
Abstract
Our group previously reported that 6-h fasting increased both insulin II mRNA expression and insulin level in rat hypothalamus. Given that insulin effects on central glucose metabolism are insufficiently understood, we wanted to examine if the centrally produced insulin affects expression and/or regional distribution of glucose transporters, and glycogen stores in the hypothalamus during short-term fasting. In addition to determining the amount of total and activated insulin receptor, glucose transporters, and glycogen, we also studied distribution of insulin receptors and glucose transporters within the hypothalamus. We found that short-term fasting did not affect the astrocytic 45 kDa GLUT1 isoform, but it significantly increased the amount of endothelial 55 kDa GLUT1, and neuronal GLUT3 in the membrane fractions of hypothalamic proteins. The level of GLUT2 whose presence was detected in neurons, ependymocytes and tanycytes was also elevated. Unlike hepatic glycogen which was decreased, hypothalamic glycogen content was not changed after 6-h fasting. Our findings suggest that neurons may be given a priority over astrocytes in terms of glucose supply even during the initial phase of metabolic response to fasting. Namely, increase in glucose influx into the brain extracellular fluid and neurons by increasing the translocation of GLUT1, and GLUT3 in the cell membrane may represent the first line of defense in times of scarcity. The absence of co-localization of these membrane transporters with the activated insulin receptor suggests this process takes place in an insulin-independent manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The metabolic demands of the brain amount to 20% of the body’s overall energy consumption [1]. Since brain has modest energy reserves, neurometabolism is highly dependent on a continuous supply of glucose from the systemic circulation. Glucose enters the brain through glucose transporter 1 (GLUT1), which is highly expressed along the blood brain barrier (BBB). In the BBB, there are two isoforms of GLUT1. They are both encoded by the same gene but differ in molecular weight due to posttranslational modifications [2, 3]. The glycosylated isoform (55 kDa) of GLUT1 is expressed in BBB endothelial cells, while the other, lower molecular weight one (45 kDa) is located in astrocyte end-feet [3]. The latter is also present in ependymal cells surrounding cerebral ventricles and choroid plexus [4].
Another abundantly expressed GLUT in the central nervous system (CNS) is GLUT3 [3, 5,6,7]. GLUT3 is exclusively expressed in neurons [8] and has high glucose affinity and transport capacity [9, 10]. These features of GLUT3 ensure a continuous influx of glucose into neurons even if the concentration of glucose in interstitial fluid is low.
To a lesser extent, other GLUT isoforms are also present in the CNS. For example, GLUT2 is detected in various brain regions including the paraventricular nucleus (PVN), lateral hypothalamus, nucleus arcuatus and the nucleus tractus solitarius [11] where it was detected in neurons, astrocytes, endothelial cells and tanycytes [12, 13]. Just like in pancreatic β cells, GLUT2 in the hypothalamus is considered to act as ‘glucose sensor’ [14]. GLUT4 is mainly found in cerebellum [15], but is also detected in hypothalamus, thalamus, olfactory bulb and cerebral cortex [5]. The overlapping distribution of GLUT4 and insulin receptor in various brain regions suggests that GLUT4 may be dependent on insulin [16].
Traditional view of neuronal oxidative glucose metabolism was questioned when the astrocyte-neuron lactate shuttle hypothesis was proposed [17, 18]. According to this hypothesis astrocytes take up more glucose than needed for their energy demands and produce lactate which is released in extracellular space through monocarboxylate transporter (MCT) 1 and 4. Lactate is taken up by neurons via MCT2 and used an energy substrate [17, 19]. However, neurons too take up glucose and use for oxidative metabolism. It was shown that neuronal glucose and lactate transporters (GLUT3 and MCT2) have higher affinity for their substrates than astrocytic ones (GLUT1, MTC1 and MTC4) [20]. Additionally, some reports suggest there is a larger glucose transport capacity in neurons than in astrocytes [9]. Lundgaard and colleagues showed that glucose is taken up preferentially by neurons during increased neural activity [21]. Besides, they showed that neurons express higher levels of hexokinases than astrocytes in mouse and human brain [21]. The similar results were obtained using metabolic biosensors in hippocampal slices and brains of awake mice [22].
