversed these changes at 8, 12, and 24 h. Finally, we exposed bEnd.3 cells to Nar for 8 h with 50 µM for further analysis of the anti-apoptotic effects. Flow cytometric analysis revealed that pretreatment with Nar significantly decreased the apoptotic index of bEnd.3 cells (Fig. 5A, B) compared to that in the OGD/R group. Moreover, the elevated Caspase-3 and Bax expression and reduced Bcl-2 expression observed in OGD/R cells were reversed by Nar administration (Fig. 5C–F). Taken together, these results show that pre-administration of Nar prevents neuronal apoptosis after OGD/R in vitro.

Pretreatment with narirutin increases the viability of bEnd.3 cells following OGD/R injury and reduces LDH release. A Normal bEnd.3 cells were cultured with different concentrations of narirutin (12.5, 25, 50, 100, or 200 µM), and cell viability was assessed using a CCK-8 assay. B CCK-8 assay showing the effect of narirutin treatment (12.5, 25, 50, 100, or 200 µM) for 24 h before exposure to OGD/R injury on the viability of bEnd.3 cells. C Cells were treated with narirutin (50 µM) for 4, 8, 12, or 24 h before exposure to OGD/R injury. D LDH release assay showing the effect of narirutin treatment (50 µM) for 4, 8, 12, or 24 h before exposure to OGD/R injury on cytotoxicity of bEnd.3 cells. n = 6; ∗∗p < 0.01 versus Control group; #p < 0.05, ##p < 0.01 versus OGD/R group, one-way ANOVA with Tukey’s multiple comparisons test

Narirutin inhibits OGD/R-induced apoptosis of bEnd.3 cells. A Flow cytometric analysis of apoptosis in bEnd.3 cells 24 h after OGD/R injury. B Statistical analysis of the percentage of apoptotic cells. C–F Western blotting analysis of apoptosis-related proteins Caspase-3 (D), Bax (E), and Bcl-2 (F) in bEnd.3 cells 24 h after OGD/R injury. n = 6; ∗∗p < 0.01 versus Control group; ##p < 0.01 versus OGD/R group; ns not significant, one-way ANOVA with Tukey’s multiple comparisons test
Nar Improves BBB Permeability After MCAO/R Injury In Vivo and OGD/R Injury In Vitro
After cerebral I/R injury, secondary brain injury due to BBB disruption is the main cause of death [32]. Thus, BBB repair may be an effective strategy for treating cerebral I/R injury. Tight junctions (TJs) are critical structural components of the BBB; hence, we investigated whether pretreatment with Nar could protect the damaged BBB after MCAO/R by measuring the expression of TJ-related proteins. Compared to the Sham and Sham + Nar groups, the expression of P-glycoprotein (P-gp), ZO-1, occludin, and claudin3 was remarkably decreased in the MCAO/R group, and pretreatment with Nar effectively reversed these changes (Fig. 6A–E). In addition, we further confirmed the effect of Nar on BBB permeability by performing a FITC-dextran leakage test (Fig. 7A) and TEER measurements (Fig. 7B) in bEnd.3 cells after OGD/R. The results showed that the administration of Nar reversed the OGD/R-induced low FITC-dextran penetration and high TEER values. Moreover, the decreased expression of P-gp, ZO-1, occludin, and claudin3 in the OGD/R group was significantly increased by Nar treatment (Fig. 7C–G). These data suggest that pretreatment with Nar can regulate BBB permeability and maintain BBB integrity after MCAO/R in vivo and OGD/R in vitro.

Narirutin upregulates the expression of TJ-related proteins after MCAO/R injury. A Representative western blot bands of TJ-related proteins. B–E Quantitative analysis of the expression levels of P-gp (B), ZO-1 (C), Occludin (D), and Claudin3 (E) in the infarct brain tissue. n = 6 mice per group; ∗∗p < 0.01 versus Sham group; ##p < 0.01 versus MCAO/R group; ns not significant, one-way ANOVA with Tukey’s multiple comparisons test

Narirutin improves BBB permeability and increases the expression of TJ-related proteins after OGD/R injury. A FITC-dextran leakage test in OGD/R-injured bEnd.3 cells after pretreatment with narirutin (50 µM) for 8 h. B TEER values of a monolayer of OGD/R-injured bEnd.3 cells after pretreatment with narirutin (50 µM) for 8 h. C–G Western blotting analysis of TJ-related proteins P-gp (D), ZO-1 (E), Occludin (F), and Claudin3 (G) in the different groups. n = 6; ∗∗p < 0.01 versus Control group; ##p < 0.01 versus OGD/R group; ns not significant, one-way ANOVA with Tukey’s multiple comparisons test
Nar Inhibits TXNIP/NLRP3 Pathway Activation Following MCAO/R and OGD/R
To assess whether Nar regulates the TXNIP/NLRP3 pathway in MCAO/R- and OGD/R-induced injury, we analyzed the expression levels of relevant pathway molecules using western blotting. The results demonstrated that pretreatment with Nar effectively inhibited the expression of TXNIP, NLRP3, ASC, Caspase-1, IL-1β, and IL-18 in MCAO/R-injured mice, and no significant difference was observed between the Sham and Sham + Nar groups (Fig. 8). Similar results were observed in bEnd.3 cells (Fig. S2). The expression of these proteins increased significantly after OGD/R and was effectively blocked by Nar exposure. Therefore, these results suggest that Nar inhibits the activation of the TXNIP/NLRP3 signaling pathway after MCAO/R and OGD/R injuries.

