
Abstract
Neuroinflammation is a predisposing factor for the development of cognitive impairment and dementia. Among the new molecules that are currently being studied, ellagic acid (EA) has stood out for its neuroprotective properties. The present study investigated the effects of ellagic acid in the object recognition test, oxidative stress, cholinergic neurotransmission, glial cell expression, and phosphorylated Tau protein expression. For this, 32 male Wistar rats received an intraperitoneal (IP) application of lipopolysaccharides (LPS) at a dose of 250 µg/kg or 0.9% saline solution (SAL) for 8 days. Two hours after the IP injections, the animals received 100 mg/kg of EA or SAL via intragastric gavage. Behavioral parameters (open field test and object recognition) were performed on days 5, 6, and 7 of the experimental periods. The results showed that the treatment with EA in the LPS group was able to inhibit cognitive impairment, modulate the immune system response by significantly reducing glial cell expression, attenuating phosphorylated Tau and oxidative damage with consequent improvement in the antioxidant system, as well as preventing the increase of acetylcholinesterase activity. Thus, the neuroprotective effects of EA and its therapeutic potential in cognitive disorders secondary to neuroinflammation were demonstrated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuroinflammation is a characteristic of several neurological disorders, including Alzheimer's disease (AD), Parkinson's disease, multiple sclerosis, and acute traumatic brain injury [1,2,3,4]. Systemic administrations of lipopolysaccharides (LPS) have been described as experimental models that mimic the pathological disorders of these diseases, including AD-associated cholinergic neuronal degeneration. LPS can impair the consolidation of specific memory processes. Acute administration of LPS before training impairs the contextual fear conditioning test, a learning paradigm dependent on the hippocampus [5], while chronic LPS infusions affect spatial memory [6] and induce impairments in memory and learning analogous to cognitive impairment observed in AD [7]. In contrast, systemic administration of LPS results in damage to the hippocampus-dependent memory on object discrimination, but not on spatial memory [8].
Intraperitoneal (IP) injections of LPS cause cognitive impairment in laboratory animals through the activation of microglia, which stimulates the production of pro-inflammatory mediators. This mechanism is apparently due to the communication pathways between the immune system and the brain [9]. In response to the production of pro-inflammatory cytokines, several reactive oxygen species (ROS) are produced, which culminates in oxidative stress [10, 11]. Increased production of ROS promotes rapid changes in the antioxidant system, through the induction or depletion of cellular antioxidant reserves [12]. Also, excessive activation of the microglia perpetuates the inflammatory cycle [13], prolonging inflammation [14], which predisposes to the development of several neurodegenerative diseases [15], damage to the vascular endothelium, depletion of redox-glutathione, and mitochondrial respiratory dysfunction, which culminates in a reduction in the consumption of ATP and O2 [16].
The tau protein (Tau) is related to several physiological processes in neurons. When hyperphosphorylated, Tau monomers detach from microtubules and tend to aggregate into neurofibrillary tangles. This process is observed in several neurodegenerative disorders, called tauopathies [17]. The neurodegenerative process in these diseases is characterized by an amyloid cascade with consequent formation of amyloid plaques, Tau phosphorylation, neuroinflammation, and neuronal death. It is believed that the formation of amyloid oligomer (A) is the first step towards neurodegeneration, initiating the amyloid cascade [18]. In a brain inflammatory microenvironment, the production of cytokines by microglia and astrocytes can potentiate the amyloid cascade, which demonstrates the relationship between tauopathies and neuroinflammation [19, 20].
Drugs for improving cognition such as memantine, aniracetam, piracetam and cholinesterase inhibitors such as galantamine are used to improve memory, mood, and behavior, but their side effects limit the use of these agents. Thus, other possibilities, including plant derivatives, have been considered and evaluated as therapeutic alternatives [21]. There are several evidences to support the potential of antioxidants in the prevention and treatment of neurodegenerative diseases, such as Parkinson's disease and Alzheimer's disease. Furthermore, evidences in the literature confirms the ability of components with antioxidant properties to protect neurons against the harmful effects of ROS, preventing, or delaying the development of neurodegenerative diseases [22, 23]. Among these antioxidants, ellagic acid (EA) stands out, which is relatively stable under physiological conditions in the stomach and can be a potential phytotherapeutic candidate for the development of neuroprotective drugs that can be administered orally. This antioxidant has multiple pharmacological properties that are useful in the treatment and maintenance of disorders of the central nervous system. It can regulate several molecular signaling pathways, in order to normalize mitochondrial dysfunctions that result in the generation of free radicals and thus attenuate neurodegeneration [24]. The antioxidant action of EA occurs due to its direct property of free radicals scavenging and potentiating endogenous antioxidants [10]. EA can protect the brain from inflammation through down-regulation of the expression of several pro-inflammatory cytokines (such as TNF-α) [11]. The suppression of microglial responses represents the therapeutic effect of EA in AD. Also, in vivo and in vitro studies have shown a reduction in the release of inflammatory cytokines by microglia and amyloid plaques induced by EA [25].
Thus, the present study aimed to evaluate the action of EA in the cerebral cortex and hippocampus by recognizing memory and oxidative stress parameters such as ROS, lipid peroxidation, protein carbonylation, and T-SHs and GSH levels in an experimental model of neuroinflammation induced by multiple applications of LPS in rats. The study also aimed to investigate the effect of EA on acetylcholinesterase (AChE) activity and expression of neural and phosphorylated proteins in this experimental model.
Materials and Methods
Animals
This work was approved by the Ethics Committee on the Use of Animals of the Federal University of Santa Maria under number 5580160118. Thirty-two male Wistar rats with 6 to 7 weeks old (200–230 g), from the Central Bioterium of the Federal University of Santa Maria, were used. Animals in this age group have been chosen as they are more anxious and show more exploratory behavior than rats aged 16 weeks (300–320 g) commonly used in several experimental models [26].
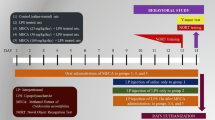
Four animals were housed per box with food and water available ad libitum. The rats were kept in an environment with controlled temperature and humidity (22–24 °C; 70% RH), light/dark cycle (7:00 a.m.–7:00 p.m.), and previously acclimated for 2 weeks. The animals were randomly divided into four groups, containing eight animals each: control (CTR + SAL), control treated with ellagic acid (CTRL + EA), lipopolysaccharide (LPS + SAL) and lipopolysaccharide treated with ellagic acid (LPS + EA). The animals in the LPS groups (LPS + SAL and LPS + EA) received, for eight consecutive days, a daily application (IP) of LPS at a dose of 250 µg/kg dissolved in 0.9% saline, while the control groups (CTRL + SAL and CTRL + EA) received only injections (IP) of 0.9% saline solution (SAL) in the same volume and period. One hour after the IP injections, the animals received via intragastric gavage (IG) EA at a dose of 100 mg/kg (CTRL + EA and LPS + EA) or 0.9% of saline in the same volume and route (CTRL + SAL and LPS + SAL). The animals were weighed daily to adjust the dose of the compounds to be used (Fig. 1).

Experimental protocol
Lipopolysaccharide
Systemic administration of LPS is a model widely used to induce neuroinflammation, as it results in increased levels of cerebral cytokines and activation of microglia [27, 28]. In this context, to induce the neuroinflammatory response, lipopolysaccharides from Escherichia coli (Sigma-Aldrich, O111-B4) diluted in saline and injected intraperitoneally at a dose of 250 μg/kg, once a day, for 8 days were used. This dose was selected according to previous studies [7, 29].