Even though the brain has a moderate capacity to store energy, glycogen represents an important short-term energy source that can be exploited in times of disturbed energy homeostasis. Despite the fact that neurons express all the necessary enzymes to produce glycogen, this process in the brain seems to be limited to astrocytes [23, 24]. Moreover, glycogen accumulation in neurons was shown to induce apoptosis [25]. Therefore, under normal physiological conditions this process is suppressed, but aging enhances glycogen synthesis in neurons [26].
Glycogen turnover in astrocytes is constantly modified to match local energy status. Under conditions of limited energy availability, astrocytes break down glycogen to lactate, which is subsequently released into the brain extracellular fluid (bECF) and thus made available to neurons. In this manner, glycogen depots in astrocytes preserve neuronal function and viability in states of hypoglycemia [19, 27,28,29]. Utilization of astrocyte glycogen can also occur in normal physiological conditions to support increased energy demand during enhanced neuronal activity [24, 30]. It is noteworthy that insulin takes part in controlling glycogen metabolism in astrocytes. An increase in brain glycogen content was observed in rats 4 h following intravenous insulin administration [27].
Our group has previously reported that 6 h of fasting increases both insulin II mRNA expression and (pro)insulin content in hypothalamic parenchyma [31]. This was the first published report of an increase in insulin expression in a fasting state. It was also shown that, unlike the circulating glucose, the cerebrospinal fluid (CSF) glucose levels were not decreased after short-term food deprivation. Based on this finding and the fact that undisturbed glucose supply to the brain is of pivotal importance, we hypothesized that the purpose of fasting-induced increase in hypothalamic insulin expression was to maintain central glucose homeostasis. In particular, our goals were to determine the effects of short-term fasting on the hypothalamic expression and activation of insulin receptors, expression and regional distribution of glucose transporters, and glycogen stores.
Materials and Methods
Experimental Design
Two-month old male Wistar rats weighing around 250 g were housed two per cage in controlled light–dark condition (12 h light–12 h darkness) and under the constant ambient temperature of 21 ± 1 °C. One group of animals was subjected to 6-h fasting (6 h F), while the control rats had unlimited access to food. All animals had unrestricted access to tap water. Considering that rats are nocturnal, food was removed at 6 pm and both fasting and control rats were sacrificed at midnight. Food intake was measured from 6 pm to 12 am. Average food intake for control animals was 5.41 ± 0.65 g/6 h. The total number of animals used per group was n = 15 (n = 6 rats for western blotting, n = 6 animals for glycogen quantification and n = 3 animals for immunofluorescence and PAS staining).
All procedures were performed according to the rules for animal care proposed by the Federation of European Laboratory Animal Science. Project was approved by the Ethics Committee of the Faculty of Biology, University of Belgrade (Permit No. EK-BF-2017/05) and Serbian Ministry of Agriculture, Forestry and Water Management, Veterinary Directorate (Permit No. 323-07-05119/2017-05).
Cerebrospinal Fluid and Tissue Sampling
After intraperitoneal anesthesia with the mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg), rats were transcardially perfused with a 0.9% NaCl. Cerebrospinal fluid was aspirated from the cisterna magna with a glass micropipette (Drummond Scientific, Broomall, PA) as previously described [31] and lactate levels were measured with Accutrend® Plus system (Cobas, Roche, Mannhein, Germany). After each decapitation, brain was removed, and placed ventral side up in order to dissect out hypothalamus. Livers were also collected and the tissue was frozen at − 80 °C until further use for Western blot and tissue glycogen assay.
Western Blot
The hypothalamic membrane protein fraction was isolated using Subcellular Protein Fractionation Kit for Tissues (Thermo Scientific, cat.87790, USA) according to the manufacturer’s instructions. Total protein concentration was measured by the Lowry method [32].