Narirutin inhibits TXNIP/NLRP3 pathway activation following MCAO/R injury. A Representative western blot bands of TXNIP, NLRP3, ASC, Caspase-1, IL-1β, and IL-18 in MCAO/R-injured mice. B–G Quantitative analysis of the expression levels of TXNIP (B), NLRP3 (C), ASC (D), Caspase-1 (E), IL-1β (F), and IL-18 (G) in the infarct brain tissue. n = 6 mice per group; ∗∗p < 0.01 versus Sham group; ##p < 0.01 versus MCAO/R group; ns not significant, one-way ANOVA with Tukey’s multiple comparisons test
Discussion
Previous studies have demonstrated that pretreatment with a Nar-rich fraction (150 and 300 mg/kg) for 7 days reduced tBCCAO-induced cerebral I/R injury in rats through its free radical-scavenging capacity and ability to ameliorate oxidative damage [25]. In the present study, we further demonstrated that pretreatment with Nar (150 mg/kg) significantly reduced infarct size, neurological deficits, histopathological damage, and neuronal death in MCAO/R-injured mice, and these effects may be related to the regulation of BBB damage and the inhibition of TXNIP/NLRP3 activation. The results of in vitro experiments showed that co-incubation with Nar (25, 50, 100, and 200 µM) increased viability in OGD/R-injured bEnd.3 cells. Moreover, pre-administration with 50 µM Nar for 8 h effectively reduced neuronal apoptosis and endothelial barrier leakage, up-regulated the expression of TJ-related proteins, and regulated the TXNIP/NLRP3 inflammasome. Together, these data suggest that Nar may be considered an effective potential candidate drug for the prevention of ischemic stroke.
Increasing evidence indicates that BBB dysfunction is a critical pathological mechanism in ischemic stroke [33, 34]. The BBB is primarily composed of endothelial cells, pericytes, neurons, astrocytes, and basement membranes [35]. The key components of the BBB are the brain microvascular endothelial cells, which are linked through interactions between TJ proteins to form a physical “barrier” essential for maintaining BBB integrity [35]. Among the TJ components, P-gp, ZO-1, occluding, and claudin3 are the most important proteins [36]. Previous studies by our laboratory and others have indicated that BBB permeability and integrity are impaired after MCAO/R and OGD/R injuries [27, 37]. Our findings in the present study are consistent with previous reports, as we found that P-gp, ZO-1, occludin, and claudin3 were decreased in MCAO/R-injured mice. Similar results were observed in OGD/R-injured bEnd.3 cells, with exhibited reduced TJ-related proteins expression, low FITC-dextran penetration, and high TEER values. Notably, our findings suggest that pre-administration of Nar can restore BBB permeability and integrity after MCAO/R injury in vivo and OGD/R injury in vitro, indicating that regulation of BBB function may be an important mechanistic component underlying the protective effects of Nar, contributing to the observed reduction in cell death.
