
Abstract
Neuroinflammation is a critical driver in the pathogenesis and progression of neurodegenerative disorders. Dammarane sapogenins (DS), a deglycosylated product of ginsenoside, possess a variety of potent biological activities. The present study aimed to explore the neuroprotective effects of DS in a rat model of neuroinflammation induced by intracerebroventricular injection of lipopolysaccharide (LPS). Our study revealed that DS pretreatment effectively improved LPS-induced associative learning and memory impairments in the active avoidance response test and spatial learning and memory in Morris water maze test. DS also remarkably inhibited LPS-induced neuroinflammation by suppressing microglia overactivation, pro-inflammatory cytok ine release (TNF‐α and IL‐1β) and reducing neuronal loss in the CA1 and DG regions of the hippocampus. Importantly, pretreatment with DS reversed LPS-induced upregulation of HMGB1 and TLR4 and inhibited their downstream NF-κB signaling activation, as evidenced by increased IκBα and decreased p-NF‐κB p65 levels. Furthermore, DS ameliorated LPS-induced synaptic dysfunction by decreasing MMP-9 and increasing NMDAR1 expression in the hippocampus. Taken together, this study suggests that DS could be a promising treatment for preventing cognitive impairments caused by neuroinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cognitive impairment is a common feature of most neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and perioperative neurocognitive disorders (PND), and is characterized by a decline in learning, memory, problem solving, reasoning, or language [1,2,3]. Neuroinflammation is an important protective and defensive mechanism of central nervous system, but the persistence of inflammatory states may lead to neuronal toxicity and ultimately to cognitive impairment [4, 5]. Growing evidence suggests that neuroinflammation is considered a critical driver in the pathogenesis and progression of neurodegenerative disorders, and that anti-inflammation treatment can alleviate cognitive impairment [6, 7]. Lipopolysaccharide (LPS), a major component of gram-negative bacteria, has been proved to stimulate neuroinflammation and cognitive dysfunction, and is generally used to prepare animal models to explore neuroinflammation-induced cognitive dysfunction [8, 9].
The hippocampus is a key part of the brain that is involved in many cognitive functions, including the formation and optimization of learning and memory, and the consolidation of declarative memory [10]. Microglia are resident immune cells in the hippocampus and play a key physiological function in the regulation of inflammatory responses [11]. Microglia are essential for maintaining homeostasis in the hippocampus, but overactivated microglia may release various inflammatory mediators that cause neuronal loss and neurotoxicity, leading to neuroinflammation and neurodegenerative disorders [12, 13].
High mobility group protein box 1 (HMGB1) is a nuclear nonhistone DNA-binding protein that mainly exists in the nucleus and cytoplasm of cells [14]. As a family of damage-associated molecular pattern (DAMP), HMGB1 can control inflammatory responses to mediate neurodegenerative disorders such as AD and PD [15, 16]. During neuroinflammatory conditions, HMGB1 is actively released by neurons and microglia following inflammasome activation [17]. Once released into the extracellular space, HMGB1 initiates inflammatory process by binding to Toll-like receptor 4 (TLR4) and leads to nuclear factor-κB (NF-κB) activation, thereby promoting the expression of proinflammatory cytokines, such as interleukin 1β (IL-1β) and tumor necrosis factor-α (TNF-α) [14, 18]. Therefore, reducing neuroinflammation by modulating HMGB1/TLR4/NF-κB signaling pathway may be an effective strategy to treat neurodegenerative disorders.
Synaptic plasticity in the hippocampus is the cytological and electrophysiological basis of cognitive function [19]. It is widely recognized that alteration of synaptic function is a leading cause of several neurodegenerative diseases, including AD [20]. At the morphological level, AD brains are characterized by progressive atrophy, which is caused by neuronal loss and reduced synaptic dendritic arborization [21]. Moreover, recent reports have shown that neuroinflammation is strongly associated with synaptic dysfunction in neurodegenerative diseases. For example, in the aging brain, increased activation of microglia and proinflammatory cytokines will lead to reduced synaptic plasticity and compromised learning and memory [22].
Ginseng is a traditional medicine with various pharmacological effects, such as anticancer, anti-inflammation, antioxidant, immunomodulatory and neuroprotective effects [23]. Ginsenosides are the most widely studied ingredients of ginseng and are considered to be the key active ingredients for improving cognitive performance [24]. At present, more than 100 ginsenosides have been identified and can be classified into three types: dammarane, oleanolic acid, and ocotillol types [25]. Among them, the dammarane are the most active, accounting for more than 90% of the total saponins in raw ginseng, which can be further divided into two major types: the 20(S)-protopanaxadiol (PPD) and 20(S)-protopanaxatriol (PPT) [26, 27]. Dammarane sapogenins (DS), the deglycosylated products of ginsenoside formed by intestinal bacterial metabolism, are more easily absorbed and exhibit stronger biological activity than their glycosides [28]. Our previous study has elucidated the ameliorative effect of DS on depression-like behaviors [29], but the ability of DS to prevent neuroinflammation-induced cognitive impairment remains unclear.
In the present study, we investigated the protective effects of DS on LPS-induced cognitive impairments, neuroinflammation and synaptic dysfunction in rats. We examined whether DS improved cognitive behavior and hippocampal neuronal morphology in a rat model of neuroinflammation established by intracerebroventricular (ICV) administration of LPS. To investigate the underlying mechanisms, the microglia activation, proinflammatory cytokines release, HMGB1 dependent NF-κB signaling pathway changes and synaptic plasticity-associated proteins were also examined in the hippocampus.
