
Abstract
The brain uses mainly glucose as fuel with an index of glucose to oxygen utilization close to 6, the maximal index if all glucose was completely oxidized. However, this high oxidative index, contrasts with the metabolic traits of the major cell types in the brain studied in culture, neurons and astrocytes, including the selective use of the malate-aspartate shuttle (MAS) in neurons and the glycerol-phosphate shuttle in astrocytes. Metabolic interactions among these cell types may partly explain the high oxidative index of the brain. In vivo, neuronal activation results in a decrease in the oxygen glucose index, which has been attributed to a stimulation of glycolysis and lactate production in astrocytes in response to glutamate uptake (astrocyte–neuron lactate shuttle, ANLS). Recent findings indicate that this is accompanied with a stimulation of pyruvate formation and astrocyte respiration, indicating that lactate formation is not the only astrocytic response to neuronal activation. ANLS proposes that neurons utilize lactate produced by neighboring astrocytes. Indeed, neurons can use lactate to support an increase in respiration with different workloads, and this depends on the Ca2+ activation of MAS. However, whether this activation operates in the brain, particularly at high stimulation conditions, remains to be established.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The brain consumes mainly glucose and is the major glucose consumer organ of the body. In the resting state, most of this consumption is oxidative, as evidenced from measurements of the stoichiometry between oxygen (CMRO2) and glucose (CMRglc) utilization, or oxygen-glucose index (OGI), which is close to 6:1, the theoretical maximum for the full oxidation of glucose (reviewed in [4, 22]). A submaximal OGI value, of around 5.5–6 is found in animal or human brain at rest, the difference generally attributed to lactate formation and efflux from the brain (reviewed in [22]). However, upon activation, the OGI in the activated brain region drops further. In this short review we will examine the properties of neurons and astrocytes in culture, especially those that have been described recently, relevant to whole brain variations in OGI.
Neurons and Astrocytes: Respiration Matters
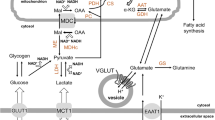
The very high oxidative consumption of glucose contrasts with the metabolic properties of the two major cell types in the brain, neurons and astrocytes. Glucose metabolism in neurons is more oxidative than in astrocytes [2, 27, 41]. Glycolysis is less active in neurons and this correlates with lower PFK1 activity than astrocytes, with low levels of the glycolysis regulating enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 (PFKFB3), and lower levels of the glycolysis activator Fructose 2,6-bisphosphate [25, 27]. In addition, neurons have high levels of the two mitochondrial carriers involved in the malate-aspartate shuttle (MAS), the aspartate-glutamate carrier Aralar/AGC1/Slc25a12 (AGC1) and the oxoglutarate-malate carrier OGC/Slc25a11 [60], which allow for a vigorous oxidation of cytosolic NADH in mitochondria and the maintenance of high respiration rate during glucose metabolism [18, 36, 48, 58]. In contrast, glycolysis and lactate production are very active in astrocytes, while oxygen consumption in mitochondria from acutely isolated brain astrocytes is lower than in those isolated from neurons [41]. Although AGC1 protein is present in astrocytes in culture [33, 53] its levels in brain astrocyte mitochondria are much lower than those in neurons, both in brain astrocytes [49], and in Müller cells in the retina [35, 66]. Its loss does not result in any difference in glucose dependent OCR in cultured astrocytes [30], indicating that the malate-aspartate shuttle does not contribute to glucose oxidation in these glial cells. In contrast, astrocytes express the two components of the glycerol-phosphate shuttle (GPS) [30], with activity values as those reported for other tissues or cell types [16, 42]. Earlier work by Nguyen et al. [46] showed that the major isoform of cytosolic glycerophosphate dehydrogenase (GPDH) had very low levels in neurons and astrocytes, suggesting that GPS might be of little importance in these cells. However, the mRNA of a second cytosolic glycerol‐P‐dehydrogenase (GPD1‐like), functionally identified in heart [40, 64], is expressed in neurons and astrocytes [12]. It is possible that the activity of cGPDH found in mouse astrocytes, which matches that of mitochondrial GPDH (45.30 ± 3.69 and 44.00 ± 2.26 μmol NADH/mg/min, respectively, Juaristi et al. [30]) corresponds to this novel isoform. These results suggest GPS in cultured mouse brain astrocytes as main NADH shuttle responsible for the transfer of redox equivalents from cytosolic NADH to mitochondria during glucose utilization and explains the lack of impact of ARALAR deficiency on lactate production and respiration rates in these cells. The finding of a glycerol-phosphate shuttle in astrocytes agrees with the mild but significant effects of inhibitors of this shuttle on the cytosolic NADH/NAD+ ratios and lactate formation examined by imaging with fluorescent probes in astrocytes [32] and with the reports of McKenna et al. [45].
