
Abstract
Methylglyoxal (MGO) is a highly reactive dicarbonyl molecule that promotes the formation of advanced glycation end products (AGEs), which are believed to play a key role in a number of pathologies, such as diabetes, Alzheimer’s disease, and inflammation. Here, Swiss mice were treated with MGO by intraperitoneal injection to investigate its effects on motor activity, mood, and cognition. Acute MGO treatment heavily decreased locomotor activity in the open field test at higher doses (80–200 mg/kg), an effect not observed at lower doses (10–50 mg/kg). Several alterations were observed 4 h after a single MGO injection (10–50 mg/kg): (a) plasma MGO levels were increased, (b) memory was impaired (object location task), (c) anxiolytic behavior was observed in the open field and marble burying test, and (d) depressive-like behavior was evidenced as evaluated by the tail suspension test. Biochemical alterations in the glutathione and glyoxalase systems were not observed 4 h after MGO treatment. Mice were also treated daily with MGO at 0, 10, 25 and 50 mg/kg for 11 days. From the 5th to the 11th day, several behavioral end points were evaluated, resulting in: (a) absence of motor impairment as evaluated in the open field, horizontal bars and pole test, (b) depressive-like behavior observed in the tail suspension test, and (c) cognitive impairments detected on working, short- and long-term memory when mice were tested in the Y-maze spontaneous alternation, object location and recognition tests, and step-down inhibitory avoidance task. An interesting finding was a marked decrease in dopamine levels in the prefrontal cortex of mice treated with 50 mg/kg MGO for 11 days, along with a ~ 25% decrease in the Glo1 content. The MGO-induced dopamine depletion in the prefrontal cortex may be related to the observed memory deficits and depressive-like behavior, an interesting topic to be further studied as a potentially novel route for MGO toxicity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Psychiatric disorders and cognitive dysfunction are complex and often comorbid, as well as related to metabolic disturbances [1, 2]. In line with this, methylglyoxal (MGO), a reactive dicarbonyl generated endogenously, is capable of inducing oxidative stress, cellular damage, apoptosis and has been associated with neuronal dysfunction [3,4,5,6]. MGO can be generated by spontaneous fragmentation and elimination of phosphate from two metabolites of the glycolytic pathway, namely glyceraldehyde-3-phosphate, and dihydroxyacetone phosphate [7, 8]. MGO can also be generated by the metabolism of glycine, tyrosine, and threonine [8,9,10]; during lipid peroxidation [11]; and by enzymatic isomerization or protein catabolism [12,13,14].
The glyoxalase system, comprised by the enzymes glyoxalase 1 (Glo1) and glyoxalase 2 (Glo2), is highly specific in detoxifying MGO [15, 16]. Initially, hemithioacetal is generated after the spontaneous reaction between MGO and glutathione (GSH). Then, hemithioacetal is converted to S-d-lactoylglutathione in a reaction catalyzed by Glo1. In the second step, Glo2 catalyzes the conversion of S-d-lactoylglutathione to d-lactate, regenerating GSH [9, 16, 17]. The absence of glyoxalase system would favor MGO to react with amino groups of basic amino acids and, by this, increasing the burden of the so-called advanced glycation end-products (AGEs) [7, 18]. AGEs, in turn, can activate the AGE receptor (RAGE) leading to the production of reactive oxygen species (ROS) and pro-inflammatory cytokines, what can promote a subset of physiological alterations contributing to illness appearance [19]. Altered MGO metabolism has been linked to diabetes [20, 21], mood disorders such as anxiety and depression [22], epilepsy [23], cancer [24], hyperalgesia, inflammation [25], and Alzheimer’s disease [26].
Modulation of Glo1 expression or MGO levels have been correlated with behavioral alterations in pre-clinical studies, including modulation of anxiety, seizure, pain and depressive-like behavior in mice [22, 27,28,29]. A possible mechanism underlying behavioral effects correlated to Glo1 levels was proposed after the identification of MGO as a competitive partial agonist on γ-aminobutyric acid A (GABAA) receptor [30]. MGO has also been shown to modulate the activity of Nav1.8 voltage-gated sodium channel, inducing hyperalgesia in diabetic neuropathy [31], and MGO-activated TRPA1 ion channel can evoke pain [32].
The levels of MGO were found to be significantly elevated in the plasma of diabetic patients and rats [16, 33, 34]. Also, a study evaluating 267 non-demented elderly humans showed that the serum concentration of MGO was positively associated with a faster rate of cognitive decline [35]. Moreover, higher serum MGO levels in older people were associated with poorer memory and executive function and grey matter atrophy, without alterations in white matter or hippocampal volume [36].
Although recent studies have investigated the role of MGO, so far, only a few animal studies evaluated the effects of MGO on learning and memory. Either cognitive impairment or absence of alterations has been shown, depending on the approach. It was observed that streptozotocin-induced diabetic rats presented increased serum levels of MGO and spatial memory impairment [33]. On the other hand, prolonged treatment with 0.5% MGO in drinking water led to increased serum MGO in Sprague–Dawley rats, but the treatment did not cause any significant cognitive impairment on spatial and working memory [37]. Another study showed that 1% MGO in drinking water for 4 weeks impaired aversive memory in mice [38]. Also, repeated intracerebroventricular (i.c.v.) MGO injections (3 µmol/µL/day) for 6 days revealed deficits in short-term recognition memory, but not working memory, when evaluated in the Y-maze [39]. This set of evidences is inconclusive and, as previously stated, the significance of MGO-induced cognitive impairment still requires further investigation [37].
Considering this scenario, Swiss mice were subjected to acute or repeated MGO treatment (5–11 days, 10–200 mg/kg), and a set of behavioral tests were undertaken: locomotor activity, vertical agility, force, motor coordination, depressive-like behavior, anxiety, and cognitive parameters to evaluate spatial, recognition, aversive and working memory, in the short- and long-term paradigms. We also assessed plasma MGO levels, antioxidant defenses and Glo1/Glo2 abundance in the prefrontal cortex and hippocampus of mice. In addition, we evaluated levels of dopamine (DA), norepinephrine (NE) and serotonin (5-HT) in the brain.
Materials and Methods
Animals and Treatments
Experiments were conducted using 3-month-old female Swiss mice (35–55 g) bred at the Federal University of Santa Catarina (UFSC), Florianópolis, Brazil. Mice were maintained in groups of 10–12 animals per cage (42 × 34 × 17 cm), under controlled temperature (22 ± 1 °C), and 12 h light cycle (lights on at 7:00 AM), with free access to food (standard chow diet) and water. Efforts were made to minimize the number of animals used and their suffering.
Experiments were performed by using female mice given the animal facility provides this specific strain, and because several studies have shown that the prevalence of stress-related psychiatric disorders (e.g., depression, post-traumatic stress disorder) are twice as prevalent in women compared to men [40]. This higher prevalence of females in depression and other stress-related disorders implies an increased sensitivity of stress-related systems or substrates, as evidenced in previous studies that documented an increased responsiveness of the hypothalamic–pituitary–adrenal axis in female vs male rats [41,42,43,44].
Mice were treated with saline 0.9% (vehicle/control) or different doses of MGO (10, 20, 25, 50, 80 or 200 mg/kg) (Sigma-Aldrich, São Paulo) diluted in vehicle. Solutions were administered once a day by intraperitoneal injection (i.p.) with a relative injection volume of 1 mL/100 g of body weight. Treatments were made once (acute treatment) or daily (repeated treatment).