Ellagic Acid
Ellagic acid (Sigma-Aldrich) was used in doses of 100 mg/kg, orally, once daily, one hour after application of LPS. The treatment lasted 8 days. The EA was suspended in saline and administered via gavage. The suspension was homogenized in a sonicator before each administration to obtain a homogeneous solution. This treatment protocol is based on previous studies with this polyphenol [30,31,32,33,34,35,36,37,38].
Open Field Test
This test was performed to identify changes in the locomotor and exploratory capacity of the animals, as previously described by [39] and was performed on day 5 (Fig. 1). The apparatus consists of a wooden box covered with waterproof material with dimensions 70 × 70 × 30 cm. The floor was divided into 16 squares measuring 12 × 12 cm each to assess the open field. The session lasted five minutes and was recorded for further processing by an automated activity monitoring system (AnyMaze, Stoelting, USA) to assess the total distance covered; mobile or immobile time; time in the central zones, walls or corners; and number of entrances or exits in the central zones, walls or corners.
Object Recognition Test
The object recognition task was used to study recognition memory in rats [40]. The animals were submitted to training on day 6 (Fig. 1), where they were individually placed in the open field containing two similar objects (A1 and A2) being allowed to explore them freely for 5 min. For the evaluation of short-term memory 2 h after the training session the animals were individually reintroduced into the open field, where one of the objects presented during training was replaced by a new object with different size and shape (A1 and B). To assess long-term memory the same procedure was performed 24 h after the training session, replacing object B with a new object of different size and shape (object C). This task consists of the spontaneous and differential exploration of familiar and new objects, and the recognition performance is derived from the time spent exploring the two stimuli. Exploration of objects was considered by animal's snout directing at a distance ≤ 2 cm from the object and sniffing or touching the object with the snout. Climbing or sitting on objects was not classified as exploratory behavior. The results were expressed as preference index (percentage of time = new object/[new object + family object] × 100) ± SEM, which evaluates the percentage of time exploring the new object, and total exploration time (total time = new object) + familiar object) ± SEM.
Brain Tissue Preparation
At the end of the behavioral assessments, the animals were euthanized by overdose of isoflurane. After opening the skull, the brain was removed and separated into the cerebral cortex and hippocampus and homogenized in a solution of 10 mM Tris–HCl (pH 7.4), under ice, in a proportion of 1:10 (weight/volume). After centrifugation, the aliquots resulting from the homogenates of the brain structures were used to determine the parameters of oxidative stress and acetylcholinesterase activity.
The protein of brain structures was previously determined through a range varying for each structure: cerebral cortex (0.7 mg/mL) and hippocampus (0.8 mg/mL), as determined by the Coomassie blue method [41].
Determination of Acetylcholinesterase Activity in the Brain
The AChE enzymatic activity was determined by the Ellman et al. [42] method as modified by Rocha et al. [43]. This method is based on formation of the yellow 5-thio-2-nitrobenzoic acid, which was measured spectrophotometrically at 412 nm for 2 min at 25 °C. The reaction mixture contained 100 mM potassium phosphate buffer (pH 7.5), 1 mM 5,5′-dithiobis (2-nitrobenzoic acid) and the AChE enzyme (40–50 μg of protein), which was pre-incubated for 2 min. The reaction was initiated by adding 0.8 mM acetylthiocholine iodide (AcSCh). The experiment was carried out in triplicate, and enzyme activity was expressed as μmol AcSCh/h/mg of protein.
Measurement of Reactive Oxygen Species (ERO)
The 2′,7′-Dichlorofluorescein fluorescence assay was used to measure the production of hydrogen peroxide and other reactive species [44]. 50 mL aliquots of the brain structure homogenate supernatant were added to a medium containing Tris–HCl buffer (0.01 mM, pH 7.4) and DCFH DA 2′,7′-Dichlorofluorescein-diacetate (1 mM). After adding DCFH-DA, the medium was incubated in the dark for 1 h until fluorescence measurement (excitation at 488 nm and emission at 525 nm, with both slit widths at 1.5 nM). Dichloro-oxidized fluorescein was determined using an oxidized dichlorofluorescein standard curve, and the results are expressed as DCFH-DA Fluorescence.
Thiobarbituric Acid Reactive Substances (TBARS) Measurement
The levels of thiobarbituric acid reactive substances (TBARS) were determined according to Jentzsch et al. [45] by measuring the concentration of malondialdehyde (MDA) as a product of lipid peroxidation through reaction with thiobarbituric acid (TBA). Briefly, the reaction mixture containing 200 µL of supernatant from the brain structure or standard homogenate (0.03mMMDA), 1 mL of 0.2 M orthophosphoric acid, and 250 µL thiobarbituric (0.1 M) was heated to 95 °C for 120 min. Absorbance was measured at 532 nm. Serum TBARS levels are expressed in nmol MDA/mg protein.
Protein Carbonyl Levels
Protein carbonyl was determined by the method of Levine et al. [46] and modified by Reznick, Packer [47] and Liebel et al. [48]. A medium containing 2,4-dinitrophenylhydrazine (DNPH) 10 mmol and hydrochloric acid (HCl) was added to the protein precipitate and incubated at room temperature for one h. During the incubation, samples of the supernatant from the brain structure homogenate were mixed vigorously every 15 min. Then, 500 µL of denaturation buffer (3% sodium dodecyl sulfate (SDS) plus 2000 µL of ethanol and 2000 µL of heptane were added. Resuspended in 1000 µL of denaturation buffer and placed in the maria for about 20 min (40 or 50 °C) until the pellets are dissolved. The reading was performed at 370 nm on the UV–VIS spectrophotometer. The results are expressed as nmol/mg of protein.
Determination of Total Thiols (T-SH) and Reduced Glutathione (GSH)
The total number of thiol groups was analyzed spectrophotometrically using the method of Ellman [49] and Boyne, Ellman [50], with some modifications. A 200 µL aliquot of the brain structures homogenate supernatant in a final volume of 900 µL of the solution was used for the reaction. The reaction product was measured at 412 nm after adding 50 μL of 10 mM 5,5-dithiobis (2-nitrobenzoic acid) (DTNB). A standard curve using cysteine was added to calculate the content of thiol groups in samples, and it will be expressed as nmol of T-SH/mL of serum. GSH was measured spectrophotometrically with Ellman's reagent. An aliquot of 200 µL of serum in a final volume of 900 µL of the solution was used for the reaction. The reaction product was measured at 412 nm after adding 50 μL of 5,5-dithiobis (2-nitrobenzoic acid) (DTNB). A standard curve using cysteine was added to calculate the content of non-protein thiol groups in samples and expressed as nmol of GSH serum/mL.
Flow Cytometry Analysis of Neural Marker Proteins and Phosphorylated Proteins
Flow cytometry experiments for measurement of p-Tau and Iba-1 were performed as previously described [51]. Briefly, cells from hippocampus were fixed for 10 min by adding 4% PFA. Primary staining was performed with monoclonal antibodies against the phosphorylated Tau (1:200; Sigma-Aldrich), glial fibrillary acidic protein (GFAP) (1:500; Sigma-Aldrich) and ionized calcium binding adaptor molecule 1 (Iba-1) (1:200; Wako) for 30 min followed by addition of secondary Alexa-Fluor-488 antibodies (1:500; Life Technologies). The measurements were performed on a Calibur Cytometer (BD Biosciences) and analyzed with Flowjo V10 software (Flowjo, Ashland, OR). The results are expressed as percentage (%) of positive cells.
