
Abstract
Alzheimer’s disease (AD) and diabetes mellitus (DM) share common pathophysiological findings, in particular, the mammalian target of rapamycin (mTOR) has been strongly implied to link to AD, while it also plays a key role in the insulin signaling pathway. However, the mechanism of how DM and AD is coupled remains elusive. In the present study, we found that streptozotocin (STZ)-induced DM mice significantly increased the levels P-mTOR Ser2448, P-p70S6K Thr389, P-tau Ser356 and Aβ levels (Aβ oligomer/monomer), as well as the levels of Drp1 and p-Drp1 S616 (mitochondrial fission proteins) are increased, whereas no change was found in the expression of Opa1, Mfn1 and Mfn2 (mitochondrial fusion proteins) compared with control mice. Moreover, the expression of 4-HNE and 8-OHdG showed an aberrant increase in the hippocampus of STZ-induced DM mice that is associated with a decreased capacity of spatial memory and a loss of synapses. Rapamycin, an inhibitor of mTOR, rescued the STZ-induced increases in mTOR/p70S6K activities, tau phosphorylation and Aβ levels, as well as mitochondria abnormality and cognitive impairment in mice. These findings imply that rapamycin prevents cognitive impairment and protects hippocampus neurons from AD-like pathology and mitochondrial abnormality, and also that rapamycin treatment could normalize these STZ-induced alterations by decreasing hippocampus mTOR/p70S6K hyperactivity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM) is defined as a complex metabolic disease, which is characterized by chronic hyperglycemia. Over 60% of DM patients experience diabetic neuropathy complications in peripheral or central nervous systems [1, 2]. Recently, several clinical findings and epidemiological studies showed that DM patients have a relatively high risk of developing cognitive dysfunction and Alzheimer’s disease (AD) [3, 4]. DM and AD share several similarities in molecular, biochemical, and mechanistic abnormalities [3,4,5]. AD is the most common cause of dementia and it is characterized by histopathological, biochemical and molecular abnormalities, including dystrophic neuritis, amyloid-β deposits, abnormal neurofibrillary tangles, constant oxidative stress, mitochondrial dysfunction and DNA damage [6]. However, the underlying mechanism of how DM and AD are connected remains relatively unexplored.
As an evolutionarily conserved serine/threonine protein kinase existing in all eukaryotic cells, the mammalian target of rapamycin (mTOR) plays a critical role in mediating protein synthesis, autophagy, glucose homeostasis, and mitochondrial function [7, 8]. mTOR is activated by growth factors (IGF1), insulin receptors (IRs), PI3K/Akt and GSK-3β. p70S6K, a downstream target for mTOR, regulates translation, and several studies indicated that mTOR could regulate the insulin response via p70S6K activation, especially in the brain. Thus, the regulation of mTOR hyperactivities is necessary for maintenance of brain homeostasis [9,10,11].
Recent studies from our and Oddo’s laboratory have spotlighted the role of mTOR in AD [12, 13], and these studies indicated that hyperactive mTOR exists in human AD brains, and that mTOR signaling is closely linked with AD pathology. Former studies also showed that the reduction of mTOR by genetic or pharmacological approaches in multiple animal models of AD could ameliorate Aβ and tau pathology while ameliorating cognitive deficits, thus suggesting an efficient therapeutic strategy for AD [12, 14]. However, whether mTOR-regulated AD-like pathology is associated with DM-related cognitive deficits remains to be elucidated. In this study, we demonstrated the activation of mTOR/p70S6K signaling and the change of AD-like pathology in STZ-induced DM mice. We also showed that rapamycin treatment could reverse AD-like pathology, mitochondrial abnormality and cognitive deficit through a mechanism of decreasing mTOR/p70S6K hyperactivity.
Methods and Material
Materials, Reagents and Antibodies
Streptozotocin (STZ), rapamycin, RIPA buffer, SDS, Tris and protease inhibitor cocktail were obtained from Sigma-Aldrich Co. (St, Louis, MO, USA). A Bradford kit was supplied by Bio-rad (California, USA). For the detailed information of the primary antibodies used in the present study, please refer to Table 1.