Materials and Methods
Reagents
DS was provided by Panagin Pharmaceuticals Inc. (USA Patent No. US patent 6,888,014 B2, 2005). DS was a hydrolyzed alkaline product of ginsenosides derived from the stem and leaf of Panax ginseng, and contained 33% 20(S)-PPT and 16% 20(S)-PPD as determined by HPLC [28]. LPS (E. coli 055: B5) was purchased from Sigma-Aldrich (St. Louis, USA). Enzyme linked immunosorbent assay (ELISA) kits for TNF-α and IL-1β were purchased from eBioscience, Inc (San Diego, USA). Primary antibodies were from several companies: anti-ionized calcium binding adapter molecule 1 (Iba1), anti-HMGB1, anti-inhibitor of NF-κBα (IκBα), anti-NF-κB p65, anti-matrix metalloprotein-9 (MMP-9), anti-N-methyl-D-aspartate receptor 1 (NMDAR1) and anti-β-actin from Cell Signaling Technology, Inc (MA, USA); anti-TLR4 from Santa Cruz Biotechnology, Inc (Heidelberg, Germany); anti-p-NF-κB p65 from Abcam (Cambridge, UK).
Animals
Sixty male Wistar rats (weighing 190–210 g) were purchased from the Vital River Laboratory (Beijing, China). Rats were housed in controlled laboratory conditions: room temperature (25 ± 2 °C), humidity (55 ± 10%), and light (12 h light/dark cycle) with free access to food and water. This study was performed in line with the guidelines for the Animal Care and Use Committee of the Institute of Medical Plant Development, Chinese Academy of Medical Sciences, and Peking Union Medical College.
Experimental Protocols
Rats were randomly assigned to six groups (n = 10 per group): (1) control group (vehicle treatment only), (2) sham group (vehicle treatment plus saline ICV injection), (3) LPS group (vehicle treatment plus LPS ICV injection), (4) DS (25 mg/kg) group (25 mg/kg DS treatment plus LPS ICV injection), (5) DS (50 mg/kg) group (50 mg/kg DS treatment plus LPS ICV injection), (6) DS (100 mg/kg) group (100 mg/kg DS treatment plus LPS ICV injection). After 3 days of acclimatization, rats were given either vehicle (distilled water) or DS (25 mg/kg, 50 mg/kg and 100 mg/kg) continuously by gavage 10 days before surgery and thereafter continued until the end of the experiment. The doses of DS were determined by our preliminary experiments [28, 29]. Experimental design was carried out according to the timeline shown in Fig. 1a.

The experimental timeline and left intraventricular injection. (a)The experimental timeline for the study. OFT, open field test; AAR test, active avoidance response test; MWM test, Morris water maze test. (b) Rat received left lateral intraventricular injection. (c) The arrow points to the brain injection site. (d) The left lateral ventricle was injected with non-toxic blue ink to indicate the injection location, and 30 min after the injection, the blue ink filled the entire cerebral ventricle, as indicated by the arrow
LPS Intracerebroventricular Injections
Rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p) and secured in a stereotaxic apparatus (Fig. 1b). A burr hole was drilled in the parietal bone posterior to bregma on left side of the midline (coordinates: 1.0 mm posterior, 1.5 mm left lateral relative to bregma and 3.8 mm below dura) for needle insertion. Prior to the formal surgical operation, we injected non-toxic blue ink into the left lateral ventricle to indicate the injection location (Fig. 1c and d). LPS (75 µg/5µl) was dissolved in normal saline and injected into left lateral ventricle of the LPS and DS groups, while the sham group injected with 5ul normal saline. Behavioral tests were assessed 3 days after the surgical operation and body weight was recorded every 3 days (9:00–10:00 a.m.) during the experiment.
Behavioral Tests
Open Field test (OFT)
The open field test was performed to evaluate locomotor activity as previously described [30]. The apparatus was a circular metal pool (100 cm in diameter and 50 cm in height) from which a camera and a 120 lx light source are hung. A computer-aided image analysis system was used for real-time detection and recording the locomotion of rats. Each rat was initially located in the center of the box and freely explored the environment for 3 min, and then tested for locomotor activity for 10 min. The total distance, average speed and exercise time of rats were recorded and analyzed.
Active Avoidance Response (AAR) test
An automated shuttle-box apparatus was used to assess the AAR as previously described in the literature with slight modifications [30]. The apparatus consists of two identical compartments (30 cm ×35 cm ×70 cm) separated by a black partition with a small door (10 cm ×10 cm) and a camera fixed in the center of the ceiling to monitor the animal’s behavior. Blue light was used as conditioned stimulus (CS) and an electric foot shock (0.4 mA) was applied as unconditioned stimulus (US). The " Random Delay Mode " was selected in the system, and each trial period consisted of 3–8 s CS, followed immediately by3-8 s US, and then 3–8 s interval, for a total experiment time of 20 min. The rats were familiarized with the apparatus for 2 min prior to the formal testing. The training session was performed for 5 consecutive days. For animal behavior, crossing to another compartment during CS and US period was considered as active avoidance and passive avoidance, respectively. The numbers of active avoidance, passive avoidance and total time to avoid electric shock were recorded and analyzed.