Statistical Analysis
All data were analyzed using two-way ANOVA followed by Tukey's post hoc test in a statistical program (GraphPad Prism 8). The data were expressed as mean ± SEM, and a statistically significant difference was considered p < 0.05.
Results
LPS Promotes a Reduction in Body Weight After the First Application
To assess the systemic effects of LPS or EA, the bodyweight of the rats was measured. We found a statistically significant difference in body weight during the experimental period by both groups (F (3, 224) = 24.81, p < 0.0001) and days (F (3, 224) = 19.87, p < 0.0001), though the interaction between these terms was not significant (F (21, 224) = 0,8424, p = 0,6655). A slight reduction in mean body weight was observed in the groups that received IP injection of LPS (LPS + SAL) on the second day of the experimental period, as shown by a Tukey’s test (Fig. 2), with a significant reduction in the bodyweight of the animals in the LPS + SAL group on days 3–5 when compared to the CTRL + SAL group. The animals in the present study showed a gradual increase in body weight during the experimental period. This fact was attributed to the growth phase of the animals.

Effect of multiple applications (IP) lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the weight of rats. The data were expressed as mean of the weights ± SEM. N = 8 animals/group. *Denotes significant difference of the LPS + SAL group compared to CTRL + SAL group. *p < 0.05
LPS and EA Did Not Alter Locomotor Activity
In this experiment, the effects of repeated applications of LPS were evaluated, as well as the treatment with EA on the locomotor activity of the rats in an open field test, since the memory test can be affected by locomotor changes. There were no significant differences between groups in the total distance travelled; mobile or immobile time; time in the central zones, walls or corners; and number of entrances or exits in the central zones, walls or corners (Table 1 and Fig. 3) indicating that the compounds did not promote changes in the animals' locomotor activity and, therefore, the results observed in the memory recognition test are not related to locomotor impairment.

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the locomotor activity of rats. The behavioral test was performed two hours after treatment (IG) with EA 100 mg/kg or saline, which occurred one hour after IP injection of LPS 250 µg/kg or saline. Data are expressed as mean ± SEM. N = 8 animals/group. There were no statistically significant differences (p < 0.05) between groups
Ellagic Acid Reverses Cognitive Impairment Induced by LPS
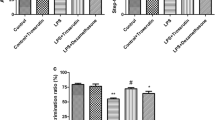
Two-way ANOVA revealed a significant influence of both time and groups (treated and not treated) on object recognition index (Table 2). All groups, except LPS + SAL, learned the localization of the object A1, as evidenced by the longer time spent exploring the new objects (Fig. 4).

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the short- and long-term memory of rats submitted to the memory recognition test. The results are expressed as % of the exploration time of the new object (percentage of time = new object/[new object + familiar object] × 100) ± SEM (a) and total exploration time of both objects (total time = new object + familiar object) ± SEM (b). N = 8 animals/group. Different symbols denote significant difference between groups. #: when compared to CTRL + SAL − O (A1), ω when compared to CTRL + EA − O (A1), σ when compared to LPS + EA − O (A1). ns p > 0.05, **p < 0.01, #p < 0.05, ω p < 0.05, σ p < 0.05
A significant reduction in the preference index of the new object was observed in 2 h (short term memory) and 24 h (long term memory) in the group that received multiple applications (IP) of LPS when compared to the control group. However, the group treated with 100 mg/kg of EA demonstrated a significant improvement in memory retention when compared to the LPS group in both short- and long-term memories, indicating that treatment with EA prevents cognitive impairments induced by LPS. Also, there was no significant difference between groups in the exploration time of both objects during the training phase, 2 h, and 24 h (Fig. 4).
EA Prevents LPS-Induced Increased AChE Activity
There was a significant influence of both control (SAL or LPS) and treatment (SAL or EA) and an interaction between these two terms on AChE activity (Table 3). Post-hoc Tuckey’s shown a significative increase (p < 0.05) in AChE activity in the CO and HP in the LPS group when compared to the control group. In contrast, treatment with EA in the LPS group (LPS + EA) was able to prevent an increase in the activity of this enzyme (Fig. 5).

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the activity of acetylcholinesterase (AChE) in the cerebral cortex (CO) and hippocampus (HP) of rats. Data are expressed as mean ± SEM. N = 8 animals/group. *Denotes significant difference between groups. *p < 0.05, **p < 0.01
EA Prevents LPS-Induced Increased Oxidative Damage
Two-way ANOVA analysis of oxidative parameters of CO and HP showed a significant effect of of both control (SAL or LPS) and treatment (SAL or EA) on the levels of ROS, TBARS and protein carbonyl (Tables 4 and 5). Also, there was a significant increase in ROS levels in the CO (Fig. 6a) and HP (Fig. 6b) in the LPS group compared to group control. As a consequence of the increased production of these reactive species, it was also possible to observe a significant increase in lipid peroxidation, demonstrated by the high levels of TBARS (Fig. 6c and d), and protein damage, evidenced by the elevation of the protein carbonyl in CO and HP (Fig. 6e and f). On the other hand, compared to the LPS group, the treatment with EA (LPS + EA) was able to inhibit the oxidative damage caused by ROS in CO and HP, as evidenced by Figs. 6a-f.

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the levels of reactive oxygen species (ROS), lipid peroxidation (TBARS) and protein carbonylation (carbonyl) in the cerebral cortex (CO) and hippocampus (HP) of rats. Data are expressed as mean ± SEM. N = 8 animals/group. *Denotes significant difference between groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
EA Prevents LPS-Induced Depletion of Total (T-SH) and Non-protein (GSH) Thiols
Since we observed a reduction in the production of ROS and related damages after treatment with EA in the group that received LPS (LPS + EA), we evaluated the levels of antioxidants to better understand the mechanisms involved in the neuroprotection performed by EA. Thus, a significant reduction in the levels of T-SH and GSH was observed in CO and HP in the LPS + SAL group when compared to the CTRL + SAL group. However, treatment with EA (LPS + EA) was able to prevent the reduction of T-SH and GSH in both brain structures when compared to the LPS + SAL group (Fig. 7). Also, it was observed influence of both control (SAL or LPS) and treatment (SAL or EA) on T-SH and GSH from cerebral cortex and hippocampus (Tables 4 and 5).

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the levels of total thiols (T-SH) and non-protein thiols (GSH) in the cerebral cortex (CO) and hippocampus (HP) of rats. Data are expressed as mean ± SEM. N = 8 animals/group. *Denotes significant difference between groups. *p < 0.05, **p < 0.01
EA Inhibits LPS-Induced Neuroinflammation
There was a significant effect of both control (SAL or LPS) and treatment (SAL or EA) and an interaction between them in the percentage of Iba-1+ and GFAP+ cells (Table 6). Post-hoc Tuckey’s shown significant increase (p < 0.05) was observed in the percentage of Iba-1+ and GFAP+ cells in the LPS + SAL group compared to the control group (CTRL + SAL) (Fig. 8). In contrast, the groups treated with EA (CTRL + EA and LPS + EA) had a low frequency of glial cells when compared to the LPS group (LPS + SAL), suggesting that this compound inhibits the neuroinflammatory process triggered by LPS.