Samples from individual animals of each experimental group were mixed so that they were equally represented with the same amount of protein. Protein lysates were then diluted by 2 × Laemmli solution sample buffer (4% SDS, 0.02% bromophenol blue, 20% glycerol, 125 mM Tris–HCl) containing 5% β-mercaptoethanol. Between 20 and 40 µg protein was loaded per well. Following electrophoresis on a 12.5% SDS-PAGE gel, proteins were transferred to polyvinylidene fluoride membrane (Bio-Rad, USA) overnight at + 4 °C, current 20 mA per gel. Membranes were blocked for 2 h in 3% non-fat dry milk (Santa Cruz Biotechnology, USA) in Tris-buffered saline containing 0.1% Tween-20 (TBST). Membranes probed with anti-insulin receptor antibody were exceptionally blocked with 5% Bovine Serum Albumin (BSA) (Sigma, USA). All primary antibodies were diluted in the blocking solution, incubated overnight at + 4 °C, rinsed in TBST and ultimately incubated with a proper HRP-conjugated secondary antibody. List of all antibodies is given in Table 1. Chemiluminescent blots were developed using ECL+ solution (Bio-Rad, USA) and captured with ChemiDoc-It Imager (Ultra-Violet Products Ltd, Cambridge, UK). Removal of primary and secondary antibodies was performed with a mild stripping buffer (pH 2.2) containing 0.2 mM glycine, 0.1% SDS and 1% Tween 20. The membranes were subsequently reprobed with anti-β-actin antibody for the purpose of a loading control. Quantification was performed in Image J program (version 1.47 m, National Institute for Health, USA). Measured optical density (OD) for target proteins was normalized according to corresponding OD of the loading control, then additional normalization to control group was performed.
Tissue Glycogen Assay
Total glycogen was extracted according to the method of Rasouli and colleagues [33]. Livers and hypothalami were measured and then digested in 30% KOH solution at 100 °C for 10 min. Samples were cooled to room temperature, ethanol was added at final concentration of 55%, and the mixture was centrifuged for 10 min at 1700 g (Sorvall, SL-50T, Super T21, Thermo Fisher Scientific). The pellet was resuspended in either 1 ml (hepatic tissue) or 100 µl (hypothalamic tissue) of distilled water. In order to determine the glycogen level we added to each well either 10 µl of standard glucose solution (5.44 mmol/l) or sample, 30 µl of water, 40 µl of 6.5% phenol and 200 µl of concentrated sulfuric-acid. The content in each well was mixed, incubated for 30 min at room temperature and absorbance was measured by microplate reader (LKB 5060-006) at 492 nm [34].
Immunofluorescence and Periodic Acid–Schiff Staining
Rats were anesthetized and perfused with 0.9% NaCl as described above and each brain was fixed with 4% paraformaldehyde (PFA) (Sigma, USA). Following an overnight post-fixation in PFA at + 4 °C, the tissue was cryoprotected in increasing concentration of sucrose solution (10%, 20%, 30%) in 0.1M phosphate buffer. Brains were frozen at − 70 °C and cut into 20 µm or 10 µm thick coronal section (for immunofluorescence or PAS staining, respectively) on cryostat Leica CM1850. Slices were placed on SuperFrost® slides (Menzel-Glasser, Germany) and stored at + 4 °C until further use.
For immunofluorescent staining, sections were washed several times in 0.01 M phosphate buffered saline (PBS), and then antigen retrieval was performed with 10 mM Sodium citrate buffer pH 6.0. After cooling to room temperature, sections were incubated with 0.1% Triton-X 100 for 15 min. Blocking was performed in 2% BSA in 0.01 M PBS for 45 min. Primary and secondary antibodies were diluted in the same solution. Primary antibodies were incubated overnight at + 4 °C. Negative control sections were prepared by omitting the addition of primary antibody. Following this incubation, sections were rinsed in PBS and incubated for 2 h with fluorescent labeled secondary antibodies. Nuclei were stained with DAPI (1:4000). The sections were washed with PBS several times and mounted with Mowiol (Sigma-Aldrich). Confocal laser microscope system LSM510 (Carl Zeiss GmbH, Jena, Germany) was used for imaging. Argon laser was utilized for excitation of Alexa 488 and helium–neon laser for Alexa 555 with excitation wavelength set at 488 nm and 543 nm, respectively.