Morris Water maze (MWM) test
MWM test was performed to evaluate the spatial learning and memory in rats [31]. A black cylindrical stainless steel pool (100 cm in diameter and 40 cm in height) was filled with water and non-toxic black ink was added to make the water opaque. The pool was divided into four equal quadrants, one of which placed the escape platform (6 cm diameter) 1.5 cm below the water surface. The test consisted of a five consecutive days of acquisition phase and one day of probe phase. During the acquisition phase, each rat was placed on the escape platform for 10 s to memorize the platform and then placed in the water to explore the hidden platform. If the rat could not approach the platform within 90 s, it was guided to the platform for an additional 10 s. The software system automatically recorded the total swimming distance, escape latency, and average swimming speed for finding the platform. During the probe phase, the platform was removed and rats were allowed to swim freely for 90 s. The number of crossings the platform and the time spent in the target quadrant were recorded.
Hematoxylin and Eosin (HE) and Immunohistochemistry (IHC) Staining
After sacrificed, brain tissue from rats was collected, embedded in paraffin and sectioned, then dewaxed and rehydrated, and incubated with 3% hydrogen peroxide. HE staining was performed according to the standard procedure. For IHC staining, brain samples were blocked with 3% bovine serum albumin and incubated with primary antibody (anti-Iba1) overnight at 4 °C, then incubated with secondary antibody for 1 h and detected with 3,3′-diaminobenzidine (DAB). Images were obtained by a light microscope (Olympus, Tokyo, Japan) and analyzed by Image-Pro Plus software (Media Cybernetics, USA).
Measurements of Inflammatory Cytokines
The hippocampus samples were homogenized in ice-cold extraction buffer and centrifuged at 12,000×g for 15 min at 4 °C. The levels of TNF-α and IL-1β were quantified by ELISA kits according to the manufacturer’s instruction.
Western Blotting Analysis
The hippocampus samples were homogenized in ice-cold extraction buffer and centrifuged at 12,000×g for 15 min at 4 °C. Protein concentrations were determined by BCA protein assay kit (Beyotime Biotechnology, Nanjing, China). Proteins were loaded and separated in SDS-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membrane. After blocked with 5% skim milk for 1 h, the membranes were incubated overnight at 4 °C with respective primary antibodies to HMGB1(1:1 000), TLR4(1:2 000), IκBα(1:1 000), NF-κB p65(1:1 000), p-NF-κB p65(1:1 000), MMP-9(1:1 000), NMDAR1(1:1 000) and β-actin (1:1 000). After three washes with TBST (5 min each), the membranes were incubated with secondary antibody for 2 h at room temperature and then washed again. Immunoreactive bands were scanned and analyzed by the Quantity One automatic imaging analysis system (Bio-Rad). Relative expression levels of proteins were normalized to β-actin.
Statistical Analysis
Statistical analysis was performed using SPSS software 26.0 (SPSS, Inc., Chicago, IL, USA), and all data were presented as mean ± standard error of mean (SEM). Data from the AAR test and acquisition phase of MWM test were analyzed using a two-way repeated measures ANOVA followed by Tukey’s post-hoc test. All other data were analyzed by one-way ANOVA followed by Tukey’s post-hoc test analysis for multiple comparisons. P < 0.05 was considered statistically significant.
Results
Effects of DS on body Weight and Locomotor Activity

Effects of DS on body weight and locomotor activity in LPS-treated rats. (a) Changes in body weight of rats after LPS injection. (b-d) Total distance, average speed and exercise time of rats in OFT. Data are presented as mean ± SEM. ##p<0.05, as compared with sham group
As shown in Fig. 2a, rats injected with LPS experienced significant weight loss from day 3 to day 9 compared with the sham group (P<0.001), while DS treatment did not result in weight improvement. To further investigate whether the locomotor activity of rats changed, the OFT was performed. The results of OFT showed no significant differences in total distance (P = 0.21; Fig. 1b), average speed (P = 0.21; Fig. 1c) and exercise time (P = 0.91; Fig. 1d) between the different groups. Despite the weight loss caused by LPS injection, the rats maintained the corresponding motor capacity, suggesting that LPS exposure and DS treatment did not affect the locomotor activity of the rats.
DS Improves LPS-induced Associative Learning and Memory Impairments in AAR test

DS improves LPS-induced associative learning and memory impairments. (a) Number of active avoidances to escape electric shock (US) when the blue light on (CS). (b) Number of passive avoidances after electric foot shock (US). (c) Total time to avoid electric foot shock. Data are presented as mean ± SEM. #p<0.05, as compared with sham group; *p<0.05, **p<0.01, as compared with LPS group
A shuttle box apparatus was used to test improvement of DS on LPS-induced associative learning and memory impairments. During the test, blue light (CS) was used as an indicator before the electric shock stimulation (US). Rats were supposed to learn the connection between different stimulation and avoid the electric shock when the blue light was on [32]. As seen in Fig. 3a and c, LPS-exposed rats showed a significant decrease in the number of active avoidance on day 2 and 4 compared with sham group (P<0.05; P<0.05), the total time to avoid electric shock also showed a decreasing trend. DS (50 mg/kg and 100 mg/kg) significantly increased the number of active avoidance from day 2 to day 4 (P<0.05 or P<0.01) and increased the total time to avoid electric shock on day 4 and 5 (P<0.05; P<0.05). Figure 3b showed that the number of passive avoidance was significantly higher in LPS group than in the sham group on day 2 and 4 (P<0.05; P<0.05), while DS (50 mg/kg or 100 mg/kg) significantly reduced this change on day 1–2 and day 4–5 (P<0.05 or P<0.01). The AAR test results showed DS treatment significantly improved the damages of associative learning and memory in rats caused by LPS exposure.