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the expression of positive GFAP (a) and positive Iba-1 cells (b) in the hippocampus (HP) of rats. Data are expressed as mean ± SEM. N = 5 animals/group. *Denotes significant difference between groups. **p < 0.01, **p < 0.001
EA Suppresses LPS-Induced Phosphorylation of Tau Protein (P-Tau)
In the present study, two-way ANOVA revealed a significant influence of both control (SAL or LPS) (F (1, 16) = 14.31, p = 0.0016) and treatment (SAL or EA) (F (1, 16) = 22.93, p = 0.0002) in the percentage of P-Tau+ cells, though the interaction between these terms was not significant (F (1, 16) = 2.069, p = 0.1696). Post-hoc Tuckey’s shown a significant reduction in the percentage of P-Tau+ cells were observed in the groups treated with EA (CTRL + EA and LPS + EA), indicating a neuroprotective effect of this compound. Although there is no statistically significant difference between the control and untreated LPS groups (CTRL + SAL and LPS + SAL), there is an increase in the frequency of P-Tau+ in the LPS + SAL group (Fig. 9).

Effect of multiple applications (IP) of lipopolysaccharide (LPS) 250 µg/kg or saline (SAL) and treatment (IG) with ellagic acid (EA) 100 mg/kg or SAL on the expression of positive P-Tau cells in the hippocampus of rats. Data are expressed as mean ± SEM. N = 5 animals/group. *Denotes significant difference between groups. **p < 0.01
Discussion
This study aimed to demonstrate the effects of EA on LPS-induced neuroinflammation through memory-related assessments, such as object recognition test and AChE activity. The percentage of Iba-1+, GFAP+, and p-Tau+ cells was quantified to evaluate the neuroinflammatory effect, the redox profile was assessed by ROS generation, lipid peroxidation and protein carbonylation, as well as levels of non-enzymatic antioxidants. Also, the effects of multiple LPS applications on the animals' body weight and locomotor activity, assessed through the open field test, were evaluated. The results of this study demonstrated that EA was able to prevent cognitive impairment caused by multiple applications of LPS, as well as modulate the immune system response by significantly reducing the expression of glial cells, attenuating oxidative damage caused by the action of endotoxins.
The animals in the present study showed a reduction in body weight from the first application of LPS (LPS + SAL and LPS + EA), becoming significant (p < 0.05) on day 3 in the LPS + SAL group (Fig. 2). From the fourth day on, there was a gradual increase in the body weight of animals in the LPS groups (LPS + SAL and LPS + EA). Also, no statistically significant differences were observed in the open field test, performed on the 6th day of the experimental period. Corroborating with the results obtained by other authors [52], which performed an IP application of LPS (100 or 200 mg/kg) on days 1, 4, and 7 in female and male rats and evaluated locomotor activity, body weight, and hormone levels. The authors reported a reduction in locomotor activity and in the body weight of the animals after the first application of LPS. In contrast, there was a reduction in the deleterious behavioral effects of LPS after a second exposure to LPS in male and female rats, being more evident in females. After the third administration of LPS, no behavioral changes were observed. The authors attributed the findings to the mechanism of tolerance to LPS, which after multiple sublethal injections, results in less responsiveness to the compound and, consequently, higher survivability to the subsequent lethal dose of endotoxins. This low responsiveness has been called tolerance [53,54,55] and comprises an adaptation of the organism to limit excessive inflammation, trough less production of pro-inflammatory cytokines [55]. Consequently, there is a reduction in sickness behavior, since this mechanism is mediated mainly by the action of macrophages and cytokines on the periphery, as well as mechanisms of transduction of inflammation from the periphery to the brain [56]. Thus, it is suggested that the weight gain observed from the 3rd day of the experimental period is a consequence of the inhibition of sickness behavior, which possibly resulted in higher food and water intake by the groups the groups that received multiple applications of LPS (IP) (LPS + SAL and LPS + EA). The same can be attributed to the absence of changes in the locomotor activity of the animals, evidenced by the open field test (Fig. 3).
Although the effect of tolerance to multiple IP applications of LPS has been well described in the literature [57], several authors have reported cognitive impairment [58,59,60,61] and elevation in pro-inflammatory cytokines in the central nervous system. Chen et al. [62] demonstrated, after multiple applications of LPS, that the expression of cytokines in response to this endotoxin can be regulated in different ways between the peripheral immune system and the CNS. The increase in the production of pro-inflammatory cytokines is associated with an increase in the activation of microglia and astrocytes [63]. Usually, microglia cells act phagocyting dead cells and cellular debris to maintain CNS homeostasis, while astrocytes are responsible for preserving neurological function [64]. However, when stimulated in excess, microglia and astrocytes significantly increase neuroinflammation, resulting in pathogenesis by the secretion of several pro-inflammatory mediators [64,65,66].
In the present study, a significant increase in the percentage of positive glial cells (Iba-1+ and GFAP+) was observed in the LPS + SAL group (Fig. 8). These findings can be attributed to the action of LPS, a potent stimulator of microglia and astrocyte activation that can cause harmful neuroinflammatory responses through the production of TNF-α, IL-6, IL-1β, iNOS and COX-2 [67, 68]. In contrast, in the group treated with EA (LPS + EA), less expression of Iba-1+ and GFAP+ cells were observed. These results are in agreement with that described by other authors [25], who observed that the EA is able to inhibit microglial activation via attenuation of Nuclear factor of activated T-cells (NFAT) activity. Still, it is believed that polyphenols acts extracellularly by capturing cytokines to attenuate the stimulation of glial cells, thus exerting their anti-inflammatory function [69]. Thus, an anti-inflammatory effect of EA was observed, since this antioxidant reduced the expression of Iba-1+ and GFAP+ cells in the hippocampus of the LPS + EA group rats, which suggests that this compound can mitigate the deleterious effects observed in neurodegenerative disorders.
As previously described, the activation of microglia and astrocytes results in the cerebral release of cytokines. These pro-inflammatory interleukins directly affect neuronal function, such as long-term potentiation (LTP), glutamate release, AMPA receptor trafficking, and activation of cell-signaling pathways [70,71,72], which are related to synaptic plasticity and neurotransmission. Therefore, there may be impairment of neuronal processes related to cognition.
In the present study, the animals in the LPS + SAL group showed significantly lower performance in object recognition in the short- and long-term memory tests when compared to the other groups (Fig. 4). This cognitive impairment is due to the high density of receptors for cytokines in the hippocampus, particularly in the dentate gyrus [73], indicating that this structure may be particularly vulnerable during neuroinflammation [8]. Consequently, the administration of immunogenic stimuli, such as LPS, can compromise hippocampus-dependent memory and learning processes [74]. In contrast, there was a protective effect of EA in the short and long-term memory test, in which the LPS + EA group had a significantly higher performance than the LPS + SAL group. Several authors have reported the beneficial effects of EA on memory in models of cognitive impairment [30, 38, 75, 76], which occurs from the action of this antioxidant at the molecular level through the attenuation of oxidative stress, reduced AChE activity and modulation of the pathway of nuclear factor kappa B (NF-κB), nuclear factor erythroid 2–related factor 2 (Nfr2) and Toll-like receptor (TLR4) signaling, which are related to the neuroinflammation mechanism induced by LPS. This endotoxin binds to TLR4 on the surface of the microglia. It activates several transduction pathways, which result in the activation of NF-κB, which will mediate the production of pro-inflammatory cytokines, chemokines and inducible enzymes, such as inducible synthase oxide (iNOS) and COX-2, culminating in neuroinflammation [21, 77], as observed by the increased expression of positive glial cells (Iba-1+ and GFAP+) in the LPS + SAL group. These findings demonstrate the potential of EA to reverse cognitive impairments secondary to neuroinflammatory processes. This hypothesis is supported by the reduction in the expression of positive glial cells observed in the LPS + EA group observed in the present study and improved performance in the object recognition test compared to the untreated group (LPS + SAL).