The presence of glycogen in the hypothalamic tissue was determined using PAS staining, with diastase digestion as a confirmation of the specificity of staining. After rinsing with distilled water, brain sections were incubated with diastase for 40 min at room temperature, and then incubated with periodic acid for 10 min, and with Schiff’s reagent for 2 min. Following the counterstaining with hematoxylin, sections were thoroughly rinsed under tap water and mounted with DPX (Sigma).
Data Analysis
Statistical evaluation was performed by GraphPad Prism software (version 6.01). Student t test was used for comparison of the experimental groups. The values were expressed as means ± SEM and the level of significance was set at p < 0.05.
Results
The Effect of Short-Term Fasting on Hypothalamic Expression, Activation and Distribution of Insulin Receptors
Western blot analysis of total insulin receptor (IR) and insulin receptor phosphorylated at tyrosine residue 1361 (IRp) showed that six hour fasting significantly increased the content of both IR (Fig. 1a, **P < 0.001) and IRp (Fig. 1b, ***P < 0.0001). Representative Western blot probed with insulin-receptor, phosphorylated-insulin receptor and β-actin (as loading control) are shown it the Fig. 1c. The hypothalamic ratio of IRp/IR was also increased in the fasted state (Fig. 1d, ***P = 0.006).

Relative levels of insulin receptor and phosphorylated-insulin receptor content in hypothalamus of control and fasted animals. a Insulin receptor expression was significantly increased in fasting rats as compared to control: **P = 0.0053, b also phosphorylated-insulin content was significantly increased: ***P < 0.0001. c Representative Western blot probed with insulin-receptor, phosphorylated-insulin receptor and β-actin (as loading control). d Also the ratio of IRp/IR was significantly increased after short term fasting: P = 0.0006. All blots were normalized to β-actin. Data are expressed as mean ± SEM (n = 6 per group). C control group; 6 h F 6-h fasting group, IR insulin receptor, IRp phosphorylated insulin receptor
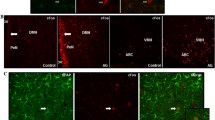
Immunofluorescence analysis was used to obtain additional information regarding the location of IR in the hypothalamus. Phosphorylated insulin receptor immunoreactivity was detected in the periventricular nucleus (PeV), both at the roof and floor of the third ventricle (3v) (Fig. 2a), as well as in the vicinity of the nucleus arcuatus (Fig. 2b) of fasted animals. In control rats, IRp immunoreactivity could be observed at the top of the 3v (Fig. 2c).

Immunofluorescence localization of phosphorylated-insulin receptor in the hypothalamus of control and fasted rats. a Phosphorylated-insulin receptor immunoreactivity was found at top of third ventricle in the periventricular region of the hypothalamus, b but also at bottom of 3v. c In control rats IRp-immunoreactivity was observed only at top of third ventricle in the periventricular nucleus of the hypothalamus. C control group, 6 h F 6-h fasting group, IR insulin receptor, IRp phosphorylated insulin receptor, 3v the third ventricle
The Effect of Short-Term Fasting on Hypothalamic Expression and Distribution of Glucose Transporters
In order to investigate the amount of functional forms of glucose transporters, hypothalamic membrane fractions were analyzed. After short term fasting, expression of GLUT1 and GLUT3, two major glucose transporters in the hypothalamus, was increased (Fig. 3a, **GLUT1 P = 0.0099; Fig. 3e, GLUT3, ***P < 0.0001). Additionally, GLUT1 forms were separately analyzed and a significant increase was recorded only in the microvascular 55 kDa GLUT1 isoform (Fig. 3c, **P = 0.0015), but not in the astrocytic 45 kDa GLUT1 isoform (Fig. 3b, P = 0.171). The amount of GLUT2 was also increased after 6-h food deprivation (Fig. 3d, **P = 0.0073), while the expression of insulin-sensitive GLUT4 remained unchanged (Fig. 3f, P = 0.3112). The representative blots of different GLUT isoforms as well as loading control are shown on the Fig. 3g.