DS Improves LPS-induced Spatial Learning and Memory Impairments in MWM test

DS improves LPS-induced spatial learning and memory impairments. (a-c) Total swimming distance, escape latency and average swimming speed to find the hidden platform in the acquisition phase. (d) Representative search strategy of rats on fifth day in the acquisition phase. (e) Number of crossings over the former platform location in the probe phase. (f) The percentage of time spent in target quadrant where the platform was once placed in the probe phase. Data are presented as mean ± SEM. #p<0.05, as compared with sham group; *p<0.05, **p<0.01, as compared with LPS group
MWM test was performed to evaluate the improvement of DS on LPS-induced spatial learning and memory impairments. The spatial learning capacity of rats was evaluated during the acquisition phase, as shown in Fig. 4a and b, rats exposed to LPS spent longer total swimming distance and escape latency on day 3 and 4 than in sham-operated rats (P<0.05; P<0.05). DS (50 mg/kg and 100 mg/kg) significantly decreased the total swimming distance and escape latency on day 2 and 4 (P<0.05 or P<0.01). However, on day 5, there were no significant differences in the total swimming distance and escape latency between groups, suggesting that although neuroinflammation impaired the spatial learning ability of the rats, it was not completely lost. There was no significant difference in the swimming speed between groups, suggesting that LPS injection and DS treatment did not affect motor activity in MWM test (Fig. 4c). Figure 4d showed the search strategy of rats on the last day of the acquisition phase. Rats in different DS treatment groups showed the tendency to seek the platform more directly, however, LPS group showed a longer search strategy.
During the probe phase, the platform was removed and the degree of spatial memory consolidation was evaluated. As shown in Fig. 4e and f, compared with the sham group, there was a decreasing trend in the number of crossing the platform and the percentage of time spent in target quadrant in LPS group, but there were no significant differences. Compared to the LPS group, DS (50 mg/kg and 100 mg/kg) significantly increased the number of crossing the platform (P<0.01; P<0.05), and DS (50 mg/kg) treated rats spent more time in the target quadrant (P<0.05). Results revealed that DS treatment partially ameliorated LPS-induced spatial learning and memory impairments.
DS Ameliorates LPS-induced Neuronal Damages in the hippocampus

DS ameliorates LPS-induced neuronal damage in the hippocampus. (a) The representative HE staining images and (b–d) number of neuron cells in the CA1, CA3, and DG regions of hippocampus (×400 magnification; n = 3 per group). Data are presented as mean ± SEM. #p<0.05, ##p<0.01, as compared with sham group; *p<0.05, **p<0.01, as compared with LPS group
HE staining was performed to observe the neuronal damages of hippocampus. As shown in Fig. 5a, in the control and sham groups, the neurons were compact and tidy, with many layers, and there were more nerve cells with clear staining. In the LPS group, images showed sparse neurons with large gaps, and many nerve cells vacuole were also observed, especially in the CA1 and DG regions of the hippocampus. Figure 5b and d showed that, the number of normal neurons in CA1 and DG regions of rats exposed to LPS was significantly decreased (p<0.05; p<0.01). And supplementation with different doses of DS significantly increased the number of normal neuron cells in the CA1 and DG regions of the hippocampus (p<0.05 or p<0.01). However, there were no obvious neuronal changes in the hippocampal CA3 region (Fig. 5a and c). Overall, DS could ameliorate LPS‑induced neuronal damages in the hippocampus.
DS Inhibits LPS-induced Microglia Overactivation and Release of Inflammatory Cytokines in the hippocampus

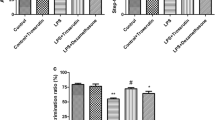
DS inhibits LPS-induced microglia overactivation in the hippocampus. (a) The representative immunohistochemical staining images and (b) mean intensity of Iba1 positive cells in the hippocampus of rats (×400 magnification; n = 3 per group). The arrow points to hippocampal region. Data are presented as mean ± SEM. ##p<0.01, as compared with sham group; **p<0.01, as compared with LPS group
Activation of hippocampal microglia simulated by LPS injection was evaluated by IHC staining of Iba1 (a microglia marker). Figure 6a showed that more microglia were activated in LPS group than in the sham group, and the activated microglia were characterized by obviously big or swelling cell body. Contrarily, different doses of DS treatment inhibited microglia activation and more resting microglia with small round or thin cell bodies were detected. The qualitative analysis of microglia activation was further confirmed by the mean intensity of total Iba1-marked area (Fig. 6b). The mean intensity of Iba1-marked area was significantly increased in the LPS group compared with sham group (p < 0.01), while treatment with DS significantly reversed microglia activation (p < 0.01).
