
Abstract
Methylglyoxal (MG) is a reactive dicarbonyl presenting both endogenous (e.g. glycolysis) and exogenous (e.g. food cooking) sources. MG induces neurotoxicity, at least in part, by affecting mitochondrial function, including a decline in the oxidative phosphorylation (OXPHOS) system activity, bioenergetics failure, and redox disturbances. Sulforaphane (SFN) is an isothiocyanate found mainly in cruciferous vegetables and exerts antioxidant and anti-inflammatory effects in mammalian cells. SFN also decreases mitochondrial vulnerability to several chemical stressors. SFN is a potent activator of the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), which is a master regulator of the mammalian redox biology. Here, we have investigated whether and how SFN would be able to prevent the MG-induced mitochondrial collapse in the human neuroblastoma SH-SY5Y cells. The cells were exposed to SFN at 5 µM for 24 h prior to the administration of MG at 500 µM for additional 24 h. We found that SFN prevented the MG-induced OXPHOS dysfunction and mitochondrial redox impairment. SFN stimulated the activity of the enzyme γ-glutamylcysteine ligase (γ-GCL), leading to increased synthesis of glutathione (GSH). Inhibition of γ-GCL with buthionine sulfoximine (BSO) or silencing of Nrf2 using small interfering RNA (siRNA) against this transcription factor reduced the levels of GSH and abolished the mitochondrial protection promoted by SFN in the MG-treated cells. Thus, SFN protected mitochondria of the MG-challenged cells by a mechanism involving the Nrf2/γ-GCL/GSH axis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methylglyoxal (MG; C3H4O2) is a reactive dicarbonyl and induces cellular dysfunction by different mechanisms [1]. The endogenous sources of MG include the glycolytic pathway (from the triose phosphates glyceraldehyde-3-phosphate and dihydroxyacetone phosphate), the degradation of certain amino acids (such as glycine and threonine), and acetone (which is originated from the ketone body acetoacetate) [2, 3]. Thus, hyperglycemia (or impaired glycolysis, leading to accumulation of its intermediates) and catabolic states (resulting or not from insulin resistance or deficiency) favor the production of MG [3]. Those events are observed mainly during the onset and progression of both types of diabetes mellitus, in which the levels of MG are high [4]. Nonetheless, MG has been viewed as an important toxic agent also in the case of neurodegenerative diseases, such as in Alzheimer’s disease [5] and in Parkinson’s disease [6]. Exogenous sources of MG include food cooking and foodstuff autoxidation [7, 8], cigarette smoking [9,10,11].
MG impairs the mammalian redox biology by both direct and indirect ways. This dicarbonyl generates the reactive species radical anion superoxide (O2−·), hydrogen peroxide (H2O2), and hydroxyl radical (·OH) by undergoing autoxidation [12]. Furthermore, the conversion of pyruvate (as well as aminoacetone or acetol) to MG leads to the formation of H2O2 [13]. By impairing the mitochondrial function, MG promotes the production of reactive species by an indirect way. It has been demonstrated that the exposure to MG causes an increase in the generation of O2−· and H2O2 by the organelles [14, 15]. At least in part, the mechanism by which MG promotes mitochondrial dysfunction includes impaired oxidative phosphorylation (OXPHOS) and loss of mitochondrial membrane potential (MMP), consequently affecting the production of adenosine triphosphate (ATP) by those organelles [16, 17]. In the cellular level, MG stimulates cell death by a mitochondria-related pathway involving the release of cytochrome c to the cytoplasm [16]. Even though these events have been described in virtually any nucleated cell type exposed to MG, neurons have been seen as a major target of MG, since the glycolytic rates and the consumption of ketone bodies are high in these cells [18]. In this context, MG levels are high in the plaques and neurofibrillary tangles samples, as well as in the cerebrospinal fluid, obtained from the brain of patients suffering from Alzheimer’s disease [19, 20]. Therefore, administrating the toxicity induced by MG is pharmacologically relevant in both prevention and treatment of diseases associated with this reactive molecule [21, 22].
The detoxification of MG is dependent on the tripeptide glutathione (GSH), which is also the major non-enzymatic antioxidant in human cells [23]. GSH is involved in the conversion of MG to D-lactate by the glyoxalase system [24]. The enzyme γ-glutamylcysteine ligase (γ-GCL) is the rate-limiting step in the GSH synthesis and its expression is modulated by the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) [25]. Importantly, Nrf2 also upregulates the expression of enzymes associated with the detoxification of MG [26,27,28]. In that context, stimulating the Nrf2/γ-GCL/GSH axis would be strategic to suppress the MG-induced mitochondrial and cellular dysfunctions observed in mammalian cells. Sulforaphane (SFN, C6H11NOS2) is an antioxidant found in the cruciferous vegetables (broccoli and cauliflower, among others) and also exerts detoxifying-related effects, as observed in several experimental models [29, 30]. SFN is a potent inducer of Nrf2, leading to the upregulation of cytoprotective enzymes involved in the maintenance of the redox homeostasis and in cell survival [14]. We have demonstrated that SFN promotes mitochondrial protection against the pro-oxidant agent H2O2 [31]. However, it was not previously checked whether SFN would be able to protect mitochondria in the case of exposure to MG. Thus, we investigated here whether and how SFN would be able to counteract the mitochondrial dysfunction induced by MG in the human neuroblastoma SH-SY5Y cells.
Materials and Methods
Materials
We acquired the analytical grade reagents and Ham’s F-12 nutrient mixture from Sigma Chemical Co. (MO, USA). The plastic materials used to culture the cells were purchased from Corning, Inc. (NY, USA) and Beckton Dickson (NJ, USA). Dulbecco’s modified Eagle medium (DMEM) were obtained from Gibco/BRL (Grand Island, USA). The assay kits herein utilized were purchased from Abcam (MA, USA) or Cayman Chemical (MI, USA), as described below in details.