The use of these two different redox shuttles by neurons and astrocytes adds another layer of complexity to the known interrelations between these two cell types. Indeed, the mitochondrial component of the glycerol-phosphate shuttle, mitochondrial glycerophosphate dehydrogenase, is a powerful source of reactive oxygen species (ROS) in mitochondria, and is activated by calcium from the external face of the inner mitochondrial membrane [1, 63]. The presence of the glycerol-phosphate shuttle in astrocytes may be related to the high ROS production in astrocyte mitochondria as compared to neuronal mitochondria [41]. The high production of mitochondrial ROS has been shown to be associated with the existence of a large proportion of deactive complex I in astrocytes, and to a greater proportion of complex I assembled into supercomplexes in neurons [41, 65]. However, as an extra ROS source, glycerophosphate dehydrogenase may add to the large ROS output in astrocytes. In addition, astrocytes are equipped with a strong redox antioxidant system, mediated by the presence of active Nrf2, a transcription factor activated by ROS that controls the expression of antioxidant genes, whereas the Nrf2 pathway activity is lower in neurons [9, 11].
The very high oxidative consumption of glucose in the brain contrasts with the metabolic properties of pure primary cell cultures studied at low, physiological glucose levels (Table 1). In the case of neurons, the more oxidative cells in the brain, mouse cortical neurons in culture convert into lactate about half of the glucose utilized, the remaining half being used in cell respiration [30], a result at odds with the almost full oxidation of glucose in the brain in vivo. In the case of astrocytes in culture, which are known to be glycolytic cells, glucose consumption is much larger than for neurons and lactate production also accounts for about half of the consumed glucose. However, basal respiration is only one tenth of that expected if the remaining glucose was fully oxidized in mitochondria, again at odds with the brain OGI (Table 1).
It may be argued that some glucose utilization and lactate production in neuronal cultures is partially due to contaminating astrocytes present in the cultures, and that the actual OGI in cultured neurons is probably closer to 6. However, this argument cannot be applied to astrocyte cultures, in which respiration is ten times lower than that required for the full oxidation of glucose [30, 31]. A factor to be considered to explain the high lactate production of these brain cells is the large volume of extracellular medium in culture as compared to the brain, which would favor lactate release and dilution in the external medium, thereby enhancing glycolysis.
In the case of astrocytes, these cells in culture are thought to differ from those in brain by becoming more glycolytic, as supported by changes in mRNA levels in rapidly isolated brain cells with respect to cultures [12]. However, a proteomic study in cultured and acutely isolated brain astrocytes revealed that these differences were not so large, and involved mainly increases in proteins from the extracellular matrix in cultured astrocytes [60].
One of the mechanisms that would resolve this paradox is through interactions and exchange reactions between neurons and astrocytes which would allow a complete oxidation of brain glucose. The astrocyte to neuron lactate shuttle, ANLS [50, 51], proposes that neuronal activity results in glutamate release and uptake by astrocytes, which stimulates lactate production in these cells. Astrocytic lactate is then taken up and oxidized by neurons. Additionally, pyruvate produced by neurons may be taken up and oxidized by astrocytes [13]. This transcellular pathway of glucose oxidation would provide an almost complete oxidation of brain glucose and glycogen.