Also, the cognitive impairment produced by systemic administration of LPS may be involved with the dysregulation of the cholinergic system, evidenced by the reduction in levels of acetylcholine (Ach), a neurotransmitter involved in the processes of memory and learning [78, 79]. Previous studies have shown that LPS causes depletion in brain ACh levels as a consequence of inducing AChE activity [12, 79, 80], which degrades ACh. Also, the expression of AChE increases in response to IL-1 [81] and oxidative stress [82, 83] induced by LPS. This pattern was observed in the present study, in which the animals that received LPS (LPS + SAL) showed a significant increase in AChE activity compared to the animals in the control group (CTRL + SAL) (Fig. 5). In contrast, the increased AChE activity was prevented in animals treated with EA (LPS + EA). It is believed that this prevention occurs through changes in the gene expression profile involved in the synthesis of AChE [84]. These results corroborate with previous studies [84, 85]. Thus, it is suggested that the improvement in cognitive performance may also be related to the reduced activity of AChE in the LPS + EA group compared to the LPS + SAL group since the reduction in the activity of this enzyme promotes an increase in the concentration of ACh. This hypothesis is supported by studies that have observed that AChE inhibition promotes learning and memory improvement in animals [84, 86].
Several authors have documented the relationship between oxidative stress and inflammation. Inflammation induces oxidative stress and DNA damage, which triggers an exacerbated production of ROS by microglia and macrophages. Damage from oxidative stress, such as oxidized proteins, glycated products, and lipid peroxidation, results in neuronal degeneration frequently reported in brain disorders [87]. Cells damaged by oxidative damage produce a large number of inflammatory mediators that promote the aging of the microglia [88]. In addition to the oxidative damage of ROS in macromolecules, these reactive species can also trigger inflammatory responses by stimulating several genes that regulate the inflammatory signaling cascade. Acute and chronic inflammation and aging processes are the primary triggers for excessive ROS production.
We observed significantly high levels of ROS, TBARS, and protein carbonylation (carbonyl) in the cerebral cortex and hippocampus in the LPS + SAL group compared to the CTRL + SAL group (Fig. 6). Studies have shown that LPS activates astrocytes and microglia that secrete gliotransmitters, such as glutamate and adenosine triphosphate (ATP), which play the role of substrate for the production of extracellular adenosine and neurotoxic molecules, such as free radicals [89, 90], which justifies the results found by our group, since there was an increase in the expression of positive glial cells in the LPS + SAL group as previously described. Furthermore, there was a depletion of the intracellular antioxidant system, demonstrated by the significant reduction in the levels of GSH and T-SH in the cerebral cortex and hippocampus of the LPS + SAL group compared to the CTRL + SAL group (Fig. 7). These results suggest exhaustion of the antioxidant system, due to the progression of the inflammatory reaction, which may contribute to the neurodegeneration process [91]. In contrast, the EA promoted a reduction in oxidative parameters (ROS, TBARS, and carbonyl) in the cerebral cortex and hippocampus (Fig. 6) through its antioxidant action, which occurs due to its direct property of free radical scavenging [10]. The hydroxyl group and the lactone ring present in the EA directly detoxify superoxide, hydroxyl radicals, hydrogen peroxide, and peroxynitrite [92]. Furthermore, this compound has a potentiation effect of endogenous antioxidants such as GSH, SOD, catalase, glutathione reductase and glutathione peroxidase [10], which can be evidenced by the significant increase in the levels of GSH and T-SH in the cerebral cortex and hippocampus in the LPS + EA group compared to the LPS + SAL group (Fig. 7). Herewith, we can relate the neuroprotective effects of EA to its anti-inflammatory potential by reducing the expression of positive glial cells and its antioxidant properties, as evidenced by the increase in the antioxidant system and consequent reduction in the generation of ROS and its by-products.
A recent study has shown that synaptic pathologies and microgliosis may be the initial manifestations of neurodegeneration related to tauopathies. Furthermore, the authors observed that the prominent activation of the microglia precedes the formation of neurofibrillary tangles, and the immunosuppression of the animals reduced the pathology related to Tau and increased the life expectancy of the animals. The causal relationship between Tau phosphorylation and neuronal dysfunction is not well established, but there are two main hypotheses: the loss of function may be caused by a reduction in the binding of Tau to microtubules (MT), resulting in destabilization of MT and transport disruption axonal; Hyperphosphorylated Tau results in aggregation and toxic effects on neuronal cells. Studies in transgenic mices have indicated that neuronal loss and impairment in memory are associated with the presence of soluble and highly phosphorylated Tau (oligomers), and suppression of its expression causes improved memory and increased number of synaptic connections [93,94,95]. Thus, it was concluded that neuroinflammation is related to the early progression of tauopathies.
In this context, in the present study, a significant reduction in the percentage of p-Tau+ cells were observed in the group LPS + EA when compared to the LPS + SAL group (Fig. 9). Zhong et al. [96] demonstrated that the potential of EA to inhibit hyperphosphorylation of Tau is related to the reduction in the activity of glycogen synthase kinase 3β (GSK3β), which is involved in the phosphorylation of Tau. However, the authors point out that several other kinases may be involved in this mechanism. These results demonstrate the potential of EA to reduce the deleterious effects caused by the hyperphosphorylation of Tau, which includes the formation of neurofibrillary tangles with consequent cognitive impairment.
The results of this study demonstrated that EA was able to prevent cognitive impairment caused by multiple applications of LPS, as well as, modulate the immune system response by significantly reducing the expression of glial cells and phosphorylated Tau, attenuating oxidative damage caused by the action of endotoxins and prevent the increase in AChE activity. Thus, this study demonstrated the beneficial effects of EA on memory, neuroinflammation, and restoring redox balance. These effects are the consequence of the anti-inflammatory and antioxidant action of this compound. With these results, the therapeutic potential of EA in cognitive disorders secondary to neuroinflammation was demonstrated.