Experimental Model
We acquired the human neuroblastoma SH-SY5Y cells from the American Type Culture Collection (Manassas, VA, USA). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 HAM nutrient medium (1:1 mixture) presenting fetal bovine serum (FBS, 10%), 2 mM l-glutamine, 1000 units/mL penicillin, 1000 µg/mL streptomycin, and 2.5 µg/mL amphotericin B in a 5% CO2 humidified incubator at 37 °C.
In order to induce a 50%-decrease in cell viability, we have utilized MG at 500 µM, as previously reported by us [32] and by others [14, 33, 34]. MG at 500 µM impairs mitochondrial function and redox biology, leading to decreased function of the OXPHOS system, loss of the mitochondrial membrane potential (MMP), decreased synthesis of ATP, and increased production of reactive species, among other effects that we aimed to investigate in this work [35]. SFN was tested at 0.625–5 µM for different periods of incubation, according to the specificities of each assay. Aiming to reveal whether and how SFN would be capable to prevent the MG-induced mitochondrial collapse, SFN was administrated to the cells for 24 h before the challenge with MG for additional 24 h. The synthesis of GSH was inhibited by buthionine sulfoximine (BSO) at 400 µM, which was administrated to the cells in the presence or absence of SFN for 24 h. Silencing of the transcription factor Nrf2 was achieved by performing cell transfection using the Lipofectamine RNAiMAX Reagent (Life Technologies, CA, USA) with small interfering RNA (siRNA) against Nrf2 or with siRNA against non-target mRNA (scrambled sequence), based on the recommendations of the manufacturer (Santa Cruz, CA, USA).
Cell Viability and Cytotoxicity
The viability of the SH-SY5Y cells was assayed by utilizing an assay kit based on the recommendations of the manufacturer (Abcam, MA, USA; catalog number ab211091). In brief, the formazan originated from 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent was recorded at 590 nm in a plate spectrophotometer. The activity of the enzyme lactate dehydrogenase (LDH, which is cytoplasmic) was tested in the culture medium was used as an index of plasma membrane integrity (cytotoxicity) by using an assay kit. After the reaction of the samples with the WST reagent, the signal was recorded at 450 nm in a plate spectrophotometer (SpectraMax M5e, Molecular Devices, USA), following the protocol provided by the manufacturer (Abcam, MA, USA; catalog number ab65393).
Mitochondria-Related Apoptotic Parameters
The levels of cytochrome c in the cytoplasm (i.e., cytochrome c released from the mitochondria) were utilized as an index of the mitochondria-related apoptotic changes observed in during cell death [36]. Cytoplasmic cytochrome c was assayed based on the protocol by Borutaite et al. [37]. Briefly, mitochondria were extracted from the cells, as described below, and the cytosolic fraction was utilized to measure the levels of cytochrome c. The ascorbate-reduced-minus-ferricyanide-oxidized signal was checked via the wavelength pairs 550/535 nm (ε = 18.5 mM−1 cm−1 for cytochrome c) in a plate reader (SpectraMax M5e, Molecular Devices, USA), as previously published by Rieske [38]. We have quantified the activity of the pro-apoptotic enzymes caspase-9 and caspase-3 by utilizing assay kits according to the protocol provided by the manufacturer (Abcam, MA, USA; catalog numbers ab65607 and ab39383, respectively). Briefly, cells were lysed on ice and incubated with LEDH-AFC (AFC: 7-amino-4-trifluoromethyl coumarin; for caspase-9) or DEVD-AFC (for caspase-3), which are specific substrates of the enzymes, during 1 h at 37 °C. The signal was recorded in a plate reader at 400 nm/505 nm (EX/EM) (SpectraMax M5e, Molecular Devices, USA). The levels of cleaved poly [ADP-ribose] polymerase 1 (PARP1) were quantified by utilizing a commercial assay kit as described by the manufacturer (Abcam, MA, USA; catalog number ab174441). After the incubations, the signal was read in a plate reader (450 nm; SpectraMax M5e, Molecular Devices, USA). DNA fragmentation was assayed through the utilization of an assay kit based on the recommendations of the manufacturer (Roche, Germany; catalog number 11585045001). The 5′-bromo-2′-deoxy-uridine (BrdU) reagent, a thymidine analogue, was incorporated into the genomic DNA and the signal of the BrdU-labeled DNA fragments was recorded in the cytoplasmic fraction of the samples by using an anti-BrdU antibody at 370 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA).
Isolation of Mitochondria and Submitochondrial Membranes
Mitochondria were extracted from the SH-SY5Y cells by using the protocol published by Wang et al. [39]. Brief, cells were washed and homogenized in a buffer presenting sucrose at 250 mM, KCl at 10 mM, EGTA at 1 mM, EDTA at 1 mM, MgCl2 at 1 mM, dithiothreitol (DTT) at 1 mM, phenylmethylsulphonyl floride (PMSF) at 1 mM, benzamidine at 1 mM, pepstatin A at 1 mM, leupeptin at 10 mg/mL, aprotonin at 2 mg/mL, and HEPES at 20 mM (pH 7.4). After that procedure, the samples were centrifuged at 1000×g during 10 min at 4 °C in order to remove cellular contaminants. Then, functional mitochondria were obtained after centrifuging the supernatants at 11,000×g during 20 min at 4 °C. We have utilized mitochondria at 1 mg/mL and protein content was quantified by the Bradford method. We quantified the activity of the LDH enzyme in the mitochondrial samples to check the purity of that fraction (Fig. S1). The submitochondrial particles (SMP), i.e. the membranes of functional mitochondria containing all the components of the OXPHOS system, were obtained after freeze and thawing (three times) functional mitochondria, as reported by Poderoso et al. [40]. We utilized the SMP to assess the production of O2−· and the levels of redox impairment markers in the mitochondria.