Effects of DS on LPS-induced HMGB1/TLR4/NF-κB Signaling Pathway in the hippocampus

Effects of DS on LPS-induced HMGB1/TLR4/NF-κB signaling pathway and inflammatory cytokines release in the hippocampus. (a) Representative western blots and (b-f) quantitative analysis of HMGB1, TLR4, IκBα, p-NF-κB p65 and NF-κB p65 protein expression in the hippocampus (n = 3 per group). β-actin was used as a loading control. (g, h) The levels of TNF-α and IL-1β in the hippocampus (n = 6 per group). Data are presented as mean ± SEM. ##p<0.01, as compared with sham group; *p<0.05, **p<0.01, as compared with LPS group
As shown in Fig. 7a-c, western blotting analysis revealed that LPS injection significantly elevated protein expression of HMGB1 and its adaptor protein TLR4 (p < 0.01; p < 0.01), which were significantly suppressed by DS treatment (p < 0.01 or p < 0.05). It has been reported that the HMGB1/TLR4 is a key inflammatory regulator leading to NF-κB activation [14]. Figure 7d and f showed that LPS exposure decreased the level of IκBα (p < 0.01) and significantly increased phosphorylated p-NF-κB p65 level (p < 0.01) compared with sham group, while there was no significant difference in NF-κB p65 expression (p>0.05). These results implicated LPS exposure activated the NF-κB signaling by inducing the nuclear translocation of the p65 subunit [33]. Different doses of DS treatment significantly blocked LPS-induced NF-κB p65 signaling activation by increasing IκBα expression (p < 0.01) and decreasing p-NF-κB p65 phosphorylation (p < 0.01).
To investigate inflammatory alterations triggered by LPS stimulation, we further detected the levels of TNF-α and IL-1β in the hippocampus. ELISA results showed that TNF-α and IL-1β levels were notably increased in the LPS group compared with sham-operated group (p < 0.01; p < 0.01; Fig. 7g h). And DS (50 mg/kg or 100 mg/kg) administration significantly decreased the elevated expression of TNF-α and IL-1β in the hippocampus (p < 0.05 or p < 0.01). These findings suggest that the inhibitory effect of DS on LPS-induced neuroinflammation may be correlated with HMGB1-mediated NF-κB signaling pathway.
Effects of DS on LPS-induced synaptic-associated Proteins in the hippocampus

Effects of DS on LPS-induced synaptic-associated proteins in the hippocampus. (a) Representative western blots and (b, c) quantitative analysis of MMP-9 and NMDAR1 protein expression in the hippocampus (n = 3 per group). β-actin was used as a loading control. Data are presented as mean ± SEM. ##p<0.01, as compared with sham group; *p<0.05, **p<0.01, as compared with LPS group
Western blotting analysis detected the synaptic-associated proteins expression of MMP-9 and NMDAR1 in the hippocampus (Fig. 8a and c). Compared with sham group, LPS injection induced synaptic dysfunction by significantly increasing the expression of MMP-9 (p < 0.01) and decreasing the expression of NMDAR1 (p < 0.01), while DS treatment effectively reversed these changes (p < 0.05 or p < 0.01).
Discussion
The present study found that DS pretreatment effectively prevented cognitive impairments caused by neuroinflammation in LPS-treated rats. DS pretreatment effectively improved LPS-induced associative learning and memory impairments in active AAR test as well as spatial learning and memory in MWM test. The neuroprotective effects of DS were associated with suppression of microglia overactivation, proinflammatory cytokine expression, likely mediated by inhibiting activated HMGB1/TLR4/NF-κB signaling pathway, thereby reducing hippocampal neuronal loss and synaptic dysfunction (Fig. 9). These findings provide a new understanding of the function of DS in LPS-induced cognitive impairment, neuroinflammation, and synaptic dysfunction.

Schematic illustration of the possible neuroprotective mechanism of DS administration in LPS-induced cognitive impairment, neuroinflammation and synaptic dysfunction
The AAR test and MWM test were used to investigate the improvement of DS on cognitive impairments in LPS-exposed rats. The AAR test is a complex conditioned reflex task and widely used to study associative learning and memory [34]. Our results showed that DS treatment significantly enhanced active avoidance as well as decreased passive avoidance to escape electric shock compared with LPS-exposed rats. We further conducted the MWM test to determine the neuroprotective effect of DS, which reflecting hippocampal dependent spatial learning and memory capacity [35]. The results showed that rats received DS treatment significantly improved LPS-induced spatial learning capacity damages by reducing total swimming distance and escape latency to search for invisible platform during the acquisition phase, and enhanced spatial memory consolidation by increasing search activities in target quadrant during the probe phase. In addition, although body weight was significantly decreased after LPS injection, OFT results showed that DS and LPS treatment did not affect locomotor activity of the rats. Overall, these suggest that DS may have potential to improve cognitive impairments induced by LPS injection.
It has been broadly established that neuroinflammation plays a key role in the process of neurodegeneration. Microglia are the main source of proinflammatory mediators in the brain, and uncontrolled activation of microglia leads to the release of large amount of proinflammatory cytokines [36]. The results of present study showed that the Iba1-marked microglia were significantly increased in the hippocampus of LPS-treated rats. DS administration inhibited LPS-induced inflammatory responses by suppressing microglia overactivation.
Sustained neuroinflammatory response were detrimental to neuronal survival and ultimately leads to cognitive deficits [37]. Although the hippocampus is one of the most vulnerable brain regions to various neurobiological injuries, the effects of damage vary from region to region [38]. For example, CA1 region has been found to be the most vulnerable region of the hippocampus to hypoxic injury, while neurons in the DG region are more sensitive to stress but resistant to ischemia and hypoxia [39,40,41]. In this experiment, we found LPS exposure significantly damaged the neurons in the hippocampal CA1 and DG regions, but no significant changes were observed in the CA3 region. DS pretreatment could effectively restore LPS-induced neuron injury in the CA1 and DG regions.