Inflammation plays a key role in the pathology of ischemic stroke, and previous studies have shown that it is a major cause of BBB breakdown [38,39,40]. Therefore, targeting neuroinflammation may be a promising therapeutic strategy for treating ischemic stroke. As the most well-characterized inflammasome, the NLRP3 inflammasome is closely associated with the inflammatory response and plays an important role in the development of ischemic stroke [10, 11]. The NLRP3 inflammasome promotes the formation of active caspase-1, which leads to the release of inflammatory cytokines IL-1β and IL-18. In addition, studies have shown that the upregulation of NLRP3 is an early event occurring within the first 24 h of cerebral I/R injury, corresponding to the hyperacute and acute phases of human stroke [11, 41]. Yang et al. found that NLRP3 deficiency reduced brain damage in mice after ischemic stroke by reducing cerebral infarction and BBB damage [42]. Therefore, blocking the NLRP3 inflammasome is an important therapeutic strategy in ischemic stroke. TXNIP is an inhibitor and modulator of endogenous thioredoxin, which rapidly binds to and activates the NLRP3 inflammasome, and TXNIP inhibition directly reduces NLRP3 inflammasome activity [15]. Recent studies have shown that TXNIP expression is upregulated after cerebral I/R injury, similar to the trend observed for NLRP3 expression [18, 37]. Thus, regulation of the TXNIP/NLRP3 pathway may be beneficial for the prevention and treatment of ischemic stroke. Indeed, in this study, increased expression of TXNIP, NLRP3, ASC, Caspase-1, IL-1β, and IL-18 was observed in both injury models, and pretreatment with Nar effectively inhibited the expression of these proteins. This is consistent with the findings of Ri et al., who reported that Nar inhibits NLRP3-ASC interactions in LPS/ATP-stimulated bone marrow-derived macrophages, thereby suppressing NLRP3 inflammasome assembly and reducing the release of proinflammatory factors [26]. Taken together, these findings suggest that the reduction in BBB destruction and cell death by Nar may be associated with inhibition of TXNIP/NLRP3 pathway activation.
In summary, the present study demonstrated that pretreatment with Nar can significantly prevent MCAO/R injury in mice and OGD/R injury in bEnd.3 cells. Our findings also extend those of previous studies, suggesting that the neuroprotective effect of Nar is not only related to the inhibition of oxidative stress but also to the inhibition of the TXNIP/NLRP3 signaling pathway and involves the reduction of BBB breakdown and cell death. Further studies are needed to assess whether other signaling pathways are also involved in the protective effects of Nar against cerebral I/R injury. In short, Nar can be considered a promising compound for further development in the prevention and treatment of ischemic stroke.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Haupt M, Gerner ST, Bähr M, Doeppner TR (2023) Neuroprotective strategies for ischemic stroke-future perspectives. Int J Mol Sci 24(5):4334. https://doi.org/10.3390/ijms24054334
Guo H, Yang J, Liu M, Wang L, Hou W, Zhang L, Ma Y (2020) Selective activation of estrogen receptor beta alleviates cerebral ischemia neuroinflammatory injury. Brain Res 1726:146536. https://doi.org/10.1016/j.brainres.2019.146536
Qin C, Yang S, Chu YH, Zhang H, Pang XW, Chen L, Zhou LQ, Chen M, Tian DS, Wang W (2022) Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther 7(1):215. https://doi.org/10.1038/s41392-022-01064-1
Zhang P, Cui J (2021) Neuroprotective effect of fisetin against the cerebral ischemia-reperfusion damage via suppression of oxidative stress and inflammatory parameters. Inflammation 44(4):1490–1506. https://doi.org/10.1007/s10753-021-01434-x
Guo JM, Shu H, Wang L, Xu JJ, Niu XC, Zhang L (2017) SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci Ther 23(4):360–369. https://doi.org/10.1111/cns.12686
Duris K, Jurajda M (2020) Evolutionary concept of inflammatory response and Stroke. J Neurosci Res 98(1):98–104. https://doi.org/10.1002/jnr.24392
Barrington J, Lemarchand E, Allan SM (2017) A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol 27(2):205–212. https://doi.org/10.1111/bpa.12476