Relative levels of glucose transporters in the hypothalamic membrane protein fraction of control and fasted animals. a GLUT1 expression in membrane fraction of hypothalamus was increased after short term fasting: **P = 0.0099, b but additional analysis of different GLUT1 isoforms showed that increase is only in microvascular 55 kDa isoform: **P = 0.0015, c but not in astrocytic 45 kDa isoform: P = 0.171. d Expression of GLUT2 in membrane fraction was increased in fasted animals in comparison to control: **P = 0.0073. e GLUT3 showed similar increase in protein content: ***P < 0.0001, f while GLUT4 expression remain unchanged after short-term food deprivation: P = 0.3112. g Representative Western blots. All blots were normalized to β-actin. Data are expressed as mean ± SEM (n = 6 per group). C control group, 6 h F 6-h fasting group
The distribution of various glucose transporters was analyzed by immunofluorescence. GLUT1-immunoreactivity was found in the parenchyma of the entire hypothalamus of both control and fasted animals, as well as in the ependymocytes (Fig. 4b) and tanycytes surrounding the third ventricle (Fig. 4a, d). The GLUT1-immunoreactivity was also observed in endothelial cells of blood vessels (Fig. 4a, c), which were surrounded with astrocyte end-foot processes (Fig. 4c). At the floor of the 3v, GLUT1-immunopositivity was detected in the apical membrane of tanycytes (Fig. 4a, d).

Hypothalamic regional distribution of GLUTs in fasted animals. GLUT1-positivity was found in the entire hypothalamic section of both fasting and control animals. GLUT1-immunoreactivity was observed in ependymal cells (b) and tanycytes (a, d) surrounding the third ventricle. Tanycytes express GLUT1 at apical membrane (d). GLUT1-positive endothelial cells (a white arrows) were surrounded with astrocyte end-foot processes (c). GLUT2-positive neurons were detected throughout the hypothalamus, especially in the paraventricular nucleus (e) and in the ventromedial hypothalamus (f). Ependymocytes lining the third ventricle showed GLUT2-immunoreactivity (g). Vicinal GLUT2 positive neurons seem to be in contact with tanycytes processes (h insert-white arrows) which also express GLUT2 (h). GLUT3-immunoreactivity was seen in plasma membrane and neuronal processes (i) in the subventricular area of hypothalamus, particularly in the region of periventricular and paraventricular nucleus (k). GLUT3-positive neuronal processes seems to be in contact with expansions of GFAP-positive astrocytes (j). GLUT4-positivity was found in ependymal cells along the cerebral ventricles (l), but also in hypothalamic parenchyma (m)
GLUT2-positivity was detected in NeuN-positive cells in the paraventricular nucleus (PVN) (Fig. 4e) and ventromedial hypothalamus (VMH) (Fig. 4f). Ependymocytes and tanycytes were also immunoreactive to GLUT2 (Fig. 4g, h) whose processes seem to be in contact with GLUT2-positive neurons detected in the nucleus arcuatus (Fig. 4h insert).
GLUT3-positivity was detected in the subventricular area of hypothalamus, particularly in the region of the PeV and PVN (Fig. 4k). GLUT3-immunoreactivity was seen in plasma membrane and neuronal processes (Fig. 4i), which seems to be in contact with extensions of GFAP-positive astrocytes; however, there is no co-localization of GLUT3-positivity with GFAP (Fig. 4j).
Lastly, GLUT4-immunoreactivity was seen in ependymal cells (Fig. 4l) and sporadically throughout the hypothalamic parenchyma (Fig. 4m).
Double-labeled immunofluorescence revealed there was no major co-localization of IRp with either GLUT1, GLUT2 or GLUT3 in the PeV (Fig. 5a–f). However, some co-localization of IRp with certain glucose transporters was sporadically observed [e.g. GLUT2 at the roof (Fig. 5c, d) and GLUT3 at the floor of 3v, (Fig. 5e, f)].