Redox Impairment Markers
Both total and mitochondrial levels of malondialdehyde (MDA) were assessed here by using a commercial assay kit according to the instructions of the manufacturer (Abcam, MA, USA; catalog number ab233471). Briefly, the cells (2 × 106) were harvested and washed in phosphate buffered saline (PBS, pH 7.4). The cells were homogenized in a lysis buffer provided in the kit and centrifuged at 13,000×g for 10 min (4 °C) to remove debris. MDA from the samples interacts with a specific color reagent leading to a product with blue color, whose signal is recorded at 695 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA). Similarly, the levels of protein carbonyl groups were measured in both total and mitochondrial samples after reacting with dinitrophenylhydrazine (DNPH) by using an ELISA assay kit, following the instructions of the manufacturer (Abcam, MA, USA; catalog number ab238536). The signal was recorded at 450 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA). A reduced/oxidized bovine serum albumin (BSA) standard curve was utilized to calculate the amounts of carbonylated proteins in the samples. Total and mitochondrial levels of 3-nitrotyrosine were assayed through the utilization of an indirect ELISA assay [32]. Before quantifying the levels of DNA-incorporated 8-hydroxy-2′-deoxyguanosine (8-OHdG), DNA was purified by using an extraction kit as recommended by the manufacturer of an assay kit (Roche, Germany; catalog number 11,814,770,001). After this step, the levels of 8-OHdG were assessed in the samples by using an ELISA assay kit following the protocol provided by the manufacturer (Abcam, MA, USA; catalog number ab201734).
Reactive Oxygen and Nitrogen Species
As described above, the SMP were obtained after extracting functional mitochondria from the cells. The production of O2−· was assayed by using the SMP, as reported by Poderoso et al. [40]. In brief, the SMP were incubated in a reaction buffer presenting mannitol at 230 mM, sucrose at 70 mM, HEPES-KOH at 10 mM, succinate at 4.2 mM, KH2PO4 at 0.5 mM, catalase at 0.1 µM, and adrenaline at 1 mM (pH 7.4). The generation of adrenochrome from adrenaline after undergoing autoxidation (in a process that gives rise to O2−·) was recorded at 480 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA). NO· production was assessed through the utilization of an assay kit based on the instructions of the manufacturer (Abcam, MA, USA; catalog number ab65327). Nitrate reductase generates nitrite, that reacts with the probe DAN (2,3-diaminonaphthalene) and the signal is recorded in a fluorescence plate reader at 360 nm/450 nm (EX/EM) (SpectraMax M5e, Molecular Devices, USA). In order to assess the production of H2O2 in the samples, we have utilized the probe OxiRed in the presence of the enzyme horseradish peroxidase as recommended by the manufacturer of the assay kit (Abcam, MA, USA; catalog number ab102500). The signal was recorded in a fluorescence plate reader at 535 nm/587 nm (EX/EM) (SpectraMax M5e, Molecular Devices, USA).
Mitochondrial Function
Different assays were utilized to test mitochondrial function in this experimental model. The activity of the Complex I was checked by using an assay kit based on the protocol provided by the manufacturer (Abcam, MA, USA; catalog number ab109721). A microplate pre-coated with capture antibodies against Complex I was utilized to ensure specificity. Complex I converts NADH into NAD+, reducing a dye (ε = 25.9/nM/well) whose signal is read at 450 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA). The activity of Complex V was assessed by using an assay kit following the instructions of the manufacturer (Cayman Chemical, MI, USA; catalog number 701,000). This assay kit uses the conversion of ATP into ADP by Complex V to detect the activity of this complex. Pyruvate kinase, which is provided in the assay kit, converts phosphoenolpyruvate into pyruvate, generating ATP from that ADP. Then, lactate dehydrogenase, which is also present in the assay kit, converts this pyruvate into lactate, consuming NADH and generating NAD+. The signal resulting from the oxidation of NADH into NAD+ is recorded at 340 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA). Alterations in the MMP were investigated through the utilization of the cationic dye tetraethylbenzimidazolylcarbocyanine iodide (JC-1) following the instructions of the manufacturer of an assay kit (Abcam, MA, USA; catalog number ab113850). Aiming to quantify the levels of ATP, the samples were deproteinized following the protocol recommended by the manufacturer of an assay kit (Abcam, MA, USA; catalog number ab83355). After specific incubations, the signal was read at 570 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA).
GSH Levels
The levels of GSH were assessed through the utilization of an assay kit following the recommendations of the manufacturer of an assay kit (Abcam, MA, USA; catalog number ab138881). In this assay, the GSG present in the samples reacts with a dye and the signal is read at 490/520 nm (EX/EM) in a fluorescence microplate reader (SpectraMax M5e, Molecular Devices, USA).
γ-GCL Activity
We have quantified the activity of γ-GCL, which is the rate-limiting step in the production of GSH in mammalian cells, based on the protocol reported by White et al. [41]. In summary, after homogenization of the cells in a buffer (20 mM Tris–HCl, 1 mM EDTA, 250 mM sucrose, 10 mM sodium borate, and 2 mM serine at pH 7.4), the samples were centrifuged at 10,000×g during 10 min at 4 °C. We have collected the supernatants and centrifuged it again at 15,000×g during 20 min at 4 °C. The supernatants were collected and used in the γ-GCL assay. The signal resulting from formation of an 2,3-naphthalenedicarboxaldehyde (NDA)-γ-glutamylcysteine (γ-GC) compound was read in a fluorescent plate reader at 472 nm/528 nm (EX/EM) (SpectraMax M5e, Molecular Devices, USA).
Nucleus Isolation
Cell nucleus was isolated after collecting the cells by scraping as recommended by the manufacturer of an assay kit (Cayman Chemical, MI, USA; catalog number 10,009,277). Purified nuclei were used to check the activity of the transcription factor Nrf2. Protein determination was performed according to the Bradford assay protocol.