Cao BQ, Tan F, Zhan J, Lai PH (2021) Mechanism underlying treatment of ischemic stroke using acupuncture: transmission and regulation. Neural Regen Res 16(5):944–954. https://doi.org/10.4103/1673-5374.297061
Prakash R, Vyawahare A, Sakla R, Kumari N, Kumar A, Ansari MM, Kanika, Jori C, Waseem A, Siddiqui AJ, Khan MA, Robertson AAB, Khan R, Raza SS (2023) NLRP3 inflammasome-targeting nanomicelles for preventing ischemia-reperfusion-Induced Inflammatory Injury. ACS Nano 17(9):8680–8693. https://doi.org/10.1021/acsnano.3c01760
Wang L, Ren W, Wu Q, Liu T, Wei Y, Ding J, Zhou C, Xu H, Yang S (2022) NLRP3 inflammasome activation: a therapeutic target for cerebral ischemia-reperfusion Injury. Front Mol Neurosci 15:847440. https://doi.org/10.3389/fnmol.2022.847440
Franke M, Bieber M, Kraft P, Weber ANR, Stoll G, Schuhmann MK (2021) The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav Immun 92:223–233. https://doi.org/10.1016/j.bbi.2020.12.009
Ismael S, Zhao L, Nasoohi S, Ishrat T (2018) Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci Rep UK. https://doi.org/10.1038/s41598-018-24350-x
Bellut M, Papp L, Bieber M, Kraft P, Stoll G, Schuhmann MK (2022) NLPR3 inflammasome inhibition alleviates hypoxic endothelial cell death in vitro and protects blood-brain barrier integrity in murine stroke. Cell Death Dis. https://doi.org/10.1038/s41419-021-04379-z
Li L, Cheng SQ, Guo W, Cai ZY, Sun YQ, Huang XX, Yang J, Ji J, Chen YY, Dong YF, Cheng H, Sun XL (2021) Oridonin prevents oxidative stress-induced endothelial injury via promoting Nrf-2 pathway in ischaemic Stroke. J Cell Mol Med 25(20):9753–9766. https://doi.org/10.1111/jcmm.16923
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11(2):136–140. https://doi.org/10.1038/ni.1831
Ishrat T, Mohamed IN, Pillai B, Soliman S, Fouda AY, Ergul A, El-Remessy AB, Fagan SC (2015) Thioredoxin-interacting protein: a novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol Neurobiol 51(2):766–778. https://doi.org/10.1007/s12035-014-8766-x
Abais JM, Xia M, Li G, Chen Y, Conley SM, Gehr TWB, Boini KM, Li PL (2014) Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J Biol Chem 289(39):27159–27168. https://doi.org/10.1074/jbc.M114.567537
Wang X, Li R, Wang X, Fu Q, Ma S (2015) Umbelliferone ameliorates cerebral ischemia-reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci Lett 600:182–187. https://doi.org/10.1016/j.neulet.2015.06.016
Olas B (2021) A review of in vitro studies of the anti-platelet potential of citrus fruit flavonoids. Food Chem Toxicol. https://doi.org/10.1016/j.fct.2021.112090
Funaguchi N, Ohno Y, La BL, Asai T, Yuhgetsu H, Sawada M, Takemura G, Minatoguchi S, Fujiwara T, Fujiwara H (2007) Narirutin inhibits airway inflammation in an allergic mouse model. Clin Exp Pharmacol Physiol 34(8):766–770. https://doi.org/10.1111/j.1440-1681.2007.04636.x
Ha SK, Park HY, Eom H, Kim Y, Choi I (2012) Narirutin fraction from citrus peels attenuates LPS-stimulated inflammatory response through inhibition of NF-kappaB and MAPKs activation. Food Chem Toxicol 50(10):3498–3504. https://doi.org/10.1016/j.fct.2012.07.007
Chakraborty S, Basu S (2017) Multi-functional activities of citrus flavonoid narirutin in Alzheimer’s disease therapeutics: an integrated screening approach and in vitro validation. Int J Biol Macromol 103:733–743. https://doi.org/10.1016/j.ijbiomac.2017.05.110
Mitra S, Lami MS, Uddin TM, Das R, Islam F, Anjum J, Hossain MJ, Emran TB (2022) Prospective multifunctional roles and pharmacological potential of dietary flavonoid narirutin. Biomed Pharmacother 150:112932. https://doi.org/10.1016/j.biopha.2022.112932
Wong ES, Li RW, Li J, Li R, Seto SW, Lee SM, Leung GP (2021) Relaxation effect of narirutin on rat mesenteric arteries via nitric oxide release and activation of voltage-gated potassium channels. Eur J Pharmacol 905:174190. https://doi.org/10.1016/j.ejphar.2021.174190
Patel P, Barve K, Bhatt LK (2022) Narirutin-rich fraction from grape fruit peel protects against transient cerebral ischemia reperfusion injury in rats. Nutr Neurosci 25(5):920–930. https://doi.org/10.1080/1028415X.2020.1821518
Ri MH, Li MY, Xing Y, Zuo HX, Li G, Li C, Ma J, Jin X (2023) Narirutin exerts anti-inflammatory activity by inhibiting NLRP3 inflammasome activation in macrophages. Phytother Res 37(4):1293–1308. https://doi.org/10.1002/ptr.7686