References
Grigoriadis N, van Pesch V (2015) A basic overview of multiple sclerosis immunopathology. Eur J Neurol 22(Suppl 2):3–13. https://doi.org/10.1111/ene.12798
Latta CH, Brothers HM, Wilcock DM (2015) Neuroinflammation in Alzheimer's disease; a source of heterogeneity and target for personalized therapy. Neuroscience 302:103–111. https://doi.org/10.1016/j.neuroscience.2014.09.061
Rocha NP, de Miranda AS, Teixeira AL (2015) Insights into neuroinflammation in Parkinson's disease: from biomarkers to anti-inflammatory based therapies. Biomed Res Int 2015:628192. https://doi.org/10.1155/2015/628192
Bergold PJ (2016) Treatment of traumatic brain injury with anti-inflammatory drugs. Exp Neurol 275(Pt 3):367–380. https://doi.org/10.1016/j.expneurol.2015.05.024
Pugh CR, Kumagawa K, Fleshner M, Watkins LR, Maier SF, Rudy JW (1998) Selective effects of peripheral lipopolysaccharide administration on contextual and auditory-cue fear conditioning. Brain Behav Immun 12(3):212–229. https://doi.org/10.1006/brbi.1998.0524
Hauss-Wegrzyniak B, Vannucchi MG, Wenk GL (2000) Behavioral and ultrastructural changes induced by chronic neuroinflammation in young rats. Brain Res 859(1):157–166. https://doi.org/10.1016/s0006-8993(00)01999-5
Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT (2008) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflamm 5:37. https://doi.org/10.1186/1742-2094-5-37
Czerniawski J, Miyashita T, Lewandowski G, Guzowski JF (2015) Systemic lipopolysaccharide administration impairs retrieval of context-object discrimination, but not spatial, memory: evidence for selective disruption of specific hippocampus-dependent memory functions during acute neuroinflammation. Brain Behav Immun 44:159–166. https://doi.org/10.1016/j.bbi.2014.09.014
DeLegge MH, Smoke A (2008) Neurodegeneration and inflammation. Nutr Clin Pract 23(1):35–41. https://doi.org/10.1177/011542650802300135
Cozzi R, Ricordy R, Bartolini F, Ramadori L, Perticone P, De Salvia R (1995) Taurine and ellagic acid: two differently-acting natural antioxidants. Environ Mol Mutagen 26(3):248–254. https://doi.org/10.1002/em.2850260310
Mashhadizadeh S, Farbood Y, Dianat M, Khodadadi A, Sarkaki A (2017) Therapeutic effects of ellagic acid on memory, hippocampus electrophysiology deficits, and elevated TNF-α level in brain due to experimental traumatic brain injury. Iran J Basic Med Sci 20(4):399–407. https://doi.org/10.22038/IJBMS.2017.8581
Tyagi E, Agrawal R, Nath C, Shukla R (2008) Influence of LPS-induced neuroinflammation on acetylcholinesterase activity in rat brain. J Neuroimmunol 205(1–2):51–56. https://doi.org/10.1016/j.jneuroim.2008.08.015
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson's disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208(1):1–25. https://doi.org/10.1016/j.expneurol.2007.07.004
Schmid CD, Melchior B, Masek K, Puntambekar SS, Danielson PE, Lo DD, Sutcliffe JG, Carson MJ (2009) Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo. J Neurochem 109(Suppl 1):117–125. https://doi.org/10.1111/j.1471-4159.2009.05984.x
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76(2):77–98. https://doi.org/10.1016/j.pneurobio.2005.06.004
Sugino K, Dohi K, Yamada K, Kawasaki T (1987) The role of lipid peroxidation in endotoxin-induced hepatic damage and the protective effect of antioxidants. Surgery 101(6):746–752
Luppi M, Hitrec T, Di Cristoforo A, Squarcio F, Stanzani A, Occhinegro A, Chiavetta P, Tupone D, Zamboni G, Amici R, Cerri M (2019) Phosphorylation and dephosphorylation of Tau protein during synthetic torpor. Front Neuroanat 13:57. https://doi.org/10.3389/fnana.2019.00057
Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256(5054):184–185. https://doi.org/10.1126/science.1566067
Dzamba D, Harantova L, Butenko O, Anderova M (2016) Glial cells—the key elements of Alzheimer s disease. Curr Alzheimer Res 13(8):894–911. https://doi.org/10.2174/1567205013666160129095924
Acosta C, Anderson HD, Anderson CM (2017) Astrocyte dysfunction in Alzheimer disease. J Neurosci Res 95(12):2430–2447. https://doi.org/10.1002/jnr.24075
Park SE, Sapkota K, Kim S, Kim H, Kim SJ (2011) Kaempferol acts through mitogen-activated protein kinases and protein kinase B/AKT to elicit protection in a model of neuroinflammation in BV2 microglial cells. Br J Pharmacol 164(3):1008–1025. https://doi.org/10.1111/j.1476-5381.2011.01389.x
Gilgun-Sherki Y, Melamed E, Offen D (2003) Antioxidant treatment in Alzheimer's disease: current state. J Mol Neurosci: MN 21(1):1–11. https://doi.org/10.1385/jmn:21:1:1
Kelsey NA, Wilkins HM, Linseman DA (2010) Nutraceutical antioxidants as novel neuroprotective agents. Molecules 15(11):7792–7814. https://doi.org/10.3390/molecules15117792
Ahmed T, Setzer WN, Nabavi SF, Orhan IE, Braidy N, Sobarzo-Sanchez E, Nabavi SM (2016) Insights into effects of ellagic acid on the nervous system: a mini review. Curr Pharm Des 22(10):1350–1360. https://doi.org/10.2174/1381612822666160125114503
Rojanathammanee L, Puig KL, Combs CK (2013) Pomegranate polyphenols and extract inhibit nuclear factor of activated T-cell activity and microglial activation in vitro and in a transgenic mouse model of Alzheimer disease. J Nutr 143(5):597–605. https://doi.org/10.3945/jn.112.169516
Ray J, Hansen S (2005) Temperamental development in the rat: the first year. Dev Psychobiol 47(2):136–144. https://doi.org/10.1002/dev.20080
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55(5):453–462. https://doi.org/10.1002/glia.20467
Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP (2008) Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation 5:15–15. https://doi.org/10.1186/1742-2094-5-15
Zhu B, Wang ZG, Ding J, Liu N, Wang DM, Ding LC, Yang C (2014) Chronic lipopolysaccharide exposure induces cognitive dysfunction without affecting BDNF expression in the rat hippocampus. Exp Ther Med 7(3):750–754. https://doi.org/10.3892/etm.2014.1479
Farbood Y, Sarkaki A, Dianat M, Khodadadi A, Haddad MK, Mashhadizadeh S (2015) Ellagic acid prevents cognitive and hippocampal long-term potentiation deficits and brain inflammation in rat with traumatic brain injury. Life Sci 124:120–127. https://doi.org/10.1016/j.lfs.2015.01.013
Jagadeesan G, Bharathi E (2014) In vivo restoration of hepatic and nephro protective potential of hesperidin and ellagic acid against mercuric chloride intoxicated rats. Biomed Aging Pathol 4(3):219–222. https://doi.org/10.1016/j.biomag.2014.01.008
Bharathi E, Jagadeesan G (2014) Antioxidant potential of hesperidin and ellagic acid on renal toxicity induced by mercuric chloride in rats. Biomed Prev Nutr 4(2):131–136. https://doi.org/10.1016/j.bionut.2013.12.007