Experimental Design
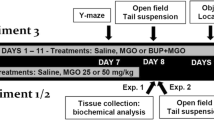
To evaluate the behavioral and neurochemical effects of MGO treatment, two administration approaches were used with independent groups of mice (Fig. 1):

Schematic representation of the experimental design. a Fifteen minutes after a single injection with MGO mice were tested in the tail suspension test followed by the open-field test. Four hours after a single injection with MGO, different groups of mice were tested in the open field and tail suspension test, or marble burying test and object location task. b Mice were subjected to repeated treatment, receiving a single daily injection of MGO (0, 10, 25 and 50 mg/kg). Twenty-four hours after the last injection mice were evaluated in the open field, object location and recognition tasks, Y-maze spontaneous alternation, triple horizontal bars, step-down inhibitory avoidance task, and pole test. Blood and brain samples were used for biochemical analyses after MGO treatment
(A) Acute administration (Fig. 1a): Based on previous studies that showed 800 mg/kg MGO was lethal [4, 45], we initially used up to 200 mg/kg MGO to investigate the lowest effective dose causing motor impairment in the open field test. MGO at 80 and 200 mg/kg (i.p.) produced a marked decrease in the locomotor activity in the open field, and were not further tested (Fig. S1). Mice receiving a single MGO at 10, 25 and 50 mg/kg injection did not present any noticeable change in the ambulatory activity 4 h after treatment, as evaluated in the open field (Fig. S1). A single MGO dose of 10, 25 and 50 mg/kg MGO was applied, and 15 min or 4 h later independent groups of mice were submitted to behavioral tests: tail suspension test (TST), marble burying test (MBT), and object location task (OLT).
(B) Repeated treatment (Fig. 1b): Mice received a single daily dose of MGO during 11 consecutive days, and the behavioral tests were performed at indicated time points, always 24 h after the last MGO injection (Fig. 1b), The testing sequence was: handling (day 0); administration of MGO (days 1–11); OF (day 5); OLT or object recognition task (ORT, days 5–9); Y-maze spontaneous alternation rate (Y-SA, day 7); triple horizontal bars test (THB, day 8); pole test (PT, day 10); step-down inhibitory avoidance task (IAT, days 10 and 11). To avoid acute effects and possible withdrawal syndrome, mice were treated with MGO 2 h after the behavioral testing. On day 12, and 24 h after the behavioral tests, mice were anesthetized with isoflurane and euthanized by cervical dislocation. The prefrontal cortex, hippocampus, and plasma were immediately removed and stored at − 80 °C until use for measurement of MGO plasma levels. Monoamines levels, and Glo1 and Glo2 content were evaluated in the prefrontal cortex and hippocampus.
General Procedures for Behavioral Testing
Behavioral tests were conducted between 09:30 and 16:30 h in a dimly lit and sound-isolated room: 15 lx for the OFT, OLT, and ORT; 30 lx for the rest of the behavioral tests. The experiments were recorded by a video camera system and images analyzed using the Any-Maze® software (Stoelting Co., Wood Dale, IL, USA). Mice were acclimatized to the experimental room for 2 h before the beginning of the tests. In tests that involved multiple sessions, once the mouse was exposed to a session paradigm, it was not mixed with non-exposed mice when it returned to its home cage. In the acute treatments, independent groups of mice were used for each behavioral test: OFT 32; MBT 26; TST 36; PT 46; OLT 20. In the repeated treatment protocol, three cohorts of mice were used to allow the execution of all behavioral tests and to avoid excessively animal stressing: (I) A group of 48 mice performed OFT, Y-SA, THB and step-down inhibitory avoidance task (IAT); (II) A group of 46 mice performed OF, OLT, and PT; (III). Another group of 46 mice performed OF, ORT, and PT. In addition, two other groups of mice underwent repeated treatment without participating in the other tests to perform only the TST (n = 16) or OLT (n = 20). The procedure was undertaken to exclude any possible interference of evaluating mice in more than one test. The total number of animals used to perform all tests in both protocols was 336 mice. The procedures used in the present study complied with the guidelines on the animal care of the UFSC Ethics Committee on the Use of Animals (CEUA/UFSC, Protocol Number 7245210616), which follows the “ARRIVE guidelines” and the “Guide for the Care and Use of Laboratory Animals” from NIH.
Open Field Test
The spontaneous locomotor activity and anxiety-like behavior of mice were evaluated in an open field arena. Each animal was placed in the center of the arena to freely explore the apparatus (40 × 40 × 30 cm) for 5 min [46, 47].
Tail Suspension Test
The immobility time was measured in the TST according to the method previously described [48]. Briefly, each animal was suspended by the tail at 60 cm from the ground for 6 min and the total immobility time was recorded. To exclude a possible interference, the locomotor activity of the mice was also evaluated in the OFT.
Marble Burying Test
A box (39.5 cm length × 33 cm width × 17 cm height) was filled with a 5 cm layer of husk bedding material that was evenly distributed across the whole cage. Fourteen glass spheres (marbles, 1.4 cm diameter) were spaced evenly in an 8 × 5 grid on the bed surface. During the test session, each mouse was placed in the box and allowed to explore it for 30 min. The number of marbles buried at least 2/3 of their depth was counted [49]. Rodents use bedding material to bury noxious and harmless objects, and the inhibition of marble burying can be considered as an anxiolytic-like effect [50].
Object Location Task
The spatial memory of mice was assessed using the OLT to evaluate both short- and long-term memory, based on protocols previously described [51, 52]. Briefly, 24 h after a habituation session in the open field arena (5 min), mice were replaced in the same arena for 5 or 10 min facing two identical objects (5 × 3 cm; training session) and after an interval of 90 min (short-term memory) or 24 h (long-term memory), one of the objects was moved to a new location (test session) and the time spent exploring the objects in the new (novel) and old (familiar) locations were recorded for 5 min. Visual cues were added in the test room as spatial reference.
Object Recognition Task
The long-term recognition memory was evaluated using the ORT. This task followed the OLT protocol. The differences included: in the test session, one of the two identical objects were replaced for another object (novel object) placed in the same location. The novel object had the same dimensions but with a different shape. No visual cues were provided in the test room to limit spatial reference by clearing off objects/cues [52].
Spontaneous Alternation Test
Evaluation of working memory was carried out measuring the Y-SA test. When moving from one place to another, rodents exhibit the natural tendency to explore the least visited area or a previously known area which has changed (novelty), this behavior is referred to as spontaneous alternation [53]. Y-SA was assessed using a Y-shaped maze, with three equal arms (30 × 10 × 25 cm height). During 5 min, the total number of arm entries with all four paws (N) was used as a parameter of locomotor activity. The number of ‘correct’ triplets (M, consecutive choices of each of the three arms without re-entries) was registered as a measure of spontaneous alternation [54, 55].
Step-Down Inhibitory Avoidance Task
To assess short- and long-term aversive memory, mice were exposed to the IAT. Based on previously described procedures [56, 57], each mouse was placed on a platform and its latency to step down on the grid with all four paws was measured. During the training session, immediately after stepping down on a grid, the mouse received a 2 s long scrambled foot shock (0.3 mA), then it was transferred to a home cage. To evaluate memory retention, test sessions were performed 1.5 h (short-term) and 24 h (long-term) after the training session. Test sessions were identical to the training session, except that no foot shock was given. A maximum of 180 s per session was waited to the mouse stepping down on the grid.
Plasma Methylglyoxal
MGO levels were determined by high-performance liquid chromatography coupled to a fluorescent detector (HPLC-FD). MGO was derivatized with 1,2-diamino-4,5-methylenedyoxybenzene (DMB) to allow fluorometric detection, as previously described by [58]. HPLC apparatus consisted of a Jasco LC-2000 Plus System coupled to fluorescent detector, using an Inerstil ODS-4®; 4.6 × 150 mm column, at a flow rate of 0.5 mL/min. The DMB-MGO derivative was eluted in an isocratic solution consisted of methanol, acetonitrile, and water at a 35:10:55 ratio. The fluorescence was monitored using an excitation at 355 nm and an emission at 399 nm.