Nrf2 Activity
An assay kit was used to quantify the activity of the transcription factor as recommended by the manufacturer (Abcam, MA, USA; catalog number ab207223). Isolated nuclear extracts were added to the wells of a plate containing the specific double stranded DNA sequence with the Nrf2 consensus binding site (5′-GTCACAGTGACTCAGCAGAATCTG-3′). The signal was recorded at 450 nm in a plate reader (SpectraMax M5e, Molecular Devices, USA).
Statistical Analyses
We have utilized the software GraphPad 5.0 to analyze our data statistically. Data presented here are shown as the mean ± standard error of the mean (SEM) of three or five independent experiments (performed in triplicate). The p values were considered significant when p < 0.05. The differences between the experimental groups were assessed after performing the one-way ANOVA analysis followed by the post hoc Tukey’s test.
Results
SFN Prevented Loss of Viability, Cytotoxicity, and Apoptosis in MG-Exposed SH-SY5Y Cells
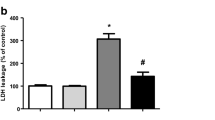
As depicted in Fig. 1a, the pretreatment with SFN at 5 µM alleviated the effect of MG on the viability of SH-SY5Y cells (p < 0.01). Besides, SFN pretreatment attenuated the MG-induced cytotoxicity in this experimental model (p < 0.01; Fig. 1b). It was also checked whether SFN would be able to prevent the MG-elicited mitochondria-related apoptosis in the SH-SY5Y cells. SFN prevented the MG-induced release of cytochrome c from the mitochondria (p < 0.01; Fig. S2A). SFN pretreatment also suppressed the the MG-elicited upregulation in the activity of the pro-apoptotic caspases-9 and -3 (p < 0.01; Fig. S2B and C, respectively). SFN pretreatment also reduced the levels of cleaved PARP and of DNA fragmentation (p < 0.001; Fig. S2D and E, respectively).

Sulforaphane (SFN) prevented the loss of cell viability (a) and cytotoxicity (b) induced by methylglyoxal (MG) in SH-SY5Y cells. Cells were treated with SFN at 5 µM for 24 h before the administration of MG at 500 µM for additional 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells
SFN Prevented the MG-Induced Redox Impairment in SH-SY5Y Cells
It was next evaluated whether SFN would be able to induce an antioxidant effect in SH-SY5Y cells exposed to MG. We found that SFN pretreatment decreased the levels of lipid peroxidation (Fig. 2a), protein carbonylation (Fig. 2b), protein nitration (Fig. 2c), and DNA oxidation (Fig. 2d) in the SH-SY5Y cells exposed to MG (p < 0.01 for all). A similar effect was observed in the membranes of mitochondria extracted from the SH-SY5Y cells challenged with MG. SFN prevented the MG-induced lipid peroxidation (Fig. 3a), protein carbonylation (Fig. 3b), and protein nitration in the mitochondrial membranes isolated from SH-SY5Y cells (p < 0.01 for all). SFN pretreatment also reduced the production of reactive species in the MG-challenged cells. SFN prevented the MG-induced increase in the production of O2−· (Fig. 4a), H2O2 (Fig. 4b), and NO· (Fig. 4c) in the SH-SY5Y cells (p < 0.01 for all).

Sulforaphane (SFN) prevented lipid peroxidation (a), protein carbonylation (b), protein nitration (c), and DNA oxidation (d) induced by methylglyoxal (MG) in SH-SY5Y cells. Cells were treated with SFN at 5 µM for 24 h before the administration of MG at 500 µM for additional 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells

Sulforaphane (SFN) prevented lipid peroxidation (a), protein carbonylation (b), and protein nitration (c) induced by methylglyoxal (MG) in the membranes of mitochondria isolated from the SH-SY5Y cells. Cells were treated with SFN at 5 µM for 24 h before the administration of MG at 500 µM for additional 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells

Sulforaphane (SFN) prevented the MG-induced increase in the production of radical anion superoxide (O2−·) (a), hydrogen peroxide (H2O2) (b) and nitric oxide (NO·) (c) in the SH-SY5Y cells. The production of O2−· was assayed using submitochondrial membranes (SMP), as described in “Materials and methods” section. Cells were treated with SFN at 5 µM for 24 h before the administration of MG at 500 µM for additional 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, * p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells
SFN Prevented the MG-Induced Mitochondrial Dysfunction in SH-SY5Y Cells
We next examined whether SFN would be able to prevent the mitochondrial impairment in the cells that were exposed to MG. We found that a pretreatment with SFN suppressed the MG-induced decline in the activities of the complexes I and V (p < 0.01; Fig. 5a and b, respectively). SFN also prevented the MMP collapse in the MG-treated cells (p < 0.01; Fig. 5c). The MG-elicited decrease in the production of ATP was also abolished in the cells that were pretreated with SFN (p < 0.01; Fig. 5d).

Sulforaphane (SFN) prevented the MG-induced decrease in the activities of complexes I (a) and V (b), and in the levels of MMP (c) and ATP (d). Cells were treated with SFN at 5 µM for 24 h before the administration of MG at 500 µM for additional 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells
SFN Depends on GSH to Prevent Mitochondrial Impairment in MG-Treated SH-SY5Y Cells
Aiming to reveal the mechanism by which SFN prevented mitochondrial collapse in the MG-challenged SH-SY5Y cells, we checked whether the inhibition of the synthesis of GSH with BSO would alter the SFN-induced effects seen here. According to Fig. 6a, BSO abolished the effect of the pretreatment with SFN on the activity of complex I in MG-exposed cells (p < 0.01). Moreover, the SFN-induced prevention of the MMP collapse was suppressed by BSO in the MG-treated cells (p < 0.01; Fig. 6b). Similarly, BSO abrogated the effects of SFN on the levels of ATP in the MG-challenged cells (p < 0.01; Fig. 6c). BSO also abolished the effects of SFN on the release of cytochrome c from the mitochondria and on lipid peroxidation in the membranes of the organelles obtained from the MG-treated SH-SY5Y cells (p < 0.01; Fig. S3A and 3B, respectively). Besides, BSO blocked the effect of the pretreatment with SFN on the viability of the SH-SY5Y cells exposed to MG (p < 0.01; Fig. 7).