Double immunofluorescence labeling of phosphorylated insulin receptor and various GLUTs in fasted rats. Neither GLUT1 (a, b), GLUT2 (c, d) or GLUT3 (e, f) was not co-localized with phosphorylated insulin receptor. However, some overlapping of signals detected for GLUT4 and phosphorylated insulin receptor was observed in ependymal cells of 3v (g white arrows)
The Effect of Short-Term Fasting on Hepatic and Hypothalamic Glycogen Content and Distribution
Short term food deprivation did not affect glycogen content in the hypothalamus (Fig. 6b, P = 0.8276), despite the fact that hepatic glycogen was reduced by 57% in fasted animals (Fig. 6a, *P = 0.0224). Lactate levels in the CSF were used as a measure of glycogen degradation rate. We found that this remained unchanged after 6 h of fasting. (Fig. 6c, P = 0.2435). PAS staining showed glycogen was homogeneously distributed throughout hypothalamus, and no difference in glycogen regional distribution was observed between the control (Fig. 7a) and fasted animals (Fig. 7c). Pretreatment of sections with diastase to digest glycogen was used to confirm the specificity of staining and reduced staining was observed (Fig. 7b, d).

Glycogen and lactate content of control and fasted animals. a Hepatic glycogen content was decreased: *P = 0.0224, b but hypothalamic was not changed after 6 h food removal: P = 0.8214. c Lactate concentration in cerebrospinal fluid remain unchanged: P = 0.2435. Data are expressed as mean ± SEM (n = 6 per group). C control group, 6 h F 6-h fasting group, HPT hypothalamus, CSF cerebrospinal fluid

Regional distribution of glycogen in the hypothalamus control and fasted rats. a, c PAS staining of hypothalami section showed homogeneous distribution of glycogen in the hypothalamus of both control and fasted rats. Difference in glycogen distribution between control and fasted animals was not observed (Orig. magnification × 5). b, d Pretreatment with diastase was used to confirm specificity of PAS staining. e, f At × 20 magnification glycogen staining in the hypothalamus. g × 40 magnification. Black arrows pointing to cells with intense glycogen staining (e–g). C control group, 6 h F 6-h fasting group
Discussion
As glucose is the main source of energy for the brain, a sufficient glucose supply must be available at all times. Consequently, numerous counter-regulatory responses have developed in order to protect neuronal tissue from sudden fluctuations of circulating glucose levels [35]. With this in mind, we wanted to determine if short-term fasting induced increase in hypothalamic insulin expression [31] may serve a purpose of maintaining glucose homeostasis in this brain region. In an effort to do so, we first checked whether increased hypothalamic insulin expression coincided with an increase in the activation of IR in the hypothalamus. Our results showed that the levels of both total and IR phosphorylated at tyrosine (Tyr) residue 1361 (IRp) were significantly upregulated in membrane fractions of hypothalamic proteins after 6-h fasting. Phosphorylation of IR at Tyr 1361 mediates the pleiotropic effects of insulin on target cells [36] and is considered to be a universal hallmark of this receptor’s activation. Additionally, the ratio of IRp/IR was also elevated which further suggests that increase in the activation of IR overlapped in time with an increase in locally produced insulin. In regard to its distribution, IRp immunoreactivity was detected at the roof of the third ventricle in the region of PeV (Fig. 2a) and in the part of nucleus arcuatus near the third ventricle (Fig. 2b). This observation is in agreement with previous reports of IR being expressed in brain regions such as hypothalamus, hippocampus, olfactory bulb, and cerebellum. In the hypothalamus, conspicuous signal was observed in dorsomedial and ventromedial parts of nucleus arcuatus [37,38,39], but some IR-immunoreactivity was also detected the PeV [39, 40]. The fact that in our study both insulin [31] and IRp reactivity was detected in PeV suggests that centrally produced insulin most likely acts in a paracrine fashion.
We subsequently wanted to examine if a connection might be established between the increase in the IR activation and the expression of various GLUTs’ isoforms. Although the GLUT isoforms predominantly expressed in the brain (i.e., GLUT1 and GLUT3) are typically considered not to be dependent to the action of insulin [41, 42], it is known that glucose transporters can change their sensitivity to insulin. For example, in the resting state, insulin stimulates skeletal muscle glucose uptake by translocation of GLUT4 form the cytosol to the cell membrane [43]. However, GLUT4 translocation during exercise is achieved by mechanism which involves a variety of signaling molecules including AMPK and Ca2+, but is entirely independent of insulin action [44]. Moreover, there are studies which demonstrated that glucose transport and/or metabolism in some types of brain cells may be insulin-sensitive. Namely, it was shown that the 2-deoxy-d-glucose (2DG) uptake by glial cultures from the rat brain can be stimulated by insulin [45].