Animals and Treatment
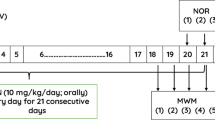
Male C57BL/6J mice (6–8 weeks old) were obtained from Guizhou experimental Animal Center in China, and pre-approved by the regional Animal Care center and Ethics Committee. The humidity and temperature kept ranged from 30 to 55% and 22–25 °C, respectively. A total of 24 mice were randomly separated into three groups (n = 8): control group, STZ group, and STZ + Rapamycin group. All mice were deprived of water for 12 h before treatment. 200 mg/kg STZ was intraperitoneally injected into C57BL/6J mice to induce DM as previously described [15]. The control mice were injected with an equal volume of 0.9% citrate buffer. Ten days after STZ administration, the mice tail vein blood glucose levels were confirmed using a digital glucometer, and a blood glucose concentration > 16 mM was considered to represent DM. After that, STZ-induced DM mice were randomly divided into STZ and STZ + Rapamycin groups, whereby the STZ + Rapamycin mice were administered with rapamycin 1.5 mg/kg, three times for one week by intraperitoneal (ip) injection, while the STZ mice group were intraperitoneally injected with 0.9% citrate buffer at the same times and volume. Behavioral tests were performed on the last week with rapamycin treatment. After the behavioral test was done, mice were then sacrificed to collect the brain tissue for western blot and immunofluoresencent staining.
Behavioral Testing
A round tank of 160 cm diameter and 50 cm height was used in this study. For the Morris water maze test (MWM) training, the mice were transferred into the water maze room at 9:00 am to acclimatize for 1 h. The training included 24 trials for 6 consecutive days. In each trial, mice were trained to seek a hidden platform (20 cm diameter) submerged 1 cm under the water surface for 60 s, and after that, mice remained on the platform for 20 s. If these mice couldn’t find the platform within the 60 s, the mice were gently guided to find the platform within the 60 s and land on the platform for 20 s. On day 7, the platform was removed and a spatial probe trial lasting 60 s was performed. The escape latency, the number of platform crossings and swimming speed were monitored by ANY-maze video tracking software (USA).
Preparation of Brain Samples and Protein Measurement
After MWM testing, all mice were immediately anesthetized by 10% chloral hydrate via intraperitoneal injection and euthanized. Immediately, the hippocampus was homogenized in RIPA buffer with 0.1% protease inhibitors cocktail on ice. Protein concentration of samples was determined by a Bradford kit.
Western Blotting
The Forty microgram protein was separated by 7.5–12% (w/v) SDS-polyacrylamide gels, and the separated proteins were further blotted onto 0.22–0.45µ m PVDF membranes (Millipore MA, USA). The membranes were blocked in 5% (w/v) nonfat milk for 1 h, after which the membranes were incubated with primary antibodies (Table 1) at 4 °C, overnight. After washing by Tris-buffered saline supplemented with 0.1% (v/v) Tween-20 (TBST), the membranes were incubated with anti-mouse or anti-rabbit secondary antibodies (1:5000, Bio-rad, California, USA) at room temperature for 1 h [13]. Immunoreactive bands were visualized by a ChemiDoc™ MP imaging system (Bio-rad, USA). The same membrane was used to detect other proteins with very different molecular weights, probed filters were stripped by using stripping buffer (100 mM 2-Mercaptoethanol, 2% SDS, 62.5 mM, Tris–HCl pH 6.8) at 50 °C for 30 min, and washing 3 times (15 min) with TBST, then incubated with the primary antibody. The protein levels of total and respective phosphorylated were examined in two separate membranes. The following procedures were adopted: (1) the same sample was used to evaluate total and phosphorylated protein levels; (2) the two gels were prepared and ran at the same time; (3) proteins have been transferred onto PVDF membranes and stained with specific antibodies, each band of interest was normalized with respect to β-tubulin.