This shuttle is supported by the distribution of specific glucose transporters and plasma membrane pyruvate/lactate transporters in neurons and astrocytes [60] which favor lactate production and efflux from astrocytes and lactate uptake and oxidation to pyruvate in neurons (see [8]). In addition, the malate aspartate NADH shuttle, which is essential for lactate and glucose oxidation by neurons [38] does not play any major role in glucose oxidation in astrocytes, either in basal or in stimulated conditions [30, 31].
Notably, in vivo studies have shown that disruption of astrocytic or neuronal lactate transporters in hippocampus leads to amnesia, suggesting the requirement of ANLS components for memory formation [61]. However, the exact connection between the lactate transporters and the process of memory remains to be established.
On a cellular level, the astrocyte response to neuronal activation has been studied in mixed cultures of astrocytes and neurons each derived from a different species, a procedure allowing to follow the transcriptomic changes in astrocytes that take place after chemically induced neuronal activation [26]. Neuronal stimulation caused upregulation of the transporters involved in ANLS, glucose and glutamate transporters and lactate dehydrogenase (LdhA), suggesting an important influence of neuronal activity on the expression of ANLS. However, these transcriptomic changes are late events which took place after several hours of neuronal activation. Importantly, they were attributed to a transcriptional pathway involving cAMP–PKA–CREB rather than to the action of glutamate on astrocytes. In fact, the ANLS is still a matter of debate ([7, 8, 22]; and references therein).
Astrocyte and Neuronal Energy Metabolism Upon Neuronal Activation
Central to ANLS is the link between neuronal activity and lactate production by astrocytes. According to the prevailing hypothesis, glutamate capture by astrocytes results in an increase in cytosolic Na+ which drives different ion pumps in the plasma membrane, to restore resting Na+ levels. The ATP required for this Na+-dependent workload is obtained by aerobic glycolysis from blood glucose or from endogenous glycogen. The other task carried out by brain astrocytes is the clearance of K+ from the extracellular space, a process dependent on the activity of the sodium bicarbonate cotransporter NBCe1/Slc4a4 [57].
Glutamate stimulation of glucose consumption and lactate formation in astrocytes was initially reported by Pellerin, Magistretti and coworkers [14, 17, 50] and other groups [39] but not by other laboratories who found that glutamate uptake in astrocytes was not accompanied by increases in lactate production or glucose consumption [21, 28, 34, 52, 62]. The reasons for this discrepancy probably lie on the culture conditions, media and/or glucose concentrations [22]. Recent findings by a number of laboratories in cultured astrocytes, indicate that upon exposure to glutamate, both K+ and glutamate stimulate glycolysis and lactate formation [10, 23], pyruvate production [31] and an increase in the NADH/NAD+ ratio [32] as determined with genetically coded sensors.
Another important consideration is whether this response to glutamate is accompanied by an increase in OXPHOS or not. In the case of K+, OXPHOS does not participate in the response as K+ does not stimulate astrocyte respiration; this is due to a variant of the classical Crabtree effect [23]. The uptake of bicarbonate through NBCe1/Slc4a4 which follows astrocyte K+-depolarization, causes cytosolic alkalinization and stimulation of glycolysis to increase ATP beyond its use by the Na+ pump, explaining the lack of a respiratory response to K+ [23]. However, in contrast to previous observations [5] OXPHOS does participate in the response to glutamate [31, 54]. The stimulation of astrocyte respiration by glutamate (about 1.6 fold, Table 1) responds to the strictly Na+-dependent workload caused by glutamate uptake, and does not depend on the Ca2+ signals elicited by glutamate in these cells [31]. It is accompanied by an increase in glycolytic pyruvate production, indicating that the response to glutamate is a stimulation of glycolysis with pyruvate production, and also oxidation in mitochondria. In addition, some of the glutamate taken up is also oxidized in astrocyte mitochondria [31] as proposed previously [29], contributing to the increase in astrocyte respiration. Interestingly, when faced to the double challenge of K+ and glutamate removal, respiratory stimulation persists, suggesting that in the in vivo situation astrocytes possibly respond to neuronal stimuli by increasing respiration [31] and, depending on the stimulation conditions, with lactate production.