Measurement of Brain Monoamines
The monoamine levels were determined by HPLC-FD according to [59]. Hippocampus and prefrontal cortex of mice were homogenized in 0.2 M perchloric acid containing 3 mM cysteine at 1:5 (w:v). Homogenate was centrifuged (12,000×g, 10 min, 4 °C) and the resulting supernatant was frozen (− 80 °C) for analysis. HPLC apparatus was from Jasco (LC-2000 Plus System), using an ACE® C18 Ultra-Inert column, at a flow of 0.6 mL/min. Monoamines were eluted in an isocratic solution of acetate (12 mM acetic acid, 0.26 mM EDTA)/methanol (86:14, v/v) solution. The fluorescence was monitored using excitation at 279 nm and emission at 320 nm.
Western Blotting
Protein levels of Glo1 and Glo2 were estimated by Western blotting in the prefrontal cortex and hippocampus of mice. The tissue samples were homogenized in ice-cold lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1% protease inhibitor cocktail, 1 mM PMSF). Samples were submitted to SDS-PAGE electrophoresis, and proteins electrotransferred to PVDF membranes and blocked with 5% non-fat dried milk. Membranes were probed with specific primary antibodies for Glo1 (SC 67351, 1:1000) and Glo2 (SC 51092, 1:1000), GCL (SC-22755, 1:1000), and TrxR1 (SC-20147, 1:1000) from Santa Cruz Biotechnology (Dallas). Band intensity was normalized by Ponceau S staining, which presented optical density linearity relative to protein load. ECL images were obtained using standard chemiluminescence techniques, then recorded by Chemidoc apparatus (BioRad, La Jolla) and quantified by ImageJ software (https://imagej.nih.gov/ij/).
Statistical Analysis
The OLT and ORT were analyzed using Student’s t test to compare indexes of exploration of the relocated or novel objects against 50% to exclude random exploration. Data presenting normal distribution were analyzed by one-way analysis of variance (ANOVA) followed by the Newman-Keuls post hoc test. Data that failed the normality test were analyzed by the Kruskal–Wallis non-parametric test, followed by Dunn’s comparison. The accepted level of statistical significance was p ≤ 0.05. Data are expressed as mean ± standard error of the mean (SEM) or median (interquartile range). All statistical tests were carried out using the Statistica software package, version 7.0 (StatSoft Inc., Tulsa, OK, EUA).
Results
Acute and Repeated Effects of MGO on Motor Function
Mice were tested in the OFT 4 h after a single MGO administration (20, 80, 200 mg/kg) as a preliminary evaluation to access locomotor activity (see Supplementary data, Fig. S1). Overall, severe impairment in the locomotor activity (distance, decreased entries and time in the center, grooming, rearings) was observed at 80 and 200 mg/kg. None of these alterations were observed at 20 or 50 mg/kg MGO (Fig. S1).
The OFT was also performed after 4 daily MGO administrations (0, 10, 25 or 50 mg/kg) 24 h after the last administration (Table S1). Mice treated with MGO did not show any significant alteration in the OFT, as compared to the control group, including total distance traveled, average speed or immobility time (Table S1). The anxiety-like parameters (number of entries and time spent in the central area) were not altered.
Force and motor coordination of mice, as evaluated by the pole test and triple horizontal bars were not altered up to 200 mg/kg in the acute treatment (Fig. S2). In the repeated treatment for 7–9 days, the doses of 10, 25 and 50 mg/kg MGO did not produce alterations on the same parameters in the pole test and triple horizontal bars (Fig. S2). Since no alterations on motor function were observed with 10, 25 and 50 mg/kg MGO, these doses were employed for further testing.
MGO Modulates Anxiety-Like Behavior
In order to characterize whether MGO induces anxiolytic effect, as previously published [27, 60], mice were tested in the MBT and in the OFT 4 h after treatment with MGO. Control group buried around 90% of spheres in the MBT test, which was around the same percentage of marbles buried by the group of animals treated with 25 mg/kg MGO (96%), while animals treated with 10 mg/kg MGO buried significantly fewer marbles (68%) [F(2,24) = 9.72; p > 0.05] (Fig. 2a). The time spent in the central area of the OFT can be used as an indicator of anxiety-like behavior [47]. In this regard, at doses 20 and 50 mg/kg (data not shown) no alterations in the time mice spent in the central area was observed in the OFT, while at 10 mg/kg mice remained almost twice as much time in the central area, when compared to the saline treated control [F(2,21) = 9.18; p > 0.05] (Fig. 2b). The decreased number of buried marbles and the increased time in the center of the open field point to an anxiolytic-like effect of MGO at the lowest dose tested, thus, confirming literature data showing an anxiolytic effect of MGO [27, 60].

Effects of MGO on anxiety-like behavior. Mice were treated with a single injection of MGO (10, 25 mg/kg) and 4 h later mice were tested. The percentage of hidden spheres in the MBT (a); and the time spent in the central area in the OFT (b) were recorded. The bars represent the mean ± SEM of 8–9 animals per group. *p < 0.05 as compared to the control group (Newman-Keuls post hoc test)
MGO Induces a Rapid Depressive-Like Behavior
Fifteen minutes after MGO injection, the immobility time was significantly increased in the TST at dose of 25 mg/kg (Fig. 3a), while after 4 h both doses (10 and 25 mg/kg) were effective in increasing the immobility time in the TST (Fig. 3b). At both time points, locomotor activity remained unaltered (Fig. 3d, e). Also, experiments in the repeated protocol with MGO (20 mg/kg) showed that 5 daily doses of MGO increased the immobility time in the TST, as evaluated 24 h after the last injection [F(1,14) = 11.27; p > 0.05] (Fig. 3c). No alterations in the spontaneous locomotor activity of mice were detected in the OFT at this time point (Fig. 3f).

Effects of MGO on depressive-like behavior. Immobility time was tested in the TST 15 min (a), or 4 h (b) after a single injection with MGO (10 and 25 mg/kg), and 24 h after 5 daily injections with MGO (20 mg/kg) (c). The distance each mouse travelled was measured in the open field 15 min (d) or 4 h (e) after a single MGO injection (10 and 25 mg/kg), and 24 h after 5 daily injections of treatment with MGO (20 mg/kg) (f). The bars represent the mean ± SEM of 5–8 animals per group. *p < 0.05 as compared to the control group (Newman-Keuls post hoc test)
Effects of MGO on Cognition
We choose 20 mg/kg MGO to test the short-term (90 min) spatial memory in the OLT, 3.6 h after mice received an acute MGO administration (Fig. 4a). Control mice explored the object B (relocated) for a significantly longer period [t(6) = 2.590; p < 0.05]. However, mice acutely treated with MGO showed memory impairment, indicated by the location index that was around 50% [t(9) = 0.011; p > 0.05] (Fig. 4a). In the habituation sessions (5 min) of the OLT on the open field, all groups presented similar locomotor activity (data not shown). In the training sessions, significant differences were not observed on the exploration time of the objects, thus, indicating no bias due to exploratory preference (Fig. 4d).