Immunofluoresencent Staining and Confocal Image
For immunofluorescent staining, we followed the method previously described (14) with minor modification. Dewaxed and rehydrated hippocampal sections were blocked in TBST with 5% normal goat serum for 60 min. Sections were then incubated with primary antibodies: 4-HNE (rabbit, 1:100) or 8-OHdG (mouse, 1:100) at 4 °C overnight. After washing in TBS, the immunoreaction was detected using Alexa Fluor 488 (1:500, Life Technology, Carlsbad, CA, USA) or Alexa Fluor 549 (1:500, Life Technology, Carlsbad, CA, USA) conjugated secondary antibodies, respectively. Sections were mounted by vector anti-fading mounting medium (vector laboratories, Burlingame, CA, USA), and observed using Leica SP8 confocal microscopy. For 4-HNE staining, all images were captured on at 40 × objective with constant exposure time for per mouse (every section of the brain, n = 8 mice/group); To count the number of 8-OHdG positive cells, which was counted manually from 5 serial 40⋅ magnification per mouse (every section of the brain, n = 8 mice/group).
Statistical Analysis
Statistical analysis was performed using SPSS software, version 19.0, and the results were presented as mean ± SEM. Before calculation, all data were analyzed with normal distribution analysis. MWM data were analyzed using two-way repeated measures ANOVA. The other assay parameters were analyzed by one-way ANOVA followed by Bonferroni post-hoc test for multiple comparisons.
Results
Rapamycin Attenuated Hyperactive mTOR/p70S6k Pathway in the Hippocampus of STZ-Induced DM Mice
mTOR hyperactivity is associated with hyperglycemia and insulin resistance in the periphery [16]; however, whether brain mTOR was linked to DM-related changes remains unclear. To investigate the role of mTOR/p70S6k in the hippocampus of STZ-induced DM mice, the protein kinases and phosphatase of mTOR/p70S6k were examined by western blotting. We found that levels of phosphorylated mTOR at S2448 and p70S6k at T389 were raised in the hippocampus compared with the control group (p < 0.05 Fig. 1a, b). However, no change was found in the expression of total mTOR and p70S6k among the three groups (Fig. 1a, b). While further exploring the effect of rapamycin reducing mTOR in the hippocampus, we detected that rapamycin administration significantly decreased the levels of p-mTOR S2448 and P-P70S6k T389 in the hippocampus of STZ + Rapamycin mice compared with STZ-induced DM mice, whereas no alteration was observed in the total mTOR and P70S6k levels (Fig. 1a, b).

Rapamycin ameliorated mTOR/p70S6K pathway in STZ-induced DM mice. Representative immunoblots of mTOR and p70S6K proteins in panel (a) Histogram of p-mTOR S2448, T-mTOR, p70S6K T389, and T-p70S6K in panel (b) CON control mice, STZ STZ-induced DM mice, STZ + Rapa rapamycin treated STZ-induced DM mice. Blots are representative from 8 mice; quantifications of the blots were performed by normalizing the protein of interest to β-tubulin. (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment)
Rapamycin Ameliorated Hyperphosphorylated Tau Protein and Aβ1-42 Levels in Hippocampus of STZ-Induced DM Mice
As shown in Figs. 2a and b, the STZ-induced DM mice had increased levels of hyperphosphorylated tau at Ser356, Aβ1-42 oligomer and monomer in the hippocampus (p < 0.05, Fig. 2a, b). Rapamycin treatment could decrease levels of phosphor-tau S356 and Aβ1-42 oligomer and monomer, whereas the total tau protein level had no change in STZ + Rapamycin mice.