Buthionine sulfoximine (BSO) suppressed the effects induced by a pretreatment with sulforaphane (SFN) on the activity of complex I (a) and on the levels of MMP (b) and ATP (c) in cells exposed to methylglyoxal (MG). BSO at 400 µM was administrated to the cells for 24 h in the absence or presence of SFN at 5 µM. Then, the cells were exposed to MG for further 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells, ºp < 0.01 vs. SFN + MG-treated cells

Buthionine sulfoximine (BSO) suppressed the cytoprotection induced by a pretreatment with sulforaphane (SFN) in cells exposed to methylglyoxal (MG). BSO at 400 µM was administrated to the cells for 24 h in the absence or presence of SFN at 5 µM. Then, the cells were exposed to MG for further 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. MG-treated cells, ºp < 0.01 vs. SFN + MG-treated cells
SFN Upregulates the Levels of GSH by an Nrf2/γ-GCL-Dependent Manner
We next checked whether and how SFN would modulate the levels of GSH in the SH-SY5Y cells. It was observed that SFN (at 2.5–5 µM for 24 h) stimulated the production of GSH in this experimental model (p < 0.01; Fig. 8a). Administration of BSO blocked the SFN-induced increase in the levels of GSH in the SH-SY5Y cells (p < 0.01; Fig. 8b). Moreover, silencing of the transcription factor Nrf2 suppressed the SFN-promoted upregulation in the levels of GSH (p < 0.01; Fig. 8c). As depicted in Fig. 9a, SFN also upregulated the activity of γ-GCL in the SH-SY5Y cells (p < 0.01). BSO was also efficient in inhibiting this enzyme in this experimental design (p < 0.01; Fig. 9b). The activity of γ-GCL was also significantly decreased in the SFN-treated cells in which Nrf2 was silenced (p < 0.01; Fig. 9c). We next investigated whether Nrf2 would be involved in the mitochondrial protection promoted by SFN in this experimental model. We found that silencing of Nrf2 suppressed the effects of the pretreatment with SFN on the activity of complex I (Fig. 10a), and on the levels of MMP (Fig. 10b) and ATP (Fig. 10c) in the cells exposed to MG (p < 0.01 for all). Nrf2 silencing also blocked the cytoprotection induced by a pretreatment with SFN in this experimental model (p < 0.01; Fig. 11). The effects of the siRNA targeting Nrf2 on the activity of this transcription factor may be seen in Fig. S4 (Fig. 12).

Sulforaphane (SFN) upregulated the levels of glutathione (GSH) in SH-SY5Y cells. The cells were incubated with SFN at 0.625–5 µM for 24 h in order to quantify the levels of GSH (a). Buthionine sulfoximine (BSO) at 400 µM was administrated to the cells for 24 h in the presence or absence of SFN at 5 µM (b). The effects of silencing Nrf2 on the levels of GSH in SFN-treated cells (c). Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. SFN-treated cells, a p < 0.01 vs. the control group transfected with negative control (NC) siRNA; b p < 0.01 vs. SFN-treated cells transfected NC siRNA

Sulforaphane (SFN) upregulated the activity of γ-glutamylcysteine ligase (γ-GCL) in SH-SY5Y cells. The cells were incubated with SFN at 0.625–5 µM for 24 h in order to examine the activity of γ-GCL (a). Buthionine sulfoximine (BSO) at 400 µM was administrated to the cells for 24 h in the presence or absence of SFN at 5 µM (b). The effects of silencing Nrf2 on the activity of γ-GCL in SFN-treated cells (c). Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs. the control group, #p < 0.01 vs. SFN-treated cells, a p < 0.01 vs. the control group transfected with negative control (NC) siRNA; b p < 0.01 vs. SFN-treated cells transfected NC siRNA

Silencing of the transcription factor Nrf2 suppressed the effects induced by a pretreatment with sulforaphane (SFN) on the activity of complex I (a) and on the levels of MMP (b) and ATP (c) in SH-SY5Y cells exposed to methylglyoxal (MG). Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, a p < 0.01 vs. the control cells; b p < 0.01 vs. the MG-treated cells transfected with negative control (NC) siRNA; *p < 0.01 vs. MG-treated cells transfected with NC siRNA; #p < 0.01 vs. the MG and SFN-treated cells transfected with NC siRNA

Silencing of the transcription factor Nrf2 suppressed the effects induced by a pretreatment with sulforaphane (SFN) on the viability of SH-SY5Y cells exposed to methylglyoxal (MG). Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, a p < 0.01 vs. the control cells; b p < 0.01 vs. the MG-treated cells transfected with negative control (NC) siRNA; *p < 0.01 vs. MG-treated cells transfected with NC siRNA; #p < 0.01 vs. the MG and SFN-treated cells transfected with NC siRNA

A summary of the effects seen in the present work. Sulforaphane (SFN) is found in cruciferous vegetables, such as broccoli and cauliflower. SFN is a potent activator of the nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor, which is the master regulator of the redox biology in mammalian cells. Nrf2 modulates the expression of several antioxidant enzymes, such as γ-glutamylcysteine ligase (γ-GCL). This is the rate-limiting step in the synthesis of the major non-enzymatic antioxidant in mammalian cells, glutathione (GSH). Silencing of the transcription factor Nrf2 or inhibition of the synthesis of GSH abolished the SFN-promoted mitochondrial protection in the cells exposed to methylglyoxal (MG), a pro-oxidant agent whose levels are high in several human diseases. Please, read the text for details.