Yu JY, Liu QQ, Li X, Zhao M, Sun T, Hu N, Jiang W, Zhang RT, Yang P, Yang Q (2021) Oxymatrine improves blood-brain barrier integrity after cerebral ischemia-reperfusion injury by downregulating CAV1 and MMP9 expression. Phytomedicine 84:153505. https://doi.org/10.1016/j.phymed.2021.153505
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M, Wang L, Zhang Y, Liu X (2022) Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J Ethnopharmacol 289:115021. https://doi.org/10.1016/j.jep.2022.115021
Yu L, Zhang Y, Chen Q, He Y, Zhou H, Wan H, Yang J (2022) Formononetin protects against inflammation associated with cerebral ischemia-reperfusion injury in rats by targeting the JAK2/STAT3 signaling pathway. Biomed Pharmacother 149:112836. https://doi.org/10.1016/j.biopha.2022.112836
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M, Wang L, Zhang Y, Liu X (2022) Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J Ethnopharmacol. https://doi.org/10.1016/j.jep.2022.115021
Luo L, Sun T, Yang L, Liu A, Liu QQ, Tian QQ, Wang Y, Zhao MG, Yang Q (2020) Scopoletin ameliorates anxiety-like behaviors in complete Freund’s adjuvant-induced mouse model. Mol Brain 13(1):15. https://doi.org/10.1186/s13041-020-0560-2
Gottschall PE, Barone FC (2012) Important role for endothelial calveolin-1 in focal cerebral ischemia-induced blood-brain barrier injury. J Neurochem 120(1):4–6. https://doi.org/10.1111/j.1471-4159.2011.07546.x
Khatri R, McKinney AM, Swenson B, Janardhan V (2012) Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology 79(Suppl 1):S52-57. https://doi.org/10.1212/WNL.0b013e3182697e70
Ng F, Churilov FC, Yassi L, Kleinig N, Thijs TJ, Wu V, Shah TY, Dewey DG, Sharma HM, Desmond G, Yan PM, Parsons B, Donnan MW, Davis GA, Mitchell SM, Leigh PJ, Campbell R, EXTEND-IA TNK Part 1 and 2 Investigators (2022) Microvascular dysfunction in blood-brain barrier disruption and hypoperfusion within the Infarct posttreatment are associated with cerebral edema. Stroke 53(5):1597–1605. https://doi.org/10.1161/STROKEAHA.121.036104
Cardoso FL, Brites D, Brito MA (2010) Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res Rev 64(2):328–363. https://doi.org/10.1016/j.brainresrev.2010.05.003
Reinhold AK, Rittner HL (2017) Barrier function in the peripheral and central nervous system-a review. Pflugers Arch 469(1):123–134. https://doi.org/10.1007/s00424-016-1920-8
Cao G, Jiang N, Hu Y, Zhang Y, Wang G, Yin M, Ma X, Zhou K, Qi J, Yu B, Kou J (2016) Ruscogenin attenuates cerebral ischemia-induced blood-brain barrier dysfunction by suppressing TXNIP/NLRP3 inflammasome activation and the MAPK pathway. Int J Mol Sci 17(9). https://doi.org/10.3390/ijms17091418
Kawabori M, Yenari MA (2015) Inflammatory responses in brain ischemia. Curr Med Chem 22(10):1258–1277. https://doi.org/10.2174/0929867322666150209154036
Pei J, You X, Fu Q (2015) Inflammation in the pathogenesis of ischemic Stroke. Front Biosci (Landmark Ed) 20(4):772–783. https://doi.org/10.2741/4336
Pan W, Kastin AJ (2007) Tumor necrosis factor and stroke: role of the blood-brain barrier. Prog Neurobiol 83(6):363–374. https://doi.org/10.1016/j.pneurobio.2007.07.008
Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y, Zhao J (2018) Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res 336:32–39. https://doi.org/10.1016/j.bbr.2017.06.027
Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, Shi W, Li F, Xin T, Pang Q, Yi F (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic Stroke. J Cereb Blood Flow Metab 34(4):660–667. https://doi.org/10.1038/jcbfm.2013.242
Funding
This work was supported by the Key Research and Development Plan of Shaanxi Province, China (2020SF-142) and the National Natural Science Foundation of China Tangdu Hospital (2021ZTXM-033).
Author information
Authors and Affiliations
Contributions
MZ and AL conceived the study. LL and SW contributed to the experiments. WL and ZZ performed the data analysis. LL drafted and edited the manuscript. All authors read and approved the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Luo, L., Wang, S., Liu, W. et al. Narirutin Attenuates Cerebral Ischemia-Reperfusion Injury by Suppressing the TXNIP/NLRP3 Pathway. Neurochem Res 49, 692–705 (2024). https://doi.org/10.1007/s11064-023-04062-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-04062-z