Rapamycin reduced levels of hyperphosphorylated tau protein and Aβ1-42. The levels of tau hyperphosphorylation, Aβ1-42 oligomer, and monomer are detected on the blots (panel a). Histogram of p-Tau S356, T-tau (tau5) protein Aβ1-42 oligomer, and monomer expression in panel b. CON control mice, STZ STZ-induced DM mice, STZ + Rapa rapamycin treated STZ-induced DM mice. Blots are representative from 8 mice; quantifications of the blots were performed by normalizing the protein of interest to β-tubulin. (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment)
Rapamycin Ameliorated the Impaired Mitochondrial Dynamics in Hippocampus of STZ-Induced DM Mice
It is proposed that mitochondrial dynamics play an important role in controlling mitochondrial morphology, distribution, and function. Western blotting revealed that the mitochondrial fusion proteins had no significant alterations as indicated by OPA1, Mfn1 and Mfn2 levels (Fig. 3a, b), whereas the levels of mitochondrial fission proteins Drp1 and p-Drp1S616 are obviously increased in STZ-induced DM mice compared to control mice, this effect being reversed by rapamycin treatment in STZ + Rapamycin mice (Fig. 3a, b)

Rapamycin reversed abnormal mitochondrial fission/fusion in STZ-induced DM mice. Representative immunoblots of OPA1, Mfn1, Mfn2 and Drp1 protein expression were detected in the hippocampus (Panel a). b Quantitative density analysis of mitochondrial fusion and fission proteins in panel b; CON control mice, STZ STZ-induced diabetic mice, STZ + Rapa rapamycin treated STZ-induced DM mice. Blots are representative from 8 mice; Quantifications of the blots were performed by normalizing the protein of interest to β-tubulin. (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment)
Rapamycin Attenuates Oxidative Stress Damage in Hippocampus of STZ-Induced DM Mice
To determine oxidative stress damage following STZ, we measured lipid peroxidation (4-hydroxynonenal (4-HNE)) and DNA oxidation (8-hydroxy-2′-deoxyguanosine (8-OHdG)) products via immunofluorescent staining or western blot (Figs. 4 and 5). The level of 4-HNE was measured in the hippocampal homogenates by western blot, as shown in Fig. 4a, and the level of 4-HNE was augmented in STZ-induced DM mice compared to control mice, and rapamycin treatment markedly decreased 4-HNE levels in STZ + Rapamycin mice (Fig. 4a, b). As shown in Figs. 4c and d, the intensity of 4-HNE significantly raised in the hippocampus of STZ-induced DM mice, compared to the control group (Fig. 4c, d). Rapamycin treatment obviously lowered 4-HNE intensity in STZ + Rapamycin mice (Fig. 4c, d). The 8-OHdG immunoreactive cells were counted in the hippocampus (Fig. 5a, b), and we detected that the number of 8-OHdG immunoreactive cells were augmented in the hippocampus of STZ-induced DM mice. In contrast, rapamycin treatment could significantly lower the number of cells in STZ + Rapamycin mice (Fig. 5a, b). These results imply that rapamycin reduces oxidative stress damage following STZ.

Rapamycin attenuated an increased 4-HNE expression in STZ-induced DM mice. Representative blots for 4-HNE in panel a 4-HNE protein expression of quantitative analysis is shown in panel (b) Blots are representative from 8 mice; Quantifications of the blots were performed by normalizing the protein of interest to β-tubulin. (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment). Representative immunofluoresent images for 4-HNE expression in the DG of the hippocampus in Panel (c) Red color denotes 4-HNE. Nuclei stained with DAPI (Blue color). Quantitative graph of 4-HNE fluorescent intensity is shown in panel (d) Scale bars indicate 20 µm

Rapamycin reduced an increased DNA oxidation in STZ-induced DM mice.Representative confocal images for anti-8-OHdG immunostaining of the CA1 areas of the brain (green color). Nuclei stained with DAPI (Blue color) in panel a; Quantitative graph presents the number of 8-OHdG positive cells in Panel b. (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment). Scale bars indicate 20 µm
Rapamycin Rescued Impaired Memory in STZ-Induced DM Mice
MWM test data showed that the escape latency of the mice significantly increased, while the time spent in the target quadrant and the number of platform location crossings dramatically decreased in STZ-induced DM mice compared with control mice (Fig. 6a, b, d). After application of rapamycin, the improved memory presented in STZ + rapamycin mice, exhibiting a shorter latency and an increased time spent in the target quadrant, along with increased numbers of platform location crossings (Fig. 6b, d). To examine if STZ could affect the athletic ability of mice, the swimming speed of mice in MWM were recorded, and the data showed no obvious differences among the three mice groups (Fig. 6c), suggesting that rapamycin treatment did not radically influence the motion ability of mice in the present experiment.