Effects of methylglyoxal on spatial and recognition memory. For short-term spatial memory evaluation, mice were tested in the OLT 90 min after the training session, and 3.6 h after a single injection (20 mg/kg). The location index of mice in the test session (a) and the exploration time of the two identical objects in the training session (d) are shown. For long-term spatial and recognition memory evaluation, the test sessions of object location (OLT) and recognition (ORT) tasks were performed after 8 days of treatment (10, 25 and 50 mg/kg). Mice were submitted to a 10 min training session, then returned to home cage, 4 h later mice received the last injection and the test session was performed after further 20 h. Location index in the OLT (b) and recognition index in the ORT (c) were calculated. The exploration time of the two objects in the training session of the OLT (e) and ORT (f) are also presented. The bars represent the mean ± SEM of 11–12 animals per group. *p < 0.05 as compared to 50% (Student’s t test)
For the repeated treatment, the long-term (24 h) spatial and recognition memory of mice were evaluated in the OLT and ORT, respectively, 24 h after 7 daily MGO (10, 25 and 50 mg/kg) administrations (Fig. 4b, c). In the test session of OLT, control mice showed location index (related to the exploration of the object in new location) significantly higher than 50% [t(10) = 4.343; p < 0.05], an indication of memory retention. The same memory retention was observed in mice receiving 10 mg/kg MGO [t(10) = 4.237; p < 0.05]. However, it was not the case for mice treated with 25 mg/kg [t(11) = 0.573] and 50 mg/kg [t(11) = 0.051] MGO, showing location index similar to 50% (p > 0.05), indicating spatial memory impairment (Fig. 4b). Long-term spatial memory evaluation (OLT) showed that all groups displayed similar exploration time of both objects during the training session (Fig. 4e). The short-term spatial memory was also tested. After 6 daily injections of MGO (20 mg/kg), we performed a similar experiment in which mice were only exposed to the OLT, to avoid interference of multiple testing. In this case, mice treated with MGO showed the same memory impairment (Fig. S3).
In the test session of ORT, control mice showed recognition index (related to the exploration of the novel object) significantly higher than 50% [t(11) = 5.201; p < 0.05], an indication of memory retention. A similar memory retention was observed in mice treated with 10 mg/kg [t(10) = 11.35; p < 0.05], and 25 mg/kg [t(11) = 3.645; p < 0.05] MGO. However, mice receiving 50 mg/kg MGO were unable to distinguish between the familiar and novel objects, as shown by the recognition index similar to 50% (p > 0.05), indicating recognition-memory impairment (Fig. 4c). In the training session of long-term recognition memory evaluation (ORT) the exploration time of both objects were not significantly different (Fig. 4f).
Evaluation of the working memory of mice treated with MGO was carried out by measuring the spontaneous alternation in the Y-maze. No differences in the alternation rate were observed in mice treated with 10 or 25 mg/kg MGO, in comparison to the control group (Fig. 5a). However, the number of alternations was significantly decreased in mice treated with 50 mg/kg MGO [F(3,38) = 4.36; p < 0.05], as compared to the control group (Fig. 5a), indicating deficit on working memory. The one-way ANOVA revealed no significant differences in the total number of arm entries [F(3,38) = 1.35; p > 0.05] (Fig. 5b).

Effects of methylglyoxal on working memory or short- and long-term aversive memory. Mice were treated for 6 days and tested in the Y maze 24 h after the last injection. The alternation rate (a) and the number of arm entries (b) were recorded. The bars represent the mean ± SEM of 10–11 animals per group. *p < 0.05 as compared to the control group (Newman-Keuls post hoc test). c Mice were treated for 10 and 11 days and short-term (1.5 h) and long-term (24 h) memory were evaluated in the step-down inhibitory avoidance test. The bars represent the median (interquartile range) of step-down latencies of 9–10 animals per group. *p < 0.05 as compared to the training session of the same group (Kruskal–Wallis non-parametric test followed by Dunn’s comparison)
Short- and long-term aversive memory of mice treated with MGO for 10 days were evaluated using the IAT (Fig. 5c). Statistical analysis revealed that only the control group displayed significantly higher latencies to step down the platform, in the short- (1.5 h) and long-term (24 h) memory tests (p < 0.05). All groups treated with MGO exhibited memory impairment, as shown by the absence of significant differences (p > 0.05) in the latency time to step down the platform, as compared to the respective training session latency. An exception was observed in mice treated with 25 MGO mg/kg that exhibited long-term memory retention (p < 0.05), but not short-term memory retention (Fig. 5c).
Biochemical Responses After MGO Treatment
Acute administration of MGO produced an increase in plasma MGO levels 4 h after treatment at 25, but not at 10 mg/kg (Fig. 6a), which is in accordance to literature data that reported maximal MGO plasma concentration is reached 4 h after administration [61]. In addition, plasma MGO levels were determined 24 h after the last MGO injection on 11th day of the repeated treatment. No statistical differences in the MGO levels were observed among the groups, as revealed by one-way ANOVA [F(3,25) = 1.29; p > 0.05] (Fig. 6b).

Plasma methylglyoxal. Four hours (a) after a single MGO injection, or 24 h after 11 daily injections with methylglyoxal (b), plasma levels of MGO were evaluated. Values are presented as mean ± SEM (n = 7–10). **p < 0.01 as compared to the control group (Newman-Keuls post hoc test)
Four hours after an acute MGO treatment the antioxidant defenses (GSH-t levels; GR and Glo1 activities; and Glo1 and Glo2 relative abundance) were not altered by 10 and 25 mg/kg MGO, when evaluated in the prefrontal cortex and hippocampus (Table S2 and Fig. S4).
We also assessed the Glo1 and Glo2 protein levels after repeated treatment for 11 days with MGO. Glo1 levels were decreased by 17% in the hippocampus after treatment at 10 mg/kg MGO, in comparison to the control group (Fig. 7a), without significant changes at 25 and 50 mg/kg. In the prefrontal cortex, a 27% decrease in the relative amount of Glo1 was observed at 50 mg/kg MGO, as compared to the control group (Fig. 7b). The other doses failed to produce a noticeable effect. Glo2 levels were not affected by MGO treatment in both the hippocampus and prefrontal cortex of mice (Fig. 7c, d).

Effects of methylglyoxal on the glyoxalase system. Glyoxalase 1 (Glo1; a and b) and glyoxalase 2 (Glo2; c and d) were evaluated in the hippocampus (a and c) and prefrontal cortex (b and d). Mice were treated daily with indicated doses of MGO for 11 days, and samples collected on the next day. Bars represent the mean ± SEM of n = 8–10 for the prefrontal cortex, and n = 3–6 for the hippocampus. * p < 0.01 as compared to the control group, as evaluated by the one-way ANOVA followed by Kruskal–Wallis non-parametric test followed by Dunn’s comparison
Monoamine Levels
After the repeated MGO (50 mg/kg) treatment for 11 days, a marked decrease on DA levels was observed in the prefrontal cortex, as compared to the control group [F(3,27) = 4.83; p < 0.05] (Fig. 8a). The method was unable to detect dopamine in the hippocampus. NE levels were not altered by MGO in the prefrontal cortex (Fig. 8b) or hippocampus (Fig. 8c). In addition, 5-HT levels were not altered by MGO in the prefrontal cortex (Fig. 8d) or hippocampus (Fig. 8e).

Effects of methylglyoxal on brain monoamine levels. Mice was treated with the indicated MGO doses for 11 days, the next day samples were harvested, and dopamine (DA, a), norepinephrine (NE, b and c), and serotonin (5-HT, d and e) were determined in the prefrontal cortex (a, b and d), and hippocampus (c and e). The method was unable to detect dopamine in the hippocampus. The bars represent the mean ± SEM of 6–11 animals per group. *p < 0.05 as compared to the control group (Newman-Keuls post hoc test)
Discussion
Motor Responses of Mice Treated with MGO
Preliminarily, we evaluated the effects of acute MGO treatment on locomotor activity, given it can be an essential factor in behavior investigation. A previous study showed that MGO can act as a competitive partial agonist of GABAA receptors, and at high acute doses, MGO leads to locomotor depression, ataxia, and hypothermia in mice [30]. These findings were corroborated by data herein presented, given that hypolocomotion was observed in the OFT, 4 h after a single MGO injection, at 80 and 200 mg/kg. These effects are considered characteristics of an inhibitory action to the central nervous system, which are in line with the idea that it was triggered by the activation of the GABAA receptors [30, 62], which remains to be confirmed. Given that hypolocomotion can add biases to behavioral tests, these doses of 80 and 200 mg/kg were not further explored.