Discussion
We have shown in the present work that SFN upregulated the Nrf2/γ-GCL/GSH axis preventing the MG-induced mitochondrial impairment in the SH-SY5Y cells. Inhibition of the synthesis of GSH or silencing the transcription factor Nrf2 abolished the SFN-promoted protection seen here. It was previously reported that SFN stimulates the synthesis of GSH in different cell types [42,43,44,45]. Moreover, it was also published that SFN prevented the cytotoxicity induced by certain toxicants, including MG [14, 42]. Nonetheless, it was not evaluated whether GSH would take a role in the SFN-induced mitochondrial protection in mammalian cells. GSH is an important part of the detoxification of MG, in which the enzymes glyoxalase I and II converts MG into D-lactate, a non-toxic metabolite that can be excreted from the cells [24]. Even though GSH is not excreted from the cells when utilized in that pathway, reduced levels of GSH may limit the detoxification of MG, leading to its accumulation and consequently affecting the cellular functions [3]. In that context, it was shown that MG scavengers alleviated the endothelial dysfunction caused by MG in aortic rings, indicating that strategies leading to reduced levels of MG result in tissue protection [21]. Recently, Tate et al. have demonstrated that MitoGamide, a mitochondria-targeted agent able to sequester MG, exerted cardioprotective effects in an experimental model of spontaneous diabetic cardiomyopathy, suggesting an important role for the mitochondria in the progression of the MG-induced cardiac intoxication [22]. Also, MG is a potent inducer of the formation of advanced glycation end products (AGEs), which may cause mitochondrial dysfunction in several cell types, including neurons and glia [46, 47]. Indeed, Angeloni et al. proposed that SFN would be a neuroprotective agent due to its antiglycative activity [48].
It was previously reported that SFN induced only a slight, but significant, increase in the activity of the enzyme glutathione reductase (GR), which is involved in the recycling of GSH in mammalian cells [25, 26]. Thus, the SFN-dependent increase in the synthesis of GSH observed here would be more important than the induction of the GR-mediated recycle of GSH in preventing cytoprotection (or even mitochondrial protection). Actually, it was confirmed, at least in part, by inhibiting the synthesis of GSH by the enzyme γ-GCL. Besides, Tarozzi et al. also reported that SFN did not upregulated the activities of the antioxidant enzymes superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) in the SH-SY5Y cell line [42]. SOD converts O2−· into H2O2, which generates H2O in the reaction mediated by CAT or GPx [49]. Taken together, it may be suggested that SFN strongly depends on the γ-GCL/GSH axis to prevent the MG-induced mitochondrial collapse. Importantly, the antioxidant effects seen here regarding the reduced production of both mitochondrial (O2−·) and total (H2O2 and NO·) reactive species may be a consequence of the mitochondrial protection elicited by SFN in this experimental model, since major enzymes involved in the detoxification of these pro-oxidant agents were not modulated by SFN in this cell line. Nevertheless, it remains to be better examined in further research.
Thus, we have found that SFN prevented the mitochondrial collapse induced by MG by a mechanism involving the activation of the Nrf2/γ-GCL/GSH axis in SH-SY5Y cells. Both mitochondria-related redox environment and bioenergetics status were protected by SFN, leading to general cytoprotection. Finally, the ability of SFN in promoting mitochondrial protection needs to be examined in in vivo experimental models, since the bioavailability of this natural compound should impact of its effects in the different mammalian tissues. Specially in the case of studying whether SFN would be able to protect neuronal and glial mitochondria, the blood–brain barrier should be taken into account, since it can limit the access of peripheral molecules to the central cells.
Data Availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).
References
Kalapos MP (1999) Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett 110:145–175. doi:https://doi.org/10.1016/s0378-4274(99)00160-5
Chakraborty S, Karmakar K, Chakravortty D (2014) Cells producing their own nemesis: understanding methylglyoxal metabolism. IUBMB Life 66:667–678. doi:https://doi.org/10.1002/iub.1324
Allaman I, Bélanger M, Magistretti PJ (2015) Methylglyoxal, the dark side of glycolysis. Front Neurosci 9:23. doi:https://doi.org/10.3389/fnins.2015.00023
Beisswenger PJ (2014) Methylglyoxal in diabetes: link to treatment, glycaemic control and biomarkers of complications. Biochem Soc Trans 42:450–456. doi:https://doi.org/10.1042/BST20130275
Angeloni C, Zambonin L, Hrelia S (2014) Role of methylglyoxal in Alzheimer’s disease. Biomed Res Int 2014:238485. doi:https://doi.org/10.1155/2014/238485
Hipkiss AR (2017) On the relationship between energy metabolism, proteostasis, aging and Parkinson’s disease: possible causative role of methylglyoxal and alleviative potential of carnosine. Aging Dis 8:334–345. https://doi.org/10.14336/AD.2016.1030
Kuntz S, Rudloff S, Ehl J, Bretzel RG, Kunz C (2009) Food derived carbonyl compounds affect basal and stimulated secretion of interleukin-6 and -8 in Caco-2 cells. Eur J Nutr 48:499–503. https://doi.org/10.1007/s00394-009-0035-9