Rapamycin rescued memory deficiency in STZ-induced DM mice. a MWM test for 6 days and the escape latency was recorded. b Representative number of platform crossings. c Representative time spent in the target quadrant. d Swimming speed in MWM. N = 8 mice (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment)
Rapamycin Protected the Synapses in STZ-Induced Diabetic Mice
Since synapses are the structural basis of memory, we measured the synaptic protein expression. As shown in Figs. 7a and b, markedly decreased levels of PSD95 (post-synaptic protein)and SNAP 25 (pre-synaptic proteins) were observed in the hippocampus of STZ-induced DM mice compared with control mice (P < 0.05, respectively). After rapamycin application, the levels of PSD95 and SNAP 25 had increased in the STZ + rapamycin mice. The level of synaptophysin (pre-synaptic proteins) were not significantly changed in the three mice groups. These results indicate that the synapses are protected by rapamycin.

Rapamycin protected the synapses in STZ-induced DM mice. Representative immunoblots of PSD95, SNAP 25 and synaptophysin protein expression were detected in the hippocampus (Panel a). Quantitative density analysis of SNAP 25, synaptophysin and PSD95 proteins in panel b; CON control mice, STZ STZ-induced diabetic mice, STZ + Rapa rapamycin treated STZ-induced DM mice. Blots are representative from 5 to 8 mice. (*P < 0.05 vs. control group; #P < 0.05 vs. rapamycin treatment)
Discussion
Previous experimental and epidemiological studies have shown that there is an apparent link between AD and DM [3, 5, 17]. Although DM has two major subtypes, with type 1 diabetes and type 2 diabetes having distinct initial etiologyies, dysregulation of insulin nevertheless occurs in both diseases. Severe hypoinsulinemia leads to pancreatic cell loss in type 1 DM [18], and for type 2 DM, which is driven by peripheral insulin resistance, the pancreas fails to produce sufficient insulin, resulting in both insulin resistance and hypoinsulinemia [19]. Like type 2 DM, type 1 DM patients also show signs of insulin resistance [20]. Although human epidemiology most clearly links type 2 DM with increased risk of AD [21,22,23], dysregulation of insulin signaling in the brain is a commonality for type 1 and type 2 DM [24, 25]. Previous evidence has shown that employing STZ in a high dose can induce DM-related cognitive deficits at an early stage [15, 26], due to the single high dose STZ being toxic to pancreatic beta cells, and so is frequently used to model Type 1 DM [26]. In the current study, hippocampal AD-like pathology accompanying cognitive impairments were observed in mice after STZ injection. AD-like pathology, as a common feature during brain aging, involves complicated mechanisms. The hippocampus is a critical functional area of learning and memory, while it is also vulnerable to hyperglycemia [26,27,28]. Therefore, in this study, we investigated whether mTOR attenuated STZ induced AD-like pathology and spatial memory impairments in early stages, and attempted to identify the underlying mechanisms.
Previous studies have revealed that mTOR/p70S6k signaling is hyperactive in postmortem human brains [11,12,13,14, 29], whereas up-related mTOR/p70S6k signaling was found to be linked to AD pathogenesis. Previous evidence from several studies has indicated that declining mTOR signaling could lower Aβ and tau levels. Specifically, Oddo’s group showed that employing rapamycin rescued cognitive deficits and improved Aβ and tau pathology by increasing autophagy in 3×Tg-AD mice [12, 14]. Our studies have also shown that genetic reduction in mTOR signaling could decrease tau synthesis and hyperphosphorylation, suggesting a strong linking Aβ, tau and mTOR. Meanwhile, accumulating evidence has shown that dysregulation of mTOR/p70S6k signaling also contributes to insulin resistance [30,31,32] and aggravates the progression of diabetic complications, i.e. diabetic nephropathy [2]. Recent publications have appeared to explore the physiological or pathological role of mTOR signaling in the brain and its relevance to the development of AD and links to DM [33].