Our results showed that acute or repeated treatment with MGO, up to the dose of 50 mg/kg, did not affect the spontaneous locomotor activity, vertical agility, force, and motor coordination, as accessed by the OF, Y-SA, THB and PT. Therefore, the doses of 10, 20 or 25, and 50 mg/kg were chosen for biochemical measurements and further behavioral analysis.
Anxiolytic and Depressive-Like Behavior Induced by MGO Treatment
The clear depressive-like effect observed in the TST as early as 15 min and 4 h after MGO treatment was not previously observed, instead, some reports showed antidepressive-like effect of MGO [60, 63, 64]. Lower Glo1 expression has been also positively associated with antidepressive-like behavior [63]. Also, our repeated protocol showed that MGO caused a non-acute depressive-like effect after 5 days of treatment (Fig. 3). The reasons for dissimilar results require further investigation on dose range and treatment regimens, genetic background, and other possible interfering variables. The presented data are consistent in acute and in repeated treatment, and were reproduced a number of times and, also reproduced when two other mice strains were tested (data not shown), indicating MGO can induce depressive-like behavior in mice. Furthermore, a clear decrease in dopamine levels was observed in the prefrontal cortex (Fig. 8), suggesting that, besides GABAA activation, MGO can affect DA levels, depending on the dose and treatment regimen. However, further experiments are needed to clarify this topic.
In support to our results, a clinical study showed that depressed patients presented lower Glo1 mRNA levels in peripheral white blood cells, which returned to basal levels upon remission of symptoms [65]. Animal models of depression, including repeated defeat and chronic unpredictable mild stress [66, 67], have shown diminished expression of Glo1 in the hippocampus or prefrontal cortex, which is compatible with the idea that elevated MGO levels are able to produce a depressive-like effect. In addition, a Glo1-KO mouse has been produced, in which a depressive-like behavior was evidenced [28], in agreement with our results. Overall, the literature and our data indicate that directly or indirectly MGO can be related to depressive-like behavior in Swiss mice. However, the divergent results [60, 63, 64] cannot be disregarded, and new experiments are warranted to clarify these apparent discrepancies.
Genetic deletion or pharmacological blockade of TRPA1 produces antidepressant-like effect in mouse models of depression, in accordance with a depressive-action elicited by MGO treatment [68]. Interestingly, MGO at low dose (10 mg/kg) produced anxiolytic effect in behavior tasks associated with neurochemical alterations in the hippocampus (reduction in the Glo1 levels and increased TrxR activity). The importance of hippocampus in the anxiety and anxiolytic action is related in the literature data [69]. In order to confirm the anxiolytic effect of MGO, we are planning experiments to investigate the hippocampus-dependent mechanism involved in such an effect.
Cognitive Parameters After MGO Treatment
MGO modification of biomolecules was considered a key event underpinning cognitive dysfunction [36]. However, previous studies have not systematically investigated the effects of MGO on learning and memory, nor its related mechanisms. Despite the different tests we employed, our results showed that MGO can disturb memory of mice, albeit at different extensions. MGO impaired spatial, recognition, aversive and working memory of mice in short- or long-term paradigms.
We found spatial short-term memory impairment in a brief period of time (3.6 h) after MGO injection. In general, AGEs formation occurs over a period of several hours to weeks [70, 71], and apoptosis induced by MGO takes hours to days to occur [72]. Based on these evidences, alternative mechanisms than AGE formation cannot be excluded as the leading cause for short-term memory impairment induced by MGO.
Repeated MGO treatment induced memory impairment, as evaluated by the spontaneous alternations in the Y-maze (50 mg/kg, day 7), OLT (25 and 50 mg/kg, day 9), ORT (50 mg/kg, day 9), and in the IAT (10, 25 and 50 mg/kg, day 10 and 11). A possible MGO-dependent mechanism inducing memory deficits would be related to AGEs formation, as MGO can be considered a major glycating agent [7, 70, 73]. Moreover, MGO can induce oxidative stress, cellular damage and apoptosis that can disturb neuronal function [3, 5, 6, 39], which would be related to memory disturbances mediated by RAGE activation [19, 74, 75]. Another possible mechanism inducing memory impairment would be related to GABAA receptor activation, since MGO can act as a competitive partial agonist of this receptor [30]. MGO has also been shown to activate TRPA1 ion channel evoked pain [32]. Interestingly, the ablation of TRPA1 channel may be crucial in regulating hippocampal fear-related learning and amygdala-dependent fear-related memory [76]. Authors also showed that TRPA1 deficient mice presented better recognition and spatial memory. These findings open the possibility that MGO-dependent activation of TRPA1 can be a potential mechanism leading to aversive memory impairment. New experiments are warranted to clarify the possible mechanisms underlying this memory impairment.
Biochemical Responses
Shortly after MGO treatment, brain MGO levels are increased [30], a similar increase in MGO levels was observed 4 h after a single intraperitoneal injection. However, the picture changes in the repeated treatment, as plasma levels of MGO remained comparable to control levels, when evaluated 24 h after the last MGO injection. Due to its reactivity toward basic amino acids, it is expected that MGO reacts with serum proteins, or to diffuse to other tissues [70, 77, 78]. Acute responses can, eventually, be regarded as direct effects of MGO, however, 24 h after MGO injection, as analyzed in the repeated treatment, cannot be attributed to a direct effect of MGO. This limitation prevents conclusions when comparing acute and repeated treatment responses to MGO, since the mechanism of action may not be the same. These are characteristics that should be taken into consideration when analyzing short term responses, as compared to repeated or chronic treatment.
Previously, we observed alterations in the activity and expression of Glo1, GR and TrxR, and in GSH-t levels [78, 79], when HT22 nerve cells received acute MGO treatment, which are derived from mice hippocampus. Likewise, after MGO treatment, acute hippocampal slices of mice presented a rapid increase in GR, TrxR, Glo1 and Glo2 [80]. We hypothesized that the in vivo treatment would produce similar responses, for this reason Glo and antioxidant defenses were also analyzed in mice acutely treated with MGO (Table S2 and Fig. S4). However, 4 h after MGO treatment, no alterations were observed in GSH-t levels, neither in Glo1 and GR activities, or Glo1 protein, as evaluated in the prefrontal cortex and hippocampus. It is plausible to suggest that the observed behavioral effects, within 4 h of treatment, are not related to these biochemical endpoints.
The previously reported changes in the glyoxalase system after in vivo MGO treatment are divergent, which would possibly be related to species and treatment protocol differences. Intracerebroventricular injection of MGO for 5 days in CD1 mice induced an increase in brain Glo1 mRNA and protein levels [60]. Conversely, Glo1 overexpression decreased brain MGO levels [30]. However, no changes were observed in Wistar rats after continuous intracerebroventricular injection of MGO for 3 weeks, and Glo1 activity evaluated 21 days later [39]. In our experimental setup, daily MGO treatment for 11 days caused a significant decrease in the Glo1 levels in the prefrontal cortex at 50 mg/kg. Based on this result, it is valid to think that cerebral cortex may present increased susceptibility for repeated MGO burden, since lower Glo1 levels were observed is this brain area.
The idea of a cortical vulnerability is supported by data showing that DA levels were decreased in the prefrontal cortex, but not in the hippocampus. Since we did not perform the measurements of DA synthesis, release, formation of metabolites, it cannot be concluded that MGO disrupts dopamine homeostasis in the brain. However, our results stimulate further investigation on this subject. It can be speculated that the DA decrease following MGO treatment may be associated with the decreased working memory performance displayed by mice in the Y-maze (50 mg/kg). Both the prefrontal cortex structure and DA levels are particularly important for the stabilization of current goal representations in working memory [81,82,83]. Importantly, as far as we know, this is the first evidence linking MGO treatment with decreased DA levels, whose mechanism of action remains to be elucidated.