It may be argued that the fact that astrocytes produce lactate and also obtain ATP from OXPHOS upon neuronal stimulation is not an objection to ANLS. However, recent findings from the field of neuronal metabolism, particularly during strong activation conditions, question aspects of the ANLS hypothesis.
Neuronal activation is associated with the workloads involved in the return to the resting state, a mostly postsynaptic activity [3]. The source of energy to obtain the required ATP is largely OXPHOS, as neurons are able to upregulate their own respiration in order to match ATP consumption [36, 37, 55] (Table 1). Interestingly, work in cortical neurons in culture has shown that the upregulation of OXPHOS is Ca2+ dependent, not only because part of the workload is also Ca2+ dependent, but due to a prominent role of Ca2+ in boosting the respiratory response. In the case of neurons, Ca2+ activation of the malate-aspartate shuttle (MAS), plays an important role in stimulation of respiration, and the effect of Ca2+ is thought to be due to Ca2+ binding to EF-hand Ca2+ binding motifs of the mitochondrial aspartate/glutamate carrier, AGC1, which face the intermembrane space [44, 47]. Indeed, the AGC1 catalyses the exchange of glutamate plus a proton against aspartate, so that the overall reaction is electrogenic. This makes this step irreversible in polarized mitochondria, a condition favored for a controlling step, and drives the entire MAS in the direction of redox equivalent transfer into mitochondria.
An important consequence of Ca2+ activation of AGC1-MAS is the supply of pyruvate to mitochondria [24]. In fact, the supply of exogenous pyruvate fully reverts the limited stimulation of respiration in response to different workloads observed in AGC1-deficient neurons [36, 55], suggesting that pyruvate supply controls neuronal respiration either on glucose or lactate [38]. The affinity for Ca2+ of AGC1-MAS is about 300 nM [15, 24], a value lower than the apparent affinity for Ca2+ of the mitochondrial Ca2+ uniporter (MCU), another Ca2+ target in mitochondria, and probably the major one in AGC1-deficient neurons.
In sum, the strong stimulation of respiration in cultured neurons and their ability to use external lactate via MAS for that purpose support ANLS, at least under basal and mild activation conditions. However, recent findings by Yellen’s lab have questioned the notion that in vivo activated neurons take up lactate from the extracellular medium [19, 20]. In fact, using biosensors expressed in neurons, it was shown that synaptic (electrical) stimulation of hippocampal dentate granule neurons or the whisker stimulation of neurons in layers II/III of the primary somatosensory barrel cortex of an awake mouse was accompanied with rapid increases in cytosolic NADH and, in dentate gyrus neurons, also with increases in cytosolic lactate. Importantly, the use of the specific MCT1 and MCT2 inhibitor AR-C155858 which prevented the uptake of lactate from the extracellular medium in hippocampal slices, did not prevent the stimulation-induced increase in lactate levels. This indicated that it arose from an increase in glycolysis and lactate formation in the stimulated neuron itself [19] although it was somewhat surprising that lactate over-accumulation in the presence of AR-C155858 was not observed. It would be interesting to know whether this presumably neuronal lactate production is maintained or varies depending on the stimulation conditions.
Regardless of that, as a standing lactate gradient exists, with higher lactate concentration in astrocytes than neurons [43], neuronal lactate may be extruded to nearby neurons with lower lactate concentrations or to the extracellular space. In fact, claims for the possible formation of lactate by neurons under strong stimulation conditions are not new. Indeed, studies in isolated brain mitochondria indicated that high calcium loads, below those required to induce the permeability transition, cause an inhibition of the malate aspartate shuttle which is expected to slow down glycolysis, and increase lactate and NADH formation [16]. This effect was attributed to an inhibition of the second transporter of the malate aspartate shuttle, the oxoglutarate-malate carrier (OGC/Slc25a11). Inhibition would be due to competition between oxoglutarate dehydrogenase and the OGC for the common substrate oxoglutarate (OG). Activation by matrix Ca2+ of oxoglutarate dehydrogenase lowers its Km for OG with a drop in matrix OG concentration resulting in lower OG efflux along OGC and a resulting drop in MAS activity. An inhibition of MAS under strong stimulation conditions has also been proposed in cultured neurons [6, 59], but Díaz-García et al. [19] did not find evidence for an inhibition of the shuttle. Whether this mechanism of shuttle inhibition actually operates in vivo is still an open question.