Pfeifer YV, Haase PT, Kroh LW (2013) Reactivity of thermally treated α-dicarbonyl compounds. J Agric Food Chem 61:3090–3096. doi:https://doi.org/10.1021/jf302959k
Camel V, Bermond A (1998) The use of ozone and associated oxidation processes in drinking water treatment. Water Res 32:3208–3222
Fujioka K, Shibamoto T (2006) Determination of toxic carbonyl compounds in cigarette smoke. Environ Toxicol 21:47–54. doi:https://doi.org/10.1002/tox.20153
Fu T-M, Jacob DJ, Wittrock F, Burrows JP, Vrekoussis M, Henze DK (2008) Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J Geophys Res 113:D15303
Yamaguchi T, Nakagawa K (1983) Mutagenicity of and formation of oxygen radicals by trioses and glyoxal derivatives. Agric Biol Chem 47:2461–2465
Thornalley PJ, Wolff SP, Crabbe MJ, Stern A (1984) The oxidation of oxyhaemoglobin by glyceraldehyde and other simple monosaccharides. Biochem J 217:615–622. doi:https://doi.org/10.1042/bj2170615
Angeloni C, Malaguti M, Rizzo B, Barbalace MC, Fabbri D, Hrelia S (2015) Neuroprotective effect of sulforaphane against methylglyoxal cytotoxicity. Chem Res Toxicol 28:1234–1245. doi:https://doi.org/10.1021/acs.chemrestox.5b00067
Fürstenau CR, de Souza ICC, de Oliveira MR (2019) Tanshinone I induces mitochondrial protection by a mechanism involving the Nrf2/GSH axis in the human neuroblastoma SH-SY5Y cells exposed to methylglyoxal. Neurotoxicol Res 36:491–502. https://doi.org/10.1007/s12640-019-00091-1
Pun PB, Murphy MP (2012) Pathological significance of mitochondrial glycation. Int J Cell Biol 2012:843505. doi:https://doi.org/10.1155/2012/843505
Bo J, Xie S, Guo Y, Zhang C, Guan Y, Li C, Lu J, Meng QH (2016) Methylglyoxal impairs insulin secretion of pancreatic β-cells through increased production of ros and mitochondrial dysfunction mediated by upregulation of UCP2 and MAPKs. J Diabetes Res 2016:2029854. https://doi.org/10.1155/2016/2029854
Cobley JN, Fiorello ML, Bailey DM (2018) 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 15:490–503. doi:https://doi.org/10.1016/j.redox.2018.01.008
Smith MA, Richey PL, Taneda S, Kutty RK, Sayre LM, Monnier VM, Perry G (1994) Advanced Maillard reaction end products, free radicals, and protein oxidation in Alzheimer’s disease. Ann N Y Acad Sci 738:447–454. doi:https://doi.org/10.1111/j.1749-6632.1994.tb21836.x
Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G (1994) Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A 91:5710–5714. doi:https://doi.org/10.1073/pnas.91.12.5710
Dhar A, Dhar I, Desai KM, Wu L (2010) Methylglyoxal scavengers attenuate endothelial dysfunction induced by methylglyoxal and high concentrations of glucose. Br J Pharmacol 161:1843–1856. doi:https://doi.org/10.1111/j.1476-5381.2010.01017.x
Tate M, Higgins GC, De Blasio MJ, Lindblom R, Prakoso D, Deo M, Kiriazis H, Park M, Baeza-Garza CD, Caldwell ST, Hartley RC, Krieg T, Murphy MP, Coughlan MT, Ritchie RH (2019) The mitochondria-targeted methylglyoxal sequestering compound, mitogamide, is cardioprotective in the diabetic heart. Cardiovasc Drugs Ther 33:669–674. https://doi.org/10.1007/s10557-019-06914-9
Thornalley PJ (1990) The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J 269:1–11. doi:https://doi.org/10.1042/bj2690001
Thornalley PJ (2003) Glyoxalase I—structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans 31:1343–1348. https://doi.org/10.1042/bst0311343
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830:3143–3153. doi:https://doi.org/10.1016/j.bbagen.2012.09.008
Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30:42–59. doi:https://doi.org/10.1016/j.mam.2008.05.005
Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284:13291–13295. doi:https://doi.org/10.1074/jbc.R900010200
Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW (2010) When NRF2 talks, who’s listening? Antioxid Redox Signal 13:1649–1663. doi:https://doi.org/10.1089/ars.2010.3216
Houghton CA (2019) Sulforaphane: its “coming of age” as a clinically relevant nutraceutical in the prevention and treatment of chronic disease. Oxid Med Cell Longev 2019:2716870. https://doi.org/10.1155/2019/2716870
Huang C, Wu J, Chen D, Jin J, Wu Y, Chen Z (2019) Effects of sulforaphane in the central nervous system. Eur J Pharmacol 853:153–168. doi:https://doi.org/10.1016/j.ejphar.2019.03.010
de Oliveira MR, Brasil FB, Fürstenau CR (2018) Sulforaphane attenuated the pro-inflammatory state induced by hydrogen peroxide in SH-SY5Y cells through the Nrf2/HO-1 signaling pathway. Neurotoxicol Res 34:241–249. https://doi.org/10.1007/s12640-018-9881-7
de Oliveira MR, Ferreira GC, Schuck PF, Dal Bosco SM (2015) Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem Biol Interact 242:396–406. doi:https://doi.org/10.1016/j.cbi.2015.11.003
de Arriba SG, Krügel U, Regenthal R, Vissiennon Z, Verdaguer E, Lewerenz A, García-Jordá E, Pallas M, Camins A, Münch G, Nieber K, Allgaier C (2006) Carbonyl stress and NMDA receptor activation contribute to methylglyoxal neurotoxicity. Free Radic Biol Med 40:779–790. doi:https://doi.org/10.1016/j.freeradbiomed.2005.09.038
de Arriba SG, Stuchbury G, Yarin J, Burnell J, Loske C, Münch G (2007) Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells–protection by carbonyl scavengers. Neurobiol Aging 28:1044–1050. doi:https://doi.org/10.1016/j.neurobiolaging.2006.05.007
de Oliveira MR, de Souza ICC, Fürstenau CR (2020) Mitochondrial protection promoted by the coffee diterpene kahweol in methylglyoxal-treated human neuroblastoma SH-SY5Y cells. Neurotoxicol Res 37:100–110. https://doi.org/10.1007/s12640-019-00107-w