Among the possible mechanisms, MGO-derived toxins are candidates to explain DA depletion. Under physiological conditions, MGO can generate free radicals [84], and react with DA to generate the neurotoxin 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (ADTIQ), which accumulates in the brain of Parkinson’s disease patients [85, 86]. Moreover, diabetic rats presented elevated levels of ADTIQ and MGO in the brain [87]. Salsolinol is another toxin derived from DA that was increased in neuronal cells exposed to MGO [88]. Furthermore, a number of possible metabolic routes for MGO toxicity has been previously reviewed [3]. Thus, MGO-derived toxins, among other routes, are candidates for the observed DA depletion in the prefrontal cortex of mice, nevertheless, experimental evidence supporting this possibility has yet to be produced.
The D1- and D2-class dopamine receptors are highly distributed in mammalian brain, including dopaminergic and non-dopaminergic neurons. The presynaptic inhibitory DA receptors in dopaminergic neurons generally provide an important negative feedback mechanism. These autoreceptors can downregulate DA synthesis by inhibiting tyrosine hydroxylase, the rate-limiting enzyme in DA synthesis, thus decreasing DA levels [89, 90]. Undergoing experiments are addressing this possibility.
Rather than measures of short-term memory, working memory has been associated with intellectual abilities in humans, especially with fluid intelligence [91]. Fluid intelligence or reasoning can be interpreted as the kind of thinking an individual can use when confronted with a relatively new task that cannot be performed automatically. This process is dependent on the activity of frontoparietal networks, especially the prefrontal cortex [83]. Thus, conditions that exhibit the pathological accumulation of MGO, such as diabetes, can be expected to induce cognitive deficits. Indeed, several studies showed that diabetes would favor cognitive impairments, including deficits associated with working memory [1, 92,93,94].
The acquisition phase of memory is driven by attentional processes in the prefrontal cortex, which are connected to other brain regions. Therefore, it is suggested that the short- and long-term memory impairment observed after MGO treatment, may be, at least in part, mediated by dopaminergic transmission during the acquisition phase of memory [91, 95]. The importance of DA for learning and memory, especially in the prefrontal cortex, is not deeply understood [96]. However, DA does seem to be important for working memory and cognitive processes dependent on the prefrontal cortex function. An example can be seen with Parkinson’s disease that features the loss of dopaminergic neurons and decreased DA levels [97]. In a spatial working memory task, Parkinson’s disease patients receiving levodopa (DA precursor), displayed better memory performance, as compared to pre-medication period, suggesting that DA is required for proper spatial working memory performance [98].
Conclusions
The present study showed that only acute treatment with the highest MGO doses tested (80 and 200 mg/kg) produced hypolocomotion, without altering motor capacity. Acute MGO treatment confirmed its anxiolytic effect. The depressive-like effect, induced by acute and repeated MGO treatment, has not been previously reported. Treatment with MGO induced cognitive deficits, as evaluated by the spontaneous alternation in the Y maze, OLT, ORT and IAT. NE and 5-HT levels were not altered by repeated treatment, while MGO decreased DA levels in the prefrontal cortex. We speculate that the MGO effect on cortical DA levels may be a novel form of MGO toxicity. More studies are required to unravel the routes leading to DA depletion, depressive-like behavior, and memory impairment, and to verify if they are connected.
References
Biessels GJ, Staekenborg S, Brunner E et al (2006) Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5:64–74. https://doi.org/10.1016/S1474-4422(05)70284-2
Bora E, Akdede BB, Alptekin K (2017) The relationship between cognitive impairment in schizophrenia and metabolic syndrome: a systematic review and meta-analysis. Psychol Med 47:1030–1040. https://doi.org/10.1017/S0033291716003366
Allaman I, Bélanger M, Magistretti PJ (2015) Methylglyoxal, the dark side of glycolysis. Front Neurosci 9:23. https://doi.org/10.3389/fnins.2015.00023
Choudhary D, Chandra D, Kale RK (1997) Influence of methylglyoxal on antioxidant enzymes and oxidative damage. Toxicol Lett 93:141–152
Dasuri K, Zhang L, Keller JN (2013) Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 62:170–185. https://doi.org/10.1016/j.freeradbiomed.2012.09.016
Kalapos MP (2008) Methylglyoxal and glucose metabolism: a historical perspective and future avenues for research. Drug Metabol Drug Interact 23:69–91
Thornalley PJ (2005) Dicarbonyl intermediates in the maillard reaction. Ann N Y Acad Sci 1043:111–117. https://doi.org/10.1196/annals.1333.014
Thornalley PJ (1996) Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification—a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol 27:565–573
Kalapos MP (1999) Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett 110:145–175
Lyles GA, Chalmers J (1992) The metabolism of aminoacetone to methylglyoxal by semicarbazide-sensitive amine oxidase in human umbilical artery. Biochem Pharmacol 43:1409–1414
Shibamoto T (2006) Analytical methods for trace levels of reactive carbonyl compounds formed in lipid peroxidation systems. J Pharm Biomed Anal 41:12–25. https://doi.org/10.1016/j.jpba.2006.01.047
Koop DR, Casazza JP (1985) Identification of ethanol-inducible P-450 isozyme 3a as the acetone and acetol monooxygenase of rabbit microsomes. J Biol Chem 260:13607–13612
Pompliano DL, Peyman A, Knowles JR (1990) Stabilization of a reaction intermediate as a catalytic device: definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry 29:3186–3194
Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344(Pt 1):109–116
Sousa Silva M, Gomes RA, Ferreira AEN et al (2013) The glyoxalase pathway: the first hundred years… and beyond. Biochem J 453:1–15. https://doi.org/10.1042/BJ20121743
Thornalley PJ (1993) The glyoxalase system in health and disease. Mol Aspects Med 14:287–371
Rabbani N, Thornalley PJ (2014) The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 63:50–52. https://doi.org/10.2337/db13-1606
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820. https://doi.org/10.1038/414813a
Hofmann MA, Drury S, Fu C et al (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Matafome P, Sena C, Seiça R (2013) Methylglyoxal, obesity, and diabetes. Endocrine 43:472–484. https://doi.org/10.1007/s12020-012-9795-8
Tian C, Alomar F, Moore CJ et al (2014) Reactive carbonyl species and their roles in sarcoplasmic reticulum Ca2+ cycling defect in the diabetic heart. Heart Fail Rev 19:101–112. https://doi.org/10.1007/s10741-013-9384-9
Distler MG, Palmer AA (2012) Role of glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: recent advances and mechanistic insights. Front Genet 3:250. https://doi.org/10.3389/fgene.2012.00250
McMurray KMJ, Distler MG, Sidhu PS et al (2014) Glo1 inhibitors for neuropsychiatric and anti-epileptic drug development. Biochem Soc Trans 42:461–467. https://doi.org/10.1042/BST20140027
Geng X, Ma J, Zhang F, Xu C (2014) Glyoxalase I in tumor cell proliferation and survival and as a potential target for anticancer therapy. Oncol Res Treat 37:570–574. https://doi.org/10.1159/000367800