HMGB1 selectively binds to several receptors, including RAGE, TLR2 and TLR4, of which TLR4 is widely expressed in neuron and microglia of the central nervous system [42]. Similar to previous findings [43, 44], our results confirmed a significant increase in HMGB1 and TLR4 expression after LPS injection, and administration of DS significantly reduced the increased expression of HMGB1 and TLR4. HMGB1/TLR4 mediated NF-κB signaling activation has been identified as a critical inflammatory stimulator of the cellular response to neuronal damages [45]. Once being activated, the NF-κB p65 subunit transfers to the nucleus to regulate the expression of inflammatory factors, while IκBα normally binds to NF-κB p65 subunit to form a complex and inhibits its entry into the nucleus [46]. We found that IκBα expression was significantly decreased and phosphorylated p-NF-κB p65 expression increased in LPS-treated rats, suggesting that the hippocampal NF-κB signaling pathway was activated, treatment with DS obviously reversed these changes. In addition, the expression levels of TNF-α and IL-1β were significantly increased in the LPS group compared to the sham group, while DS administration significantly decreased the expression of these inflammatory factors. Taken together, our data suggest that DS may attenuated LPS-induced inflammatory damages by inhibiting HMGB1/TLR4/NF-κB signaling pathway.
MMP-9 is an extracellular enzyme involves in synaptic plasticity by controlling the shape of dendritic spines and function of excitatory synapses, thus playing a pivotal role in learning and memory [21]. The quantity of MMP-9 is very low in the naive brain, but it is markedly activated following neuroinflammation stimuli. Increased MMP-9 also play a prominent role in neuroinflammation development by promoting inflammatory cell infiltration [47]. NMDAR is an ionotropic glutamate receptor (iGluR) that maintains normal physiological functions of neurons by regulating calcium influx [48]. The long-term potentiation (LTP), which requires NMDAR activation, has been recognized as a major model of synaptic plasticity [49, 50]. Our results showed that LPS induced a significant increase in MMP-9 expression and a decrease in NMDAR1 in rats, and that DS pretreatment ameliorated these changes. These data provide insight into the potential neuroprotective effects of DS on synaptic plasticity.
There are some limitations in this study. A limitation of this study is that only a single ICV injection of LPS-induced acute inflammation model was used in this study, and the ameliorative effects of DS in different animal models of cognitive dysfunction, such as injection of Aβ1−40, vascular dementia, surgery/anesthesia, need to be further investigated. Another limitation of this study is that only the role of microglia activation on neuroinflammatory injury was analyzed, but it has been suggested that astrocyte activation as well as microglia polarization and other glial cell changes are also involved in this process, which needs to be further verified.
Conclusions
The present study demonstrates that DS pretreatment can prevent cognitive impairments induced by ICV injection of LPS in rats. The neuroprotective effects of DS are mainly through the inhibition of microglia overactivation, reduction of proinflammatory cytokines release, suppression of the HMGB1/TLR4/NF-κB signaling pathway and modulation of synaptic-associated proteins, thus reducing hippocampal neuronal damage and synaptic dysfunction. Overall, our findings suggest that DS is a promising candidate therapy for neuroinflammation related cognitive impairments.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Azam S, Haque ME, Jakaria M, Jo SH, Kim IS, Choi DK (2020) G-protein-coupled receptors in CNS: a potential therapeutic target for intervention in neurodegenerative disorders and associated cognitive deficits. Cells 9:506. https://doi.org/10.1186/s12974-014-0151-1
Cotter J, Granger K, Backx R, Hobbs M, Looi CY, Barnett JH (2018) Social cognitive dysfunction as a clinical marker: a systematic review of meta-analyses across 30 clinical conditions. Neurosci Biobehav Rev 84:92–99. https://doi.org/10.5114/fn.2019.84423
2021 Alzheimer’s disease facts and Fig. (2021) Alzheimers Dement 17:327–406. https://doi.org/10.1002/alz.12328
Allison DJ, Ditor DS (2014) The common inflammatory etiology of depression and cognitive impairment: a therapeutic target. J Neuroinflammation 11:151. https://doi.org/10.1186/s12974-014-0151-1
Wang J, Song Y, Chen Z, Leng SX (2018) Connection between systemic inflammation and neuroinflammation underlies neuroprotective mechanism of several phytochemicals in neurodegenerative diseases. Oxid Med Cell Longev 2018, 1972714. https://doi.org/10.1155/2018/1972714
Bradburn S, Murgatroyd C, Ray N (2019) Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a meta-analysis. Ageing Res Rev 50:1–8. https://doi.org/10.1016/j.arr.2019.01.002
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement 12:719–732. https://doi.org/10.1016/j.jalz.2016.02.010