Jiang X, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73:87–106. doi:https://doi.org/10.1146/annurev.biochem.73.011303.073706
Borutaite V, Morkuniene R, Brown GC (1999) Release of cytochrome c from heart mitochondria is induced by high Ca2+ and peroxynitrite and is responsible for Ca(2+)-induced inhibition of substrate oxidation. Biochim Biophys Acta 1453:41–48. https://doi.org/10.1016/s0925-4439(98)00082-9
Rieske JS (1967) The quantitative determination of mitochondrial hemoproteins. Methods Enzymol 10:488–493
Wang K, Zhu L, Zhu X, Zhang K, Huang B, Zhang J, Zhang Y, Zhu L, Zhou B, Zhou F (2014) Protective effect of paeoniflorin on Aβ25-35-induced SH-SY5Y cell injury by preventing mitochondrial dysfunction. Cell Mol Neurobiol 34:227–234. doi:https://doi.org/10.1007/s10571-013-0006-9
Poderoso JJ, Carreras MC, Lisdero C, Riobó N, Schöpfer F, Boveris A (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92. doi:https://doi.org/10.1006/abbi.1996.0146
White CC, Viernes H, Krejsa CM, Botta D, Kavanagh TJ (2003) Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal Biochem 318:175–180. doi:https://doi.org/10.1016/s0003-2697(03)00143-x
Tarozzi A, Morroni F, Merlicco A, Hrelia S, Angeloni C, Cantelli-Forti G, Hrelia P (2009) Sulforaphane as an inducer of glutathione prevents oxidative stress-induced cell death in a dopaminergic-like neuroblastoma cell line. J Neurochem 111:1161–1171. doi:https://doi.org/10.1111/j.1471-4159.2009.06394.x
Dias IH, Chapple IL, Milward M, Grant MM, Hill E, Brown J, Griffiths HR (2013) Sulforaphane restores cellular glutathione levels and reduces chronic periodontitis neutrophil hyperactivity in vitro. PLoS ONE 8:e66407. https://doi.org/10.1371/journal.pone.0066407
Steele ML, Fuller S, Patel M, Kersaitis C, Ooi L, Münch G (2013) Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol 1:441–445. doi:https://doi.org/10.1016/j.redox.2013.08.006
Sedlak TW, Nucifora LG, Koga M, Shaffer LS, Higgs C, Tanaka T, Wang AM, Coughlin JM, Barker PB, Fahey JW, Sawa A (2018) Sulforaphane augments glutathione and influences brain metabolites in human subjects: a clinical pilot study. Mol Neuropsychiatry 3:214–222. https://doi.org/10.1159/000487639
Wang X, Yu S, Wang CY, Wang Y, Liu HX, Cui Y, Zhang LD (2015) Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell Dev Biol Anim 51:204–209. doi:https://doi.org/10.1007/s11626-014-9823-5
Patel SH, Yue F, Saw SK, Foguth R, Cannon JR, Shannahan JH, Kuang S, Sabbaghi A, Carroll CC (2019) Advanced glycation end-products suppress mitochondrial function and proliferative capacity of achilles tendon-derived fibroblasts. Sci Rep 9:12614. https://doi.org/10.1038/s41598-019-49062-8
Angeloni C, Malaguti M, Hrelia S (2015) Antiglycative activity of sulforaphane: a new avenue to counteract neurodegeneration? Neural Regen Res 10:1750–1751. doi:https://doi.org/10.4103/1673-5374.169613
Sies H, Berndt C, Jones DP (2017) Oxidative stress. Annu Rev Biochem 86:715–748. https://doi.org/10.1146/annurev-biochem-061516-045037
Acknowledgements
MRO receives a “Bolsa de Produtividade em Pesquisa 2 - PQ2” fellow from the Conselho Nacional de Pesquisa e Desenvolvimento Tecnológico (CNPq) (protocol number 301273/2018-9). This work was partially supported by CNPq (protocol numbers 400216/2016-7). RCBG receives a fellow from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11064_2020_3204_MOESM1_ESM.pdf
Figure S1 Mitochondrial isolation quality control. The activity of the cytoplasmic enzyme lactate dehydrogenase (LDH) was evaluated in the mitochondrial fraction obtained from the SH-SY5Y cells to check the purity of the samples.
11064_2020_3204_MOESM2_ESM.pdf
Figure S2 Sulforaphane (SFN) prevented the release of cytochrome c to the cytosol (A), the upregulation in the activity of the caspases-9 (B) and -3 (C), and the increase in the levels of cleaved PARP (D) and of DNA fragmentation (E) in methylglyoxal (MG)-treated SH-SY5Y cells. Cells were treated with SFN at 5 µM for 24 h before the administration of MG at 500 µM for additional 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs the control group, #p < 0.01 vs MG-treated cells.
11064_2020_3204_MOESM3_ESM.pdf
Figure S3 Buthionine sulfoximine (BSO) suppressed the effect of a pretreatment with sulforaphane (SFN) on the levels of cytosolic cytochrome c and of malondialdehyde (MDA, a molecular marker of lipid peroxidation) in the membranes of mitochondria obtained from SH-SY5Y cells exposed to methylglyoxal (MG). Buthionine sulfoximine (BSO) at 400 µM was administrated to the cells for 24 h in the presence or absence of SFN at 5 µM. Then, the cells were exposed to MG for further 24 h. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.01 vs the control group, #p < 0.01 vs MG-treated cells, º p < 0.01 vs SFN + MG-treated cells.
11064_2020_3204_MOESM4_ESM.pdf
Figure S4 The effects of silencing Nrf2 on the activity of this transcription factor. Data are presented as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, a p < 0.01 vs the control cells transfected with negative control (NC) siRNA; b p < 0.01 vs the SFN-treated cells transfected with NC siRNA
Rights and permissions
About this article

Cite this article
Brasil, F.B., Gobbo, R.C.B., de Almeida, F.J.S. et al. The Isothiocyanate Sulforaphane Depends on the Nrf2/γ-GCL/GSH Axis to Prevent Mitochondrial Dysfunction in Cells Exposed to Methylglyoxal. Neurochem Res 46, 740–754 (2021). https://doi.org/10.1007/s11064-020-03204-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03204-x