In this study, we indicated that levels of phosphorylated mTOR at Ser 2448 and phosphorylated p70S6k at Thr389 were augmented in the hippocampus of STZ-induced DM mice, implying it was hyperactivated in the diabetic brain. Rapamycin has been shown to effectively decrease levels of p-mTOR Ser 2448 and p-p70S6k Thr389 in the hippocampus of STZ + Rapamycin mice compared to STZ-DM mice. In STZ induced-DM mice, we identified increasing levels of hyperphosphorylated tau at Ser356 site and Aβ1-42 oligomer and monomer in the hippocampus after STZ injection compared with control mice. To further investigate if the mTOR/p70S6k pathway mediates phosphorylation of tau protein, and Aβ synthesis contributes to AD-like pathology in DM, we observed the effect of rapamycin on STZ-DM mice. After rapamycin administration into STZ-induced diabetic mice, we found that hyperphosphorylated tau at Ser356, but not total tau, decreased in STZ + Rapamycin mice, and levels of Aβ1-42 oligomer and monomer also decreased in STZ + Rapamycin mice. These data suggested that mTOR-regulated hyperphosphorylation of tau and Aβ synthesis are associated with DM-related cognitive deficits. With the increased expression of p-tau and Aβ oligomer and monomer in the condition of hyperglycemia, oxidative stress is greatly increased in the DM animal models and clinical DM patients [34, 35], and radically speeds up the pathological progress of the mitochondrial dynamics [34, 36, 37]. A balance of mitochondrial dynamics is vital for maintaining mitochondrial morphology and functions under physical and pathological situations such as DM [36], AD [34], and stroke [38]. In this study, we indicated increased levels of mitochondrial fission protein (Drp1 and p-Drp1 S616) in the hippocampus of STZ-induced DM mice, whereas no change had been found in mitochondrial fusion proteins (OPA1, Mfn1 and Mfn2) compared to the control group. Rapamycin treatment could lower Drp1 and p-Drp1 S616 levels but not the mitochondrial fusion proteins in STZ + Rapamycin mice, suggesting that rapamycin reverses the impairment of mitochondrial dynamics by reducing mTOR/p70S6K hyperactivity in the hippocampus of the STZ-treated DM mice. Defective mitochondria could generate more ROS products, finally causing oxidative stress damage to lipid and nucleic acids [39, 40]. In this study,we showed that the expressions of lipid peroxidation (4-HNE) and nucleic peroxidation (8-OHdG) were clearly increased in STZ-induced DM mice (Figs. 4 and 5), and subsequent rapamycin treatment could decrease the expressions of 4-HNE and 8-OHdG in the hippocampus of the STZ + rapamycin mice, which taken together suggest that hyperactive mTOR/p70S6k induced by hyperglycemia might accelerate tau hyperphosphorylation and Aβ oligomer/monomer aggregates, the impaired balance of mitochondrial fission and fusion, and cellular peroxidation, which were drastically ameliorated by rapamycin treatment. STZ-induced DM mice showed a learning and memory deficit similar to that seen in DM patients [41]. In this study, STZ-induced DM mice performed with cognitive dysfunction according to the MWM test. We further explored in this study whether inactivation of mTOR/p70S6k by rapamycin could attenuate STZ induced spatial memory impairment. Application of rapamycin with STZ-induced DM mice for one week could improve the spatial memory of the mice. It is proposed that STZ could impair spatial memory via activation of mTOR/p70S6k, whereas rapamycin might effectively reverse memory impairments in the STZ- induced DM mice. Many evidence showed that loss of synapses occurred in early AD, which is a robust link to cognitive deficits, leading to the conception that synaptic failure plays a vital role in AD [42]. Animal data have implied that memory deficiency is correlated with the distinct changes in synaptic plasticity in hippocampal slices in STZ induced diabetic rats [43, 44]. In our study, we found that rapamycin treatment reversed the decreased levels of PSD95 and SNAP 25 in the hippocampus of STZ-induced DM mice. Previous evidence has also supported that mTOR/p70S6k had played a key role in maintaining cognitive function and synaptic plasticity, and chronic suppression of mTOR/p70S6K with rapamycin improved memory in young adult mice and delayed age-related cognitive decline in older mice [12, 45].