Koivisto A, Chapman H, Jalava N et al (2014) TRPA1: a transducer and amplifier of pain and inflammation. Basic Clin Pharmacol Toxicol 114:50–55. https://doi.org/10.1111/bcpt.12138
Angeloni C, Zambonin L, Hrelia S (2014) Role of methylglyoxal in Alzheimer’s disease. Biomed Res Int 2014:238485. https://doi.org/10.1155/2014/238485
Hovatta I, Tennant RS, Helton R et al (2005) Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438:662–666. https://doi.org/10.1038/nature04250
Jang S, Kwon DM, Kwon K, Park C (2017) Generation and characterization of mouse knockout for glyoxalase 1. Biochem Biophys Res Commun 490:460–465. https://doi.org/10.1016/j.bbrc.2017.06.063
Krömer SA, Kessler MS, Milfay D et al (2005) Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J Neurosci Off J Soc Neurosci 25:4375–4384. https://doi.org/10.1523/JNEUROSCI.0115-05.2005
Distler MG, Plant LD, Sokoloff G et al (2012) Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J Clin Invest 122:2306–2315. https://doi.org/10.1172/JCI61319
Bierhaus A, Fleming T, Stoyanov S et al (2012) Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med 18:926–933. https://doi.org/10.1038/nm.2750
Andersson DA, Gentry C, Light E et al (2013) Methylglyoxal evokes pain by stimulating TRPA1. PLoS ONE 8:e77986. https://doi.org/10.1371/journal.pone.0077986
Huang X, Wang F, Chen W et al (2012) Possible link between the cognitive dysfunction associated with diabetes mellitus and the neurotoxicity of methylglyoxal. Brain Res 1469:82–91. https://doi.org/10.1016/j.brainres.2012.06.011
Kong X, Ma M, Huang K et al (2014) Increased plasma levels of the methylglyoxal in patients with newly diagnosed type 2 diabetes 2. J Diabetes 6:535–540. https://doi.org/10.1111/1753-0407.12160
Beeri MS, Moshier E, Schmeidler J et al (2011) Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech Ageing Dev 132:583–587. https://doi.org/10.1016/j.mad.2011.10.007
Srikanth V, Westcott B, Forbes J et al (2013) Methylglyoxal, cognitive function and cerebral atrophy in older people. J Gerontol A 68:68–73. https://doi.org/10.1093/gerona/gls100
Watanabe K, Okada K, Fukabori R et al (2014) Methylglyoxal (MG) and cerebro-renal interaction: does long-term orally administered MG cause cognitive impairment in normal Sprague-Dawley rats? Toxins 6:254–269. https://doi.org/10.3390/toxins6010254
Chun HJ, Lee Y, Kim AH, Lee J (2016) Methylglyoxal causes cell death in neural progenitor cells and impairs adult hippocampal neurogenesis. Neurotox Res 29:419–431. https://doi.org/10.1007/s12640-015-9588-y
Hansen F, Pandolfo P, Galland F et al (2016) Methylglyoxal can mediate behavioral and neurochemical alterations in rat brain. Physiol Behav 164:93–101. https://doi.org/10.1016/j.physbeh.2016.05.046
Wong ML, Licinio J (2001) Research and treatment approaches to depression. Nat Rev Neurosci 2:343–351. https://doi.org/10.1038/35072566
Critchlow V, Liebelt RA, Bar-Sela M et al (1963) Sex difference in resting pituitary-adrenal function in the rat. Am J Physiol 205:807–815. https://doi.org/10.1152/ajplegacy.1963.205.5.807
Kitay JI (1961) Sex differences in adrenal cortical secretion in the rat. Endocrinology 68:818–824. https://doi.org/10.1210/endo-68-5-818
Mevel LJC, Abitbol S, Beraud G, Maniey J (1979) Temporal changes in plasma adrenocorticotropin concentration after repeated neurotropic stress in male and female rats. Endocrinology 105:812–817. https://doi.org/10.1210/endo-105-3-812
Seale JV, Wood SA, Atkinson HC et al (2004) Gonadectomy reverses the sexually diergic patterns of circadian and stress-induced hypothalamic-pituitary-adrenal axis activity in male and female rats. J Neuroendocrinol 16:516–524. https://doi.org/10.1111/j.1365-2826.2004.01195.x
Kalapos MP, Schaff Z, Garzó T et al (1991) Accumulation of phenols in isolated hepatocytes after pretreatment with methylglyoxal. Toxicol Lett 58:181–191
Belzung C (1999) Measuring rodent exploratory behavior. In: Crusio WE, Gerlai RT (eds) Techniques in the behavioral and neural sciences. Elsevier, Amsterdam, pp 738–749
Prut L, Belzung C (2003) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 463:3–33
Steru L, Chermat R, Thierry B, Simon P (1985) The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85:367–370. https://doi.org/10.1007/bf00428203
Kedia S, Chattarji S (2014) Marble burying as a test of the delayed anxiogenic effects of acute immobilisation stress in mice. J Neurosci Methods 233:150–154. https://doi.org/10.1016/j.jneumeth.2014.06.012
Albelda N, Joel D (2012) Animal models of obsessive-compulsive disorder: exploring pharmacology and neural substrates. Neurosci Biobehav Rev 36:47–63. https://doi.org/10.1016/j.neubiorev.2011.04.006
Assini FL, Duzzioni M, Takahashi RN (2009) Object location memory in mice: pharmacological validation and further evidence of hippocampal CA1 participation. Behav Brain Res 204:206–211. https://doi.org/10.1016/j.bbr.2009.06.005
Vogel-Ciernia A, Wood MA (2014) Examining object location and object recognition memory in mice. Curr Protoc Neurosci. https://doi.org/10.1002/0471142301.ns0831s69
Tolman EC (1925) Purpose and cognition: the determiners of animal learning. Psychol Rev 32:285–297. https://doi.org/10.1037/h0072784
Dember WN, Fowler H (1958) Spontaneous alternation behavior. Psychol Bull 55:412–428
Kleschevnikov AM, Yu J, Kim J et al (2017) Evidence that increased Kcnj6 gene dose is necessary for deficits in behavior and dentate gyrus synaptic plasticity in the Ts65Dn mouse model of down syndrome. Neurobiol Dis 103:1–10. https://doi.org/10.1016/j.nbd.2017.03.009
Moreira ELG, de Oliveira J, Nunes JC et al (2012) Age-related cognitive decline in hypercholesterolemic LDL receptor knockout mice (LDLr-/-): evidence of antioxidant imbalance and increased acetylcholinesterase activity in the prefrontal cortex. J Alzheimers Dis JAD 32:495–511. https://doi.org/10.3233/JAD-2012-120541
Roesler R, Walz R, Quevedo J et al (1999) Normal inhibitory avoidance learning and anxiety, but increased locomotor activity in mice devoid of PrP(C). Brain Res Mol Brain Res 71:349–353
Ogasawara Y, Tanaka R, Koike S et al (2016) Determination of methylglyoxal in human blood plasma using fluorescence high performance liquid chromatography after derivatization with 1,2-diamino-4,5-methylenedioxybenzene. J Chromatogr B 1029–1030:102–105. https://doi.org/10.1016/j.jchromb.2016.07.019
De Benedetto GE, Fico D, Pennetta A et al (2014) A rapid and simple method for the determination of 3,4-dihydroxyphenylacetic acid, norepinephrine, dopamine, and serotonin in mouse brain homogenate by HPLC with fluorimetric detection. J Pharm Biomed Anal 98:266–270. https://doi.org/10.1016/j.jpba.2014.05.039
Hambsch B, Chen B-G, Brenndörfer J et al (2010) Methylglyoxal-mediated anxiolysis involves increased protein modification and elevated expression of glyoxalase 1 in the brain. J Neurochem 113:1240–1251. https://doi.org/10.1111/j.1471-4159.2010.06693.x
Ghosh M, Talukdar D, Ghosh S et al (2006) In vivo assessment of toxicity and pharmacokinetics of methylglyoxal: augmentation of the curative effect of methylglyoxal on cancer-bearing mice by ascorbic acid and creatine. Toxicol Appl Pharmacol 212:45–58. https://doi.org/10.1016/j.taap.2005.07.003
Kullmann DM, Ruiz A, Rusakov DM et al (2005) Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog Biophys Mol Biol 87:33–46. https://doi.org/10.1016/j.pbiomolbio.2004.06.003