References
Adam-Vizi V, Tretter L (2013) The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem Int 62:757–763
Almeida A, Almeida J, Bolaños JP, Moncada S (2001) Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Natl Acad Sci USA 98:15294–15299
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145
Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468:232–243
Azarias G, Perreten H, Lengacher S, Poburko D, Demaurex N, Magistretti PJ, Chatton JY (2011) Glutamate transport decreases mitochondrial pH and modulates oxidative metabolism in astrocytes. J Neurosci 31:3550–3559
Bak LK, Walls AB, Schousboe A, Ring A, Sonnewald U, Waagepetersen HS (2009) Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J Neurochem 109(Suppl 1):87–93
Bak LK, Walls AB (2018) CrossTalk opposing view: lack of evidence supporting an astrocyte-to-neuron lactate shuttle coupling neuronal activity to glucose utilisation in the brain. J Physiol 596:351–353
Barros LF, Weber B (2018) CrossTalk proposal: an important astrocyte-to-neuron lactate shuttle couples neuronal activity to glucose utilisation in the brain. J Physiol 596:347–350
Baxter PS, Hardingham GE (2016) Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100:147–152
Bittner CX, Valdebenito R, Ruminot I, Loaiza A, Larenas V, Sotelo-Hitschfeld T, Moldenhauer H, San Martín A, Gutiérrez R, Zambrano M, Barros LF (2011) Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. J Neurosci 31:4709–4713
Bolaños JP (2016) Bioenergetics and redox adaptations of astrocytes to neuronal activity. J Neurochem 139(Suppl 2):115–125
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278
Cerdán S, Rodrigues TB, Sierra A, Benito M, Fonseca LL, Fonseca CP, García-Martín ML (2006) The redox switch/redox coupling hypothesis. Neurochem Int 48:523–530
Chatton JY, Pellerin L, Magistretti PJ (2003) GABA uptake into astrocytes is not associated with significant metabolic cost: implications for brain imaging of inhibitory transmission. Proc Natl Acad Sci USA 100:12456–12461
Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrústegui J (2007) Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem 282:7098–7106
Contreras L, Satrústegui J (2009) Calcium signaling in brain mitochondria: interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J Biol Chem 284:7091–7099
Debernardi R, Magistretti PJ, Pellerin L (1999) Trans-inhibition of glutamate transport prevents excitatory amino acid-induced glycolysis in astrocytes. Brain Res 850:39–46
del Arco A, Satrústegui J (1998) Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem 273:23327–23334
Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26:361–374
Díaz-García CM, Yellen G (2018) Neurons rely on glucose rather than astrocytic lactate during stimulation. J Neurosci Res. https://doi.org/10.1002/jnr.24374
Dienel GA, Cruz NF (2004) Nutrition during brain activation: does cell-to-cell lactate shuttling contribute significantly to sweet and sour food for thought? Neurochem Int 45:321–351
Dienel GA (2019) Brain glucose metabolism: integration of energetics with function. Physiol Rev 99:949–1045
Fernández-Moncada I, Ruminot I, Robles-Maldonado D, Alegría K, Deitmer JW, Barros LF (2018) Neuronal control of astrocytic respiration through a variant of the Crabtree effect. Proc Natl Acad Sci USA 115:1623–1628
Gellerich FN, Gizatullina Z, Gainutdinov T, Muth K, Seppet E, Orynbayeva Z, Vielhaber S (2013) The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life 65:180–190
Gonçalves CA, Rodrigues L, Bobermin LD, Zanotto C, Vizuete A, Quincozes-Santos A, Souza DO, Leite MC (2019) Glycolysis-derived compounds from astrocytes that modulate synaptic communication. Front Neurosci 12:1035
Hasel P, Dando O, Jiwaji Z, Baxter P, Todd AC, Heron S, Márkus NM, McQueen J, Hampton DW, Torvell M, Tiwari SS, McKay S, Eraso-Pichot A, Zorzano A, Masgrau R, Galea E, Chandran S, Wyllie DJA, Simpson TI, Hardingham GE (2017) Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun 8:15132
Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752
Hertz L, Swanson RA, Newman GC, Marrif H, Juurlink BH, Peng L (1998) Can experimental conditions explain the discrepancy over glutamate stimulation of aerobic glycolysis? Dev Neurosci 20:339–347
Hertz L, Hertz E (2003) Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem Int 43:355–361
Juaristi I, García-Martín ML, Rodrigues TB, Satrústegui J, Llorente-Folch I, Pardo B (2017) ARALAR/AGC1 deficiency, a neurodevelopmental disorder with severe impairment of neuronal mitochondrial respiration, does not produce a primary increase in brain lactate. J Neurochem 142:132–139
Juaristi I, Llorente-Folch I, Satrústegui J, del Arco A (2019) Extracellular ATP and glutamate drive pyruvate production and energy demand to regulate mitochondrial respiration in astrocytes. Glia 67:759–774
Köhler S, Winkler U, Sicker M, Hirrlinger J (2018) NBCe1 mediates the regulation of the NADH/NAD + redox state in cortical astrocytes by neuronal signals. Glia 66:2233–2245
Li B, Hertz L, Peng L (2012) Aralar mRNA and protein levels in neurons and astrocytes freshly isolated from young and adult mouse brain and in maturing cultured astrocytes. Neurochem Int 61:1325–1332
Liao SL, Chen CJ (2003) l-glutamate decreases glucose utilization by rat cortical astrocytes. Neurosci Lett 348:81–84
Lindsay KJ, Du J, Sloat SR, Contreras L, Linton JD, Turner SJ, Sadilek M, Satrústegui J, Hurley JB (2014) Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Natl Acad Sci USA 111:15579–15584
Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, Satrústegui J (2013) Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci 33:13957–13971
Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J (2015) The regulation of neuronal mitochondrial metabolism by calcium. J Physiol 593:3447–3462
Llorente-Folch I, Rueda CB, Pérez-Liébana I, Satrústegui J, Pardo B (2016) l-lactate-mediated neuroprotection against glutamate-induced excitotoxicity requires ARALAR/AGC1. J Neurosci 36:4443–4456
Loaiza A, Porras OH, Barros LF (2003) Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J Neurosci 23:7337–7342
London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC Jr (2007) Mutation in glicerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116:2260–2268
Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, Almeida A, Bolaños JP (2016) Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. PNAS 113:13063–13068
McDonnald MJ, Marshall LK (2000) Mouse lacking NAD1-linked glycerol phosphate dehydrogenase has normal pancreatic beta cell function but abnormal metabolite pattern in skeletal muscle. Arch Biochem Biophys 384:143–153
Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martín A, Romero-Gómez I, Baeza-Lehnert F, Lengacher S, Schneider BL, Aebischer P, Magistretti PJ, Barros LF, Weber B (2016) In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23:94–102
Mármol P, Pardo B, Wiederkehr A, del Arco A, Wollheim CB, Satrústegui J (2009) Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells. J Biol Chem 284:515–524
McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U (2006) Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol 71:399–407
Nguyen NH, Brathe A, Hassel B (2003) Neuronal uptake and metabolism of glycerol and the neuronal expression of mitochondrial glycerol-3-phosphate dehydrogenase. J Neurochem 85:831–842
Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrústegui J, Palmieri F (2001) Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J 20:5060–5069
Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrústegui J (2006) Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem 281:1039–1047
Pardo B, Rodrigues TB, Contreras L, Garzón M, Llorente-Folch I, Kobayashi K, Saheki T, Cerdan S, Satrústegui J (2011) Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab 31:90–101
Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Natl Acad Sci USA 91:10625–10629
Pellerin L, Magistretti PJ (2012) Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32:1152–1166