Conclusions
In summary, our study showed that in diabetic rats, we observed mTOR/p70S6k hyperactivation, tau hyperphosphorylation, Aβ oligomer/monomer accumulations, mitochondria abnormality and memory impairment, and we demonstrated that inactivation of mTOR/p70S6k by rapamycin attenuated the AD-like pathology and DM-related cognitive deficits.
References
Akhmedzhanova LT, Barinov AN, Strokov IA (2018) [Diabetic and non-diabetic neuropathies in patients with diabetes mellitus]. Zhurnal nevrologii i psikhiatrii imeni SS Korsakova 118:113–120
Manschot SM, Biessels GJ, Rutten GE, Kessels RP, Gispen WH, Kappelle LJ, Utrecht Diabetic Encephalopathy Study G (2008) Peripheral and central neurologic complications in type 2 diabetes mellitus: no association in individual patients. J Neurol Sci 264:157–162
Bartl J, Monoranu CM, Wagner AK, Kolter J, Riederer P, Grunblatt E (2013) Alzheimer’s disease and type 2 diabetes: two diseases, one common link? World J Biol Psychiatry 14:233–240
Moreira PI (2012) Alzheimer’s disease and diabetes: an integrative view of the role of mitochondria, oxidative stress, and insulin. J Alzheimer’s Dis JAD 30(Suppl 2):S199–S215
Rosales-Corral S, Tan DX, Manchester L, Reiter RJ (2015) Diabetes and Alzheimer disease two overlapping pathologies with the same background: oxidative stress. Oxidative Med Cell Longev 2015:985845
Chen XQ, Mobley WC (2019) Alzheimer disease pathogenesis: insights from molecular and cellular biology studies of oligomeric abeta and tau species. Front NeuroSci 13:659
Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP (2013) mTOR kinase structure, mechanism and regulation. Nature 497:217–223
Takei N, Nawa H (2014) mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci 7:28
Perluigi M, Di Domenico F, Butterfield DA (2015) mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis 84:39–49
Weichhart T (2018) mTOR as regulator of lifespan, aging, and cellular senescence: a mini-review. Gerontology 64:127–134
Pei JJ, Hugon J (2008) mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med 12:2525–2532
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S (2010) Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 285:13107–13120
Tang Z, Bereczki E, Zhang H, Wang S, Li C, Ji X, Branca RM, Lehtio J, Guan Z, Filipcik P, Xu S, Winblad B, Pei JJ (2013) Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: implication for Alzheimer disease. J Biol Chem 288:15556–15570
Caccamo A, Magri A, Medina DX, Wisely EV, Lopez-Aranda MF, Silva AJ, Oddo S (2013) mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging cell 12:370–380
Wang S, Zhou SL, Min FY, Ma JJ, Shi XJ, Bereczki E, Wu J (2014) mTOR-mediated hyperphosphorylation of tau in the hippocampus is involved in cognitive deficits in streptozotocin-induced diabetic mice. Metab Brain Dis 29:729–736
Suhara T, Baba Y, Shimada BK, Higa JK, Matsui T (2017) The mTOR signaling pathway in myocardial dysfunction in type 2 diabetes mellitus. Curr Diabetes Rep 17:38
Kandimalla R, Thirumala V, Reddy PH (2017) Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim Biophys Acta Mol Basis Dis 1863:1078–1089
McCrimmon RJ, Sherwin RS (2010) Hypoglycemia in type 1 diabetes. Diabetes 59:2333–2339
DeFronzo RA (2004) Pathogenesis of type 2 diabetes mellitus. Med Clin N Am 88:787–835 (ix)
Greenbaum CJ (2002) Insulin resistance in type 1 diabetes. Diab/Metab Res Rev 18:192–200
Akomolafe A, Beiser A, Meigs JB, Au R, Green RC, Farrer LA, Wolf PA, Seshadri S (2006) Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 63:1551–1555
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P (2006) Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5:64–74
Sims-Robinson C, Kim B, Rosko A, Feldman EL (2010) How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 6:551–559
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Investig 122:1316–1338