Benton CS, Miller BH, Skwerer S et al (2012) Evaluating genetic markers and neurobiochemical analytes for fluoxetine response using a panel of mouse inbred strains. Psychopharmacology 221:297–315. https://doi.org/10.1007/s00213-011-2574-z
Wu Z, Fu Y, Yang Y et al (2018) Gating TrkB switch by methylglyoxal enables GLO1 as a target for depression. bioRxiv. https://doi.org/10.1101/435867
Fujimoto M, Uchida S, Watanuki T et al (2008) Reduced expression of glyoxalase-1 mRNA in mood disorder patients. Neurosci Lett 438:196–199. https://doi.org/10.1016/j.neulet.2008.04.024
Patki G, Solanki N, Atrooz F et al (2013) Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res 1539:73–86. https://doi.org/10.1016/j.brainres.2013.09.033
Yang Y, Yang D, Tang G et al (2013) Proteomics reveals energy and glutathione metabolic dysregulation in the prefrontal cortex of a rat model of depression. Neuroscience 247:191–200. https://doi.org/10.1016/j.neuroscience.2013.05.031
de Moura JC, Noroes MM, de Rachetti VPS et al (2014) The blockade of transient receptor potential ankirin 1 (TRPA1) signalling mediates antidepressant- and anxiolytic-like actions in mice. Br J Pharmacol 171:4289–4299. https://doi.org/10.1111/bph.12786
Jimenez JC, Su K, Goldberg AR et al (2018) Anxiety cells in a hippocampal-hypothalamic circuit. Neuron 97:670–683.e6. https://doi.org/10.1016/j.neuron.2018.01.016
Lo TW, Westwood ME, McLellan AC et al (1994) Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J Biol Chem 269:32299–32305
Singh R, Barden A, Mori T, Beilin L (2001) Advanced glycation end-products: a review. Diabetologia 44:129–146. https://doi.org/10.1007/s001250051591
Di Loreto S, Zimmitti V, Sebastiani P et al (2008) Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int J Biochem Cell Biol 40:245–257. https://doi.org/10.1016/j.biocel.2007.07.019
Thornalley PJ, Rabbani N (2011) Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev Biol 22:318–325. https://doi.org/10.1016/j.semcdb.2011.02.006
Xue J, Ray R, Singer D et al (2014) The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 53:3327–3335. https://doi.org/10.1021/bi500046t
Schmidt AM, Hori O, Chen JX et al (1995) Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 96:1395–1403. https://doi.org/10.1172/JCI118175
Lee K-I, Lin H-C, Lee H-T et al (2017) Loss of transient receptor potential ankyrin 1 channel deregulates emotion, learning and memory, cognition, and social behavior in mice. Mol Neurobiol 54:3606–3617. https://doi.org/10.1007/s12035-016-9908-0
Thornalley PJ (2008) Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems–role in ageing and disease. Drug Metabol Drug Interact 23:125–150
Dafre AL, Goldberg J, Wang T et al (2015) Methylglyoxal, the foe and friend of glyoxalase and Trx/TrxR systems in HT22 nerve cells. Free Radic Biol Med 89:8–19. https://doi.org/10.1016/j.freeradbiomed.2015.07.005
Dafre AL, Schmitz AE, Maher P (2017) Methylglyoxal-induced AMPK activation leads to autophagic degradation of thioredoxin 1 and glyoxalase 2 in HT22 nerve cells. Free Radic Biol Med 108:270–279. https://doi.org/10.1016/j.freeradbiomed.2017.03.028
Schmitz AE, de Souza LF, Dos Santos B et al (2017) Methylglyoxal-induced protection response and toxicity: role of glutathione reductase and thioredoxin systems. Neurotox Res 32:340–350. https://doi.org/10.1007/s12640-017-9738-5
van Schouwenburg M, Aarts E, Cools R (2010) Dopaminergic modulation of cognitive control: distinct roles for the prefrontal cortex and the basal ganglia. Curr Pharm Des 16:2026–2032
Cools R (2016) The costs and benefits of brain dopamine for cognitive control. Wiley Interdiscip Rev Cogn Sci 7:317–329. https://doi.org/10.1002/wcs.1401
Otero TM (2017) Brief review of fluid reasoning: conceptualization, neurobasis, and applications. Appl Neuropsychol Child 6:204–211. https://doi.org/10.1080/21622965.2017.1317484
Szent-Györgyi A, McLaughlin JA (1975) Interaction of glyoxal and methylglyoxal with biogenic amines. Proc Natl Acad Sci USA 72:1610–1611. https://doi.org/10.1073/pnas.72.4.1610
Deng Y, Zhang Y, Li Y et al (2012) Occurrence and distribution of salsolinol-like compound, 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (ADTIQ) in parkinsonian brains 1996. J Neural Transm Vienna Austria 119:435–441. https://doi.org/10.1007/s00702-011-0724-4
Hipkiss AR (2017) On the relationship between energy metabolism, proteostasis, aging and Parkinson’s disease: possible causative role of methylglyoxal and alleviative potential of carnosine. Aging Dis 8:334–345. https://doi.org/10.14336/AD.2016.1030
Song D-W, Xin N, Xie B-J et al (2014) Formation of a salsolinol-like compound, the neurotoxin, 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, in a cellular model of hyperglycemia and a rat model of diabetes. Int J Mol Med 33:736–742. https://doi.org/10.3892/ijmm.2013.1604
Xie B, Lin F, Peng L et al (2014) Methylglyoxal increases dopamine level and leads to oxidative stress in SH-SY5Y cells. Acta Biochim Biophys Sin 46:950–956. https://doi.org/10.1093/abbs/gmu094
Anzalone A, Lizardi-Ortiz JE, Ramos M et al (2012) Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. J Neurosci 32:9023–9034. https://doi.org/10.1523/JNEUROSCI.0918-12.2012
Ford CP (2014) The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282:13–22. https://doi.org/10.1016/j.neuroscience.2014.01.025
Cowan N (2008) What are the differences between long-term, short-term, and working memory? Prog Brain Res 169:323–338. https://doi.org/10.1016/S0079-6123(07)00020-9
Kodl CT, Seaquist ER (2008) Cognitive dysfunction and diabetes mellitus. Endocr Rev 29:494–511. https://doi.org/10.1210/er.2007-0034
Kopf D, Frölich L (2009) Risk of incident Alzheimer’s disease in diabetic patients: a systematic review of prospective trials. J Alzheimers Dis JAD 16:677–685. https://doi.org/10.3233/JAD-2009-1011
Miles W, Root H (1922) Psychologic tests applied to diabetic patients. Arch Intern Med 30:767–777
Gray S, Green S, Alt M et al (2017) The structure of working memory in young children and its relation to intelligence. J Mem Lang 92:183–201. https://doi.org/10.1016/j.jml.2016.06.004
Puig MV, Rose J, Schmidt R, Freund N (2014) Dopamine modulation of learning and memory in the prefrontal cortex: insights from studies in primates, rodents, and birds. Front Neural Circuits 8:93. https://doi.org/10.3389/fncir.2014.00093
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet Lond Engl 386:896–912. https://doi.org/10.1016/S0140-6736(14)61393-3
Lange KW, Robbins TW, Marsden CD et al (1992) L-dopa withdrawal in Parkinson’s disease selectively impairs cognitive performance in tests sensitive to frontal lobe dysfunction. Psychopharmacology 107:394–404
Acknowledgements
This work was supported by the Brazilian funding agency CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, #462333/2014-0, #306204/2014-2), and ALD is a research fellow (307057/2018-1). MPC received a post-doctoral PNPD, and GRLA and JCS received scholarships from CAPES (Coordination for the Improvement of Higher Education Personnel, Brazil).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Szczepanik, J.C., de Almeida, G.R.L., Cunha, M.P. et al. Repeated Methylglyoxal Treatment Depletes Dopamine in the Prefrontal Cortex, and Causes Memory Impairment and Depressive-Like Behavior in Mice. Neurochem Res 45, 354–370 (2020). https://doi.org/10.1007/s11064-019-02921-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02921-2