Bharathi E, Jagadeesan G, Vijayakumar M (2014) Hepato-ameliorative effect of hesperidin and ellagic acid on mercuric chloride intoxicated rats. Biomed Aging Pathol 4(1):17–21. https://doi.org/10.1016/j.biomag.2013.10.002
Hassaan Y, Handoussa H, El-Khatib AH, Linscheid MW, El Sayed N, Ayoub N (2014) Evaluation of plant phenolic metabolites as a source of Alzheimer's drug leads. Biomed Res Int 2014:843263. https://doi.org/10.1155/2014/843263
Ueda H, Kawanishi K, Moriyasu M (2004) Effects of ellagic acid and 2-(2,3,6-trihydroxy-4-carboxyphenyl)ellagic acid on sorbitol accumulation in vitro and in vivo. Biol Pharm Bull 27(10):1584–1587. https://doi.org/10.1248/bpb.27.1584
Uzar E, Alp H, Cevik MU, Firat U, Evliyaoglu O, Tufek A, Altun Y (2012) Ellagic acid attenuates oxidative stress on brain and sciatic nerve and improves histopathology of brain in streptozotocin-induced diabetic rats. Neurol Sci 33(3):567–574. https://doi.org/10.1007/s10072-011-0775-1
Guada M, Ganugula R, Vadhanam M, Ravi Kumar MNV (2017) Urolithin A mitigates cisplatin-induced nephrotoxicity by inhibiting renal inflammation and apoptosis in an experimental rat model. J Pharmacol Exp Ther 363(1):58–65. https://doi.org/10.1124/jpet.117.242420
Mashhadizadeh S, Farbood Y, Dianat M, Khodadadi A, Sarkaki A (2017) Therapeutic effects of ellagic acid on memory, hippocampus electrophysiology deficits, and elevated TNF-alpha level in brain due to experimental traumatic brain injury. Iran J Basic Med Sci 20(4):399–407. https://doi.org/10.22038/IJBMS.2017.8581
Zanin M, Takahashi RN (1994) Sex difference in sensitization to the locomotor effects of mazindol in rats. Brain Res Bull 34(4):385–387. https://doi.org/10.1016/0361-9230(94)90034-5
Lueptow L (2017) Novel object recognition test for the investigation of learning and memory in mice. J Vis Exp 126:e55718. https://doi.org/10.3791/55718
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1–2):248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Rocha JB, Emanuelli T, Pereira ME (1993) Effects of early undernutrition on kinetic parameters of brain acetylcholinesterase from adult rats. Acta Neurobiol Exp 53(3):431–437
Myhre O, Andersen JM, Aarnes H, Fonnum F (2003) Evaluation of the probes 2′,7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem Pharmacol 65(10):1575–1582. https://doi.org/10.1016/S0006-2952(03)00083-2
Jentzsch AM, Bachmann H, Fürst P, Biesalski HK (1996) Improved analysis of malondialdehyde in human body fluids. Free Radical Biol Med 20(2):251–256. https://doi.org/10.1016/0891-5849(95)02043-8
Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz A-G, Ahn B-W, Shaltiel S, Stadtman ER (1990) [49] Determination of carbonyl content in oxidatively modified proteins. In: Methods in enzymology, vol 186. Academic Press, New York, pp 464–478. https://doi.org/10.1016/0076-6879(90)86141-H
Reznick AZ, Packer L (1994) [38] Oxidative damage to proteins: spectrophotometric method for carbonyl assay. In: Methods in enzymology, vol 233. Academic Press, New York, pp 357–363. https://doi.org/10.1016/S0076-6879(94)33041-7
Liebel S, Oliveira Ribeiro CA, Silva RC, Ramsdorf WA, Cestari MM, Magalhães VF, Garcia JRE, Esquivel BM, Filipak Neto F (2011) Cellular responses of Prochilodus lineatus hepatocytes after cylindrospermopsin exposure. Toxicol In Vitro 25(7):1493–1500. https://doi.org/10.1016/j.tiv.2011.05.010
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82(1):70–77. https://doi.org/10.1016/0003-9861(59)90090-6
Boyne AF, Ellman GL (1972) A methodology for analysis of tissue sulfhydryl components. Anal Biochem 46(2):639–653. https://doi.org/10.1016/0003-2697(72)90335-1
Pillat MM, Lameu C, Trujillo CA, Glaser T, Cappellari AR, Negraes PD, Battastini AM, Schwindt TT, Muotri AR, Ulrich H (2016) Bradykinin promotes neuron-generating division of neural progenitor cells through ERK activation. J Cell Sci 129(18):3437–3448. https://doi.org/10.1242/jcs.192534
Engeland CG, Kavaliers M, Ossenkopp KP (2003) Sex differences in the effects of muramyl dipeptide and lipopolysaccharide on locomotor activity and the development of behavioral tolerance in rats. Pharmacol Biochem Behav 74(2):433–447. https://doi.org/10.1016/s0091-3057(02)01024-9
Cross AS (2002) Endotoxin tolerance-current concepts in historical perspective. J Endotoxin Res 8(2):83–98. https://doi.org/10.1179/096805102125000227
West MA, Heagy W (2002) Endotoxin tolerance: a review. Crit Care Med 30(1):S64–S73
Liu Y, Xie X, Xia L-P, Lv H, Lou F, Ren Y, He Z-Y, Luo X-G (2017) Peripheral immune tolerance alleviates the intracranial lipopolysaccharide injection-induced neuroinflammation and protects the dopaminergic neurons from neuroinflammation-related neurotoxicity. J Neuroinflamm 14(1):223–223. https://doi.org/10.1186/s12974-017-0994-3
Clark SM, Michael KC, Klaus J, Mert A, Romano-Verthelyi A, Sand J, Tonelli LH (2015) Dissociation between sickness behavior and emotionality during lipopolysaccharide challenge in lymphocyte deficient Rag2(-/-) mice. Behav Brain Res 278:74–82. https://doi.org/10.1016/j.bbr.2014.09.030
Seeley JJ, Ghosh S (2017) Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol 101(1):107–119. https://doi.org/10.1189/jlb.3MR0316-118RR
Wang F, Zhang ZZ, Cao L, Yang QG, Lu QF, Chen GH (2020) Lipopolysaccharide exposure during late embryogenesis triggers and drives Alzheimer-like behavioral and neuropathological changes in CD-1 mice. Brain Behav 10:e01546. https://doi.org/10.1002/brb3.1546
Ji MH, Zhang L, Mao MJ, Zhang H, Yang JJ, Qiu LL (2020) Overinhibition mediated by parvalbumin interneurons might contribute to depression-like behavior and working memory impairment induced by lipopolysaccharide challenge. Behav Brain Res 383:112509. https://doi.org/10.1016/j.bbr.2020.112509
Lee B, Yeom M, Shim I, Lee H, Hahm DH (2020) Inhibitory effect of carvacrol on lipopolysaccharide-induced memory impairment in rats. Korean J Physiol Pharmacol 24(1):27–37. https://doi.org/10.4196/kjpp.2020.24.1.27
Khan MS, Muhammad T, Ikram M, Kim MO (2019) Dietary supplementation of the antioxidant curcumin halts systemic LPS-induced neuroinflammation-associated neurodegeneration and memory/synaptic impairment via the JNK/NF-kappaB/Akt signaling pathway in adult rats. Oxid Med Cell Longev 2019:7860650. https://doi.org/10.1155/2019/7860650
Chen R, Zhou H, Beltran J, Malellari L, Chang SL (2005) Differential expression of cytokines in the brain and serum during endotoxin tolerance. J Neuroimmunol 163(1):53–72. https://doi.org/10.1016/j.jneuroim.2005.02.012
Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflamm 12:114. https://doi.org/10.1186/s12974-015-0332-6
Almad A, Maragakis NJ (2018) A stocked toolbox for understanding the role of astrocytes in disease. Nat Rev Neurol 14(6):351–362. https://doi.org/10.1038/s41582-018-0010-2