Considering these findings, we wanted to determine the effect of short-term fasting on the presence of various GLUT isoforms in the membrane fraction of hypothalamic proteins and their possible co-localization with IRp. Following the 6-h fasting period, the levels of two major brain glucose transporters, GLUT1 and GLUT3, were elevated. Different GLUT1 isoforms were analyzed separately and we found there was a significant increase in the amount of 55 kDa microvascular GLUT1 (Fig. 3c). However, the level 45 kDa GLUT1 found in astrocytes remained unchanged (Fig. 3b).
We next examined whether short term fasting, in addition to increasing their expression, affected the distribution of GLUT1 and GLUT3 in the hypothalamus as well. GLUT1-immunoreactivity was homogeneously distributed throughout the entire hypothalamic sections of both control and fasting animals. In addition to the hypothalamic parenchyma, GLUT1-immunoreactivity was observed in endothelial cells of blood vessels (Fig. 4a, c). GLUT3 positivity was detected in the subventricular area of hypothalamus, particularly in the PeV and PVN (Fig. 4k). However, double-labeling immunofluorescence did not show any significant co-localization of IRp with either GLUT1 or GLUT3 in any of the hypothalamic regions (Fig. 5a, e). Even though the lack of co-localization of the examined GLUTs with IRp in the hypothalamus suggests that short-term fasting does not affect their sensitivity to insulin, we thought it important to further explore the adaptive significance of their increased expression under the examined circumstances.
In order to enter the brain bECF, glucose must first cross the BBB. This process is mediated by GLUT1, which ensures constant glucose influx into brain independent of blood glucose concentration [46, 47]. As previously mentioned, it was the microvascular 55-kDa GLUT1, and not the astrocytic 45-kDa form that was upregulated after 6 h of fasting. This is in agreement with the results of Simpson and colleagues, who found a 23% increase in 55-kDa GLUT1, and no change in the levels of nonvascular 45-kDa GLUT1 during hypoglycemia [48]. The importance of this finding was also reflected in the fact that, even though blood glucose was significantly decreased, the CSF glucose levels remained unaffected by fasting [31]. Taken together, these data indicate that enhanced glucose transport across the BBB due to GLUT1 upregulation, contributes to the maintenance of the CSF glucose which is pivotal for undisturbed brain function in the state of compromised energy homeostasis.
In addition to endothelial cells, ependymal cells also have an important role in the acquisition of glucose by the CNS. Owing to the fact that they express both GLUT1 [49] and GLUT2 [13], these cells are involved in glucose sensing in the hypothalamus. Our results showed not only that levels of both GLUT1 and GLUT2 were increased, but they were also detected in ependymocytes and tanycytes surrounding the third ventricle (GLUT1: Fig. 4a, b, d; GLUT2: Fig. 4g, h). The fact that upregulated GLUTs were found in ependymal cells further reinforces the concept of these cells acting as a “bridge” between blood and the brain interstitial fluid [50], which in this case helps to maintain adequate glucose supply to the CSF.
After entering the bECF, glucose can be used by neurons and glial cells. Neurons take up glucose via GLUT3 and primarily use it as a source of pyruvate to fuel mitochondrial oxidative metabolism to produce ATP. On the other hand, astrocytes take up glucose via 45-kDa GLUT1 and use it for glycolysis and glycogen synthesis [51]. In our study, the 45-kDa GLUT1 isoform remained unchanged. On the other hand, the expression of neuronal GLUT3 was increased. Based mainly on mathematical modeling, it is suggested that neurons have a higher glucose transport capacity relative to astrocytes [19]. Additionally, an in vitro study showed that during glucose deprivation, the uptake of 2-DG in neuronal cells increased, whereas that of astrocytes did not change [52]. It is worth mentioning that, in addition to GLUT3, neurons express GLUT2, whose expression was also found to be upregulated in this study. GLUT2-positive neurons were detected throughout hypothalamus in the PVN and in the ventromedial hypothalamus (Fig. 4e, f). Taken together, these findings suggest that, due to their high sensitivity even to minor threats to energy homeostasis [53], neuronal glucose supply represents a priority even during short term fasting.