Feng X, Hu J, Zhan F, Luo D, Hua F, Xu G (2021) MicroRNA-138-5p regulates hippocampal neuroinflammation and cognitive impairment by NLRP3/Caspase-1 signaling pathway in rats. J Inflamm Res 14:1125–1143. https://doi.org/10.2147/JIR.S304461
Mao X, Kelty TJ, Kerr NR, Childs TE, Roberts MD, Booth FW (2021) Creatine supplementation upregulates mTORC1 signaling and markers of synaptic plasticity in the dentate gyrus while ameliorating LPS-induced cognitive impairment in female rats. Nutrients 13:2758. https://doi.org/10.3390/nu13082758
Hainmueller T, Bartos M (2018) Parallel emergence of stable and dynamic memory engrams in the hippocampus. Nature 558:292–296. https://doi.org/10.1038/s41586-018-0191-2
Cornell J, Salinas S, Huang HY, Zhou M (2022) Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res 17:705–716. https://doi.org/10.4103/1673-5374.322423
Cunningham C (2013) Microglia and neurodegeneration: the role of systemic inflammation. Glia 61:71–90. https://doi.org/10.1002/glia.22350
Szczepanik AM, Ringheim GE (2003) IL-10 and glucocorticoids inhibit Abeta (1–42) and lipopolysaccharide-induced pro-inflammatory cytokine and chemokine induction in the central nervous system. J Alzheimers Dis 5:105–117. https://doi.org/10.3233/jad-2003-5205
Paudel YN, Shaikh MF, Chakraborti A, Kumari Y, Aledo-Serrano Á, Aleksovska K, Alvim MKM, Othman I (2018) HMGB1: a common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front Neurosci 12:628. https://doi.org/10.3389/fnins.2018.00628
Fujita K, Motoki K, Tagawa K, Chen X, Hama H, Nakajima K, Homma H, Tamura T, Watanabe H, Katsuno M, Matsumi C, Kajikawa M, Saito T, Saido T, Sobue G, Miyawaki A, Okazawa H (2016) HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci Rep 6:31895. https://doi.org/10.1038/srep31895
Sasaki T, Liu K, Agari T, Yasuhara T, Morimoto J, Okazaki M, Takeuchi H, Toyoshima A, Sasada S, Shinko A, Kondo A, Kameda M, Miyazaki I, Asanuma M, Borlongan CV, Nishibori M, Date I (2016) Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp Neurol 275 Pt 1220–231. https://doi.org/10.1016/j.expneurol.2015.11.003
Ravizza T, Terrone G, Salamone A, Frigerio F, Balosso S, Antoine DJ, Vezzani A (2017) High mobility group box 1 is a novel pathogenic factor and a mechanistic biomarker for epilepsy. Brain Behav Immun 72:14–21. https://doi.org/10.1016/j.bbi.2017.10.008
Meng L, Li L, Lu S, Li K, Su Z, Wang Y, Fan X, Li X, Zhao G (2018) The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-κB and PI3K/Akt/mTOR pathways. Mol Immunol 94:7–17. https://doi.org/10.1016/j.molimm.2017.12.008
Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. https://doi.org/10.1038/361031a0
Ju Y, Tam KY (2022) Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen Res 17:543–549. https://doi.org/10.4103/1673-5374.320970
Vafadari B, Salamian A, Kaczmarek L (2016) MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem 139(2):91–114. https://doi.org/10.1111/jnc.13415
Elmore MRP, Hohsfield LA, Kramár EA, Soreq L, Lee RJ, Pham ST, Najafi AR, Spangenberg EE, Wood MA, West BL, Green KN (2018) Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell 17:e12832. https://doi.org/10.1111/acel.12832
Kim HJ, Jung SW, Kim SY, Cho IH, Kim HC, Rhim H, Kim M, Nah SY (2018) Panax ginseng as an adjuvant treatment for Alzheimer’s disease. J Ginseng Res 42:401–411. https://doi.org/10.1016/j.jgr.2017.12.008
Huang X, Li N, Pu Y, Zhang T, Wang B (2019) Neuroprotective effects of ginseng phytochemicals: recent perspectives. Molecules 24:2939. https://doi.org/10.3390/molecules24162939
Feng H, Xue M, Deng H, Cheng S, Hu Y, Zhou C (2022) Ginsenoside and its therapeutic potential for cognitive impairment. Biomolecules 12:1310. https://doi.org/10.3390/biom12091310
Mohanan P, Subramaniyam S, Mathiyalagan R, Yang DC (2018) Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J Ginseng Res 42:123–132. https://doi.org/10.1016/j.jgr.2017.01.008
Piao X, Zhang H, Kang JP, Yang DU, Li Y, Pang S, Jin Y, Yang DC, Wang Y (2020) Advances in saponin diversity of Panax ginseng. Molecules 25:3452. https://doi.org/10.3390/molecules25153452
Dong L, Yang Y, Lu Y, Lu C, Lv J, Jiang N, Xu Q, Gao Y, Chang Q, Liu X (2018) Radioprotective effects of dammarane sapogenins against 60 co-induced myelosuppression in mice. Phytother Res 32:741–749. https://doi.org/10.1002/ptr.6027
Jiang N, Zhang BY, Dong LM, Lv JW, Lu C, Wang Q, Fan LX, Zhang HX, Pan RL, Liu XM (2018) Antidepressant effects of dammarane sapogenins in chronic unpredictable mild stress-induced depressive mice. Phytother Res 32:1023–1029. https://doi.org/10.1002/ptr.6040
Wang Q, Dong L, Wang M, Chen S, Li S, Chen Y, He W, Zhang H, Zhang Y, Pires Dias AC, Yang S, Liu X (2021) Dammarane sapogenins improving simulated weightlessness-induced depressive-like behaviors and cognitive dysfunction in rats. Front Psychiatry 12:638328. https://doi.org/10.3389/fpsyt.2021.638328