Bauer J, Rauschka H, Lassmann H (2001) Inflammation in the nervous system: The human perspective. Glia 36(2):235–243. https://doi.org/10.1002/glia.1112
Yanguas-Casas N, Barreda-Manso MA, Nieto-Sampedro M, Romero-Ramirez L (2014) Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J Neuroinflamm 11:50. https://doi.org/10.1186/1742-2094-11-50
Long-Smith CM, Sullivan AM, Nolan YM (2009) The influence of microglia on the pathogenesis of Parkinson's disease. Prog Neurobiol 89(3):277–287. https://doi.org/10.1016/j.pneurobio.2009.08.001
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7(4):354–365. https://doi.org/10.1016/j.nurt.2010.05.014
Hollebeeck S, Winand J, Herent MF, During A, Leclercq J, Larondelle Y, Schneider YJ (2012) Anti-inflammatory effects of pomegranate (Punica granatum L.) husk ellagitannins in Caco-2 cells, an in vitro model of human intestine. Food Funct 3(8):875–885. https://doi.org/10.1039/c2fo10258g
Vereker E, Campbell V, Roche E, McEntee E, Lynch MA (2000) Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem 275(34):26252–26258. https://doi.org/10.1074/jbc.M002226200
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295(5563):2282–2285. https://doi.org/10.1126/science.1067859
Lynch AM, Walsh C, Delaney A, Nolan Y, Campbell VA, Lynch MA (2004) Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10–a role for IL-1 beta? J Neurochem 88(3):635–646. https://doi.org/10.1046/j.1471-4159.2003.02157.x
Schobitz B, Voorhuis DA, De Kloet ER (1992) Localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Neurosci Lett 136(2):189–192. https://doi.org/10.1016/0304-3940(92)90046-a
Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF (2002) Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res 134(1):291–298. https://doi.org/10.1016/S0166-4328(02)00043-8
Dolatshahi M, Farbood Y, Sarkaki A, Mansouri SM, Khodadadi A (2015) Ellagic acid improves hyperalgesia and cognitive deficiency in 6-hydroxidopamine induced rat model of Parkinson's disease. Iran J Basic Med Sci 18(1):38–46
Mansouri MT, Farbood Y, Naghizadeh B, Shabani S, Mirshekar MA, Sarkaki A (2016) Beneficial effects of ellagic acid against animal models of scopolamine- and diazepam-induced cognitive impairments. Pharm Biol 54(10):1947–1953. https://doi.org/10.3109/13880209.2015.1137601
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6):918–934. https://doi.org/10.1016/j.cell.2010.02.016
Houdek HM, Larson J, Watt JA, Rosenberger TA (2014) Bacterial lipopolysaccharide induces a dose-dependent activation of neuroglia and loss of basal forebrain cholinergic cells in the rat brain. Inflamm Cell Signal. https://doi.org/10.14800/ics.47
Ming Z, Wotton CA, Appleton RT, Ching JC, Loewen ME, Sawicki G, Bekar LK (2015) Systemic lipopolysaccharide-mediated alteration of cortical neuromodulation involves increases in monoamine oxidase-A and acetylcholinesterase activity. J Neuroinflamm 12:37. https://doi.org/10.1186/s12974-015-0259-y
Eduviere AT, Umukoro S, Adeoluwa OA, Omogbiya IA, Aluko OM (2016) Possible mechanisms involved in attenuation of lipopolysaccharide-induced memory deficits by methyl jasmonate in mice. Neurochem Res 41(12):3239–3249. https://doi.org/10.1007/s11064-016-2050-6
Li Y, Liu L, Kang J, Sheng JG, Barger SW, Mrak RE, Griffin WS (2000) Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci 20(1):149–155
Bond CE, Patel P, Crouch L, Tetlow N, Day T, Abu-Hayyeh S, Williamson C, Greenfield SA (2006) Astroglia up-regulate transcription and secretion of 'readthrough' acetylcholinesterase following oxidative stress. Eur J Neurosci 24(2):381–386. https://doi.org/10.1111/j.1460-9568.2006.04898.x
Bond CE, Greenfield SA (2007) Multiple cascade effects of oxidative stress on astroglia. Glia 55(13):1348–1361. https://doi.org/10.1002/glia.20547
Jha AB, Panchal SS, Shah A (2018) Ellagic acid: insights into its neuroprotective and cognitive enhancement effects in sporadic Alzheimer's disease. Pharmacol Biochem Behav 175:33–46. https://doi.org/10.1016/j.pbb.2018.08.007
Kiasalari Z, Heydarifard R, Khalili M, Afshin-Majd S, Baluchnejadmojarad T, Zahedi E, Sanaierad A, Roghani M (2017) Ellagic acid ameliorates learning and memory deficits in a rat model of Alzheimer's disease: an exploration of underlying mechanisms. Psychopharmacology 234(12):1841–1852. https://doi.org/10.1007/s00213-017-4589-6
Pepeu G, Giovannini MG (2010) Cholinesterase inhibitors and memory. Chem Biol Interact 187(1–3):403–408. https://doi.org/10.1016/j.cbi.2009.11.018
Popa-Wagner A, Mitran S, Sivanesan S, Chang E, Buga AM (2013) ROS and brain diseases: the good, the bad, and the ugly. Oxid Med Cell Longev 2013:963520. https://doi.org/10.1155/2013/963520
Wu Z, Yu J, Zhu A, Nakanishi H (2016) Nutrients, microglia aging, and brain aging. Oxid Med Cell Longev 2016:7498528. https://doi.org/10.1155/2016/7498528
Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B (2002) Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem 81(6):1285–1297. https://doi.org/10.1046/j.1471-4159.2002.00928.x
Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS (2004) NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279(2):1415–1421. https://doi.org/10.1074/jbc.M307657200
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97(6):1634–1658. https://doi.org/10.1111/j.1471-4159.2006.03907.x
Garcia-Nino WR, Zazueta C (2015) Ellagic acid: pharmacological activities and molecular mechanisms involved in liver protection. Pharmacol Res 97:84–103. https://doi.org/10.1016/j.phrs.2015.04.008
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu G-Q, Palop JJ, Noebels JL, Mucke L (2011) Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci 31(2):700–711. https://doi.org/10.1523/JNEUROSCI.4152-10.2011
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733):476–481. https://doi.org/10.1126/science.1113694
Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM (2011) Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci 31(7):2511–2525. https://doi.org/10.1523/jneurosci.5245-10.2011
Zhong L, Liu H, Zhang W, Liu X, Jiang B, Fei H, Sun Z (2018) Ellagic acid ameliorates learning and memory impairment in APP/PS1 transgenic mice via inhibition of β-amyloid production and tau hyperphosphorylation. Exp Ther Med 16(6):4951–4958. https://doi.org/10.3892/etm.2018.6860
Funding
The authors acknowledge the financial support given by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dornelles, G.L., de Oliveira, J.S., de Almeida, E.J.R. et al. Ellagic Acid Inhibits Neuroinflammation and Cognitive Impairment Induced by Lipopolysaccharides. Neurochem Res 45, 2456–2473 (2020). https://doi.org/10.1007/s11064-020-03105-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03105-z