Peng L, Swanson RA, Hertz L (2001) Effects of l-glutamate, d-aspartate, and monensin on glycolytic and oxidative glucose metabolism in mouse astrocyte cultures: further evidence that glutamate uptake is metabolically driven by oxidative metabolism. Neurochem Int 38:437–443
Ramos M, del Arco A, Pardo B, Martínez-Serrano A, Martínez-Morales JR, Kobayashi K, Yasuda T, Bogónez E, Bovolenta P, Saheki T, Satrústegui J (2003) Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res Dev Brain Res 143:33–46
Rimmele TS, de Castro Abrantes H, Wellbourne-Wood J, Lengacher S, Chatton JY (2018) Extracellular potassium and glutamate interact to modulate mitochondria in astrocytes. ACS Chem Neurosci 9:2009–2015
Rueda CB, Llorente-Folch I, Amigo I, Contreras L, González-Sánchez P, Martínez-Valero P, Juaristi I, Pardo B, del Arco A, Satrústegui J (2014) Ca2+ regulation of mitochondrial function in neurons. Biochim Biophys Acta 1837:1617–1624
Rueda CB, Traba J, Amigo I, Llorente-Folch I, González-Sánchez P, Pardo B, Esteban JA, del Arco A, Satrústegui J (2015) Mitochondrial ATP-Mg/Pi carrier SCaMC-3/Slc25a23 counteracts PARP-1-dependent fall in mitochondrial ATP caused by excitotoxic insults in neurons. J Neurosci 35:3566–3581
Ruminot I, Gutiérrez R, Peña-Münzenmayer G, Añazco C, Sotelo-Hitschfeld T, Lerchundi R, Niemeyer MI, Shull GE, Barros LF (2011) NBCe1 mediates the acute stimulation of astrocytic glycolysis by extracellular K+. J Neurosci 31:14264–14271
Satrústegui J, Pardo B, Del Arco A (2007) Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev 87:29–67
Satrustegui J, Bak LK (2015) Fluctuations in cytosolic calcium regulate the neuronal malate-aspartate NADH shuttle. Implications for neuronal energy metabolism. Neurochem Res 40:2425–2430
Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M (2015) Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci 18:1819–1831
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte–neuron lactate transport is required for long-term memory formation. Cell 144:810–823
Swanson RA, Yu AC, Chan PH, Sharp FR (1990) Glutamate increases glycogen content and reduces glucose utilization in primary astrocyte culture. J Neurochem 54:490–496
Tretter L, Adam-Vizi V (2012) High Ca2+ load promotes hydrogen peroxide generation via activation of α-glycerophosphate dehydrogenase in brain mitochondria. Free Radic Biol Med 53:2119–2130
Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ (2007) Molecular and functional characterization of novel glicerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116:2253–2259
Vicente-Gutierrez C, Bonora N, Bobo-Jimenez V, Jimenez-Blasco D, Lopez-Fabuel I, Fernandez E, Josephine C, Bonvento G, Enriquez JA, Almeida A, Bolaños JP (2019) Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat Metab 1:201–211
Xu Y, Ola MS, Berkich DA, Gardner TW, Barber AJ, Palmieri F, Hutson SM, LaNoue KF (2007) Energy sources for glutamate neurotransmission in the retina: absence of the aspartate/glutamate carrier produces reliance on glycolysis in glia. J Neurochem 101:120–131
Funding
This work has been funded by grants from the Spanish Ministry of Science, Innovation and Universities SAF2014-56929R (to JS and BP), SAF2017-82560-R (to AdelA and BP). By a grant from Fundación Ramón Areces (to JS). This work has also be funded by the CIBERER, an initiative from the ISCIII, and by an institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa. I J and I P-L are recipient of a predoctoral fellowship from Gobierno Vasco and MINECO, P G-S is the recipient of a postdoctoral research contract from Comunidad de Madrid.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special Issue: In Honor of Prof. Vera Adam-Vizi.
Rights and permissions
About this article

Cite this article
Juaristi, I., Contreras, L., González-Sánchez, P. et al. The Response to Stimulation in Neurons and Astrocytes. Neurochem Res 44, 2385–2391 (2019). https://doi.org/10.1007/s11064-019-02803-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02803-7