Dong L, Wang Y, Lv J, Zhang H, Jiang N, Lu C, Xu P, Liu X (2019) Memory enhancement of fresh ginseng on deficits induced by chronic restraint stress in mice. Nutr Neurosci 22:235–242. https://doi.org/10.1080/1028415X.2017.1373928
Liu W, Liu J, Gao J, Duan X, Zhang L (2022) Effects of subchronic aluminum exposure on learning, memory, and neurotrophic factors in rats. Neurotox Res 40:2046–2060. https://doi.org/10.1007/s12640-022-00599-z
Lee YJ, Choi DY, Choi IS, Kim KH, Kim YH, Kim HM, Lee K, Cho WG, Jung JK, Han SB, Han JY, Nam SY, Yun YW, Jeong JH, Oh KW, Hong JT (2012) Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappab in vitro and in vivo models. J Neuroinflammation 9:35. https://doi.org/10.1186/1742-2094-9-35
García-Capdevila S, Portell-Cortés I, Torras-Garcia M, Coll-Andreu M, Costa-Miserachs D (2009) Effects of long-term voluntary exercise on learning and memory processes: dependency of the task and level of exercise. Behav Brain Res 202:162–170. https://doi.org/10.1016/j.bbr.2009.03.020
D’Hooge R, De Deyn PP (2001) Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev 36:60–90. https://doi.org/10.1016/s0165-0173(01)00067-4
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Attia GM, Elmansy RA, Elsaed WM (2019) Neuroprotective effect of nilotinib on pentylenetetrazol-induced epilepsy in adult rat hippocampus: involvement of oxidative stress, autophagy, inflammation, and apoptosis. Folia Neuropathol 57:146–160. https://doi.org/10.5114/fn.2019.84423
Reagan LP, McEwen BS (1997) Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat 13:149–167. https://doi.org/10.1016/s0891-0618(97)00031-8
Owen JE, BenediktsdÓttir B, Gislason T, Robinson SR (2019) Neuropathological investigation of cell layer thickness and myelination in the hippocampus of people with obstructive sleep apnea. Sleep 42. https://doi.org/10.1093/sleep/zsy199
Mirescu C, Gould E (2006) Stress and adult neurogenesis. Hippocampus 16:233–238. https://doi.org/10.1002/hipo.20155
Kreisman NR, Soliman S, Gozal D (2000) Regional differences in hypoxic depolarization and swelling in hippocampal slices. J Neurophysiol 83:1031–1038. https://doi.org/10.1152/jn.2000.83.2.1031
Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T (2003) Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A 100:8514–8519. https://doi.org/10.1152/jn.2000.83.2.1031
Li Y, Liu T, Li Y, Han D, Hong J, Yang N, He J, Peng R, Mi X, Kuang C, Zhou Y, Han Y, Shi C, Li Z, Guo X (2020) Baicalin ameliorates cognitive impairment and protects microglia from LPS-induced neuroinflammation via the SIRT1/HMGB1 pathway. Oxid Med Cell Longev 2020, 4751349. https://doi.org/10.1155/2020/4751349
Wang L, Yang JW, Lin LT, Huang J, Wang XR, Su XT, Cao Y, Fisher M, Liu CZ (2020) Acupuncture attenuates inflammation in microglia of vascular dementia rats by inhibiting miR-93-mediated TLR4/MyD88/NF-κB signaling pathway. Oxid Med Cell Longev 2020, 8253904. https://doi.org/10.1155/2020/8253904
Fang P, Schachner M, Shen YQ (2012) HMGB1 in development and diseases of the central nervous system. Mol Neurobiol 45:499–506. https://doi.org/10.1007/s12035-012-8264-y
Hayden MS, Ghosh S (2011) NF-κB in immunobiology. Cell Res 21:223–244. https://doi.org/10.1038/cr.2011.13
Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR (1998) Matrix metalloproteinases and diseases of the CNS. Trends Neurosci 21:75–80. https://doi.org/10.1016/s0166-2236(97)01169-7
He C, Zhao X, Li H, Wang F, Zhang J, Wang Y, Han Y, Yuan C, Niu Q (2021) Regulation of mGluR1 on the expression of PKC and NMDAR in aluminum-exposed PC12 cells. Neurotox Res 39:634–644. https://doi.org/10.1007/s12640-020-00319-5
Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21. https://doi.org/10.1016/j.neuron.2004.09.012
Lau CG, Zukin RS (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8:413–426. https://doi.org/10.1038/nrn2153
Funding
This work was supported by Beijing Tongren Hospital, Capital Medical University (2020-YJJ-ZZL-050), Bethune Public Welfare Foundation (B-19-H-20200622) and Chinese Academy of Medical Sciences and Peking Union Medical College (2016-I2M-2-006).
Author information
Authors and Affiliations
Contributions
Liming Dong performed the experiments and prepared the manuscript. Ning Jiang and Jie Bai provided experimental technical support. Yiman Li and Zhihui Song performed the data analysis. Xinmin Liu and Chao Zhang conceived the experiments and revised the manuscript. All authors checked and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests.
This study was performed in line with the guidelines for the Animal Care and Use Committee of the Institute of Medical Plant Development, Chinese Academy of Medical Sciences, and Peking Union Medical College.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dong, L., Jiang, N., Bai, J. et al. Neuroprotective Effects of Dammarane Sapogenins Against lipopolysaccharide-induced Cognitive Impairment, Neuroinflammation and Synaptic Dysfunction. Neurochem Res 48, 3525–3537 (2023). https://doi.org/10.1007/s11064-023-03997-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03997-7