Biessels GJ, Reagan LP (2015) Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci 16:660–671
Beauquis J, Roig P, De Nicola AF, Saravia F (2010) Short-term environmental enrichment enhances adult neurogenesis, vascular network and dendritic complexity in the hippocampus of type 1 diabetic mice. PLoS ONE 5:e13993
Alvarez EO, Banzan AM (1996) Hippocampus and learning: possible role of histamine receptors. Medicina 56:155–160
Thomas J, Garg ML, Smith DW (2013) Altered expression of histone and synaptic plasticity associated genes in the hippocampus of streptozotocin-induced diabetic mice. Metab Brain Dis 28:613–618
An WL, Bjorkdahl C, Liu R, Cowburn RF, Winblad B, Pei JJ (2005) Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3beta in SH-SY5Y neuroblastoma cells. J Neurochem 92:1104–1115
de la Monte SM (2012) Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 72:49–66
de la Monte SM (2017) Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 77:47–65
Diehl T, Mullins R, Kapogiannis D (2017) Insulin resistance in Alzheimer’s disease. Transl Res 183:26–40
Liang H, Nie J, Van Skike CE, Valentine JM, Orr ME (2019) Mammalian target of rapamycin at the crossroad between Alzheimer’s disease and diabetes. Adv Exp Med Biol 1128:185–225
Zhu X, Perry G, Smith MA, Wang X (2013) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimer’s Dis JAD 33(Suppl 1):S253–S262
Darroudi S, Fereydouni N, Tayefi M, Ahmadnezhad M, Zamani P, Tayefi B, Kharazmi J, Tavalaie S, Heidari-Bakavoli A, Azarpajouh MR, Ferns GA, Mohammadpour AH, Esmaily H, Ghayour-Mobarhan M (2019) Oxidative stress and inflammation, two features associated with a high percentage body fat, and that may lead to diabetes mellitus and metabolic syndrome. BioFactors 45:35–42
Wada J, Nakatsuka A (2016) Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med Okayama 70:151–158
Yoon Y, Galloway CA, Jhun BS, Yu T (2011) Mitochondrial dynamics in diabetes. Antioxid Redox Signal 14:439–457
Liu W, Yamashita T, Tian F, Morimoto N, Ikeda Y, Deguchi K, Abe K (2013) Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr Neurovasc Res 10:222–230
Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G, Smith MA, Zhu X (2008) Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res 5:525–532
Jezek P, Jaburek M, Plecita-Hlavata L (2019) Contribution of oxidative stress and impaired biogenesis of pancreatic beta-cells to type 2 diabetes. Antioxid Redox Signal 31:722–751
Flood JF, Mooradian AD, Morley JE (1990) Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes 39:1391–1398
Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ (2007) Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68:1501–1508
Brenner SR (2005) A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 64:1991
Coleman P, Federoff H, Kurlan R (2004) A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63:1155–1162
Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, Austad SN, Strong R, Richardson A, Hart MJ, Galvan V (2012) Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience 223:102–113
Acknowledgements
This work was supported by Chinese National Natural Science Foundation (81560241, 81960265), the Foundation for Science and Technology projects in Guizhou ([2017]1014, LH [2017]7156, [2018]1009, [2018]5752, [2020]1Y354), the Foundation for Science and Technology projects in Guiyang ([2019]9-2-7) and the Foundation of the Education Department of Guizhou Province (KY [2016]035).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ding, Y., Liu, H., Cen, M. et al. Rapamycin Ameliorates Cognitive Impairments and Alzheimer’s Disease-Like Pathology with Restoring Mitochondrial Abnormality in the Hippocampus of Streptozotocin-Induced Diabetic Mice. Neurochem Res 46, 265–275 (2021). https://doi.org/10.1007/s11064-020-03160-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03160-6