Insulin-sensitive GLUT4 appeared not to be affected by 6-h fasting. The GLUT4-positivity was found in ependymal cells along cerebral ventricles, and sporadically in some neurons (Fig. 4o, p). However, its levels in the membrane fractions of hypothalamic proteins did not change, which once again implies that locally produced insulin in the fasting state is not involved in the regulation of glucose uptake.
Brain glycogen stores are an important source of energy used during periods of intense neuronal activity or hypoglycemia. Whenever energy demands exceed glucose supply, astrocytes break down glycogen to lactate, which is released into the CSF and thus made available to adjacent neurons to use as fuel [54]. Glycogen synthesis in liver and muscles is long known to be stimulated by insulin [55, 56]. However, less is known about the regulation of brain glycogen metabolism. Therefore we thought it would be useful looking into the effect of short-term fasting on the content of glycogen in the hypothalamus. Our results showed that hepatic glycogen content was significantly decreased after 6 h of food deprivation. Considering that serum cortisol and urea levels were not changed [57], this result indicates that rats were in the initial stage of the metabolic response to starvation at that point in time. Nonetheless, the importance of maintaining central glucose homeostasis was reflected in the fact that, unlike hepatic, hypothalamic glycogen levels remained unchanged after 6 h of fasting. This is in agreement with Garriga and Cusso, who also showed that 5-h fasting did not alter glycogen content of not only the hypothalamus, but the cerebellum, cortex, striatum, and hippocampus as well [58]. Considering the fact that brain glycogen synthesis and degradation can occur simultaneously [54], we analyzed the CSF lactate levels as a measure of glycogen degradation. We found that the concentration of lactate in the CSF did not change after 6-h food deprivation. This result further indicates that glycogen turnover was not affected by short-term fasting.
Even though fasting did not affect total glycogen hypothalamic content, we wanted to explore whether there were any differences in the distribution of glycogen between control and fasted animals. The results of the PAS staining confirmed that glycogen was homogeneously distributed throughout the hypothalamic region of both control and fasted animals and no regional differences were observed.
In summary, we found that short-term fasting did not affect the expression of the astrocytic 45 kDa GLUT1 isoform. On the other hand, there was a significant increase in the expression of endothelial 55 kDa GLUT1 and neuronal GLUT3 isoforms which facilitate glucose diffusion into the bECF and neuronal cytosol respectively. The expression of GLUT2 detected in both neurons and ependymal cells was also elevated. From a teleological point of view these findings suggest that, due to their high sensitivity to changes in energy homeostasis, neurons may be given priority over astrocytes in terms of glucose supply in times of scarcity. The fact the hypothalamic glycogen content was not decreased by short-term fasting additionally points to the conclusion that the increase in glucose influx into the bECF and neurons by an increase in GLUT1 and GLUT3 expression represents a first line of defense in the initial phase of metabolic response to food deprivation. The absence of co-localization of these membrane transporters with the activated IR suggests that this process takes place in an insulin-independent manner. Further analysis of downstream signaling pathways linked to insulin activity may reveal a role of de novo insulin synthesized in the hypothalamus during short-term fasting.
References
Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36:587–597. https://doi.org/10.1016/j.tins.2013.07.001
Sivitz W, DeSautel S, Walker PS, Pessin JE (1989) Regulation of the glucose transporter in developing rat brain. Endocrinology 124:1875–1880. https://doi.org/10.1210/endo-124-4-1875
Maher F, Vannucci SJ, Simpson IA (1993) Glucose transporter isoforms in brain: absence of GLUT3 from the blood–brain barrier. J Cereb Blood Flow Metab 13:342–345. https://doi.org/10.1038/jcbfm.1993.43
Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA (1998) GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res 797:1–11. https://doi.org/10.1016/S0006-8993(98)00103-6
Choeiri C, Staines W, Messier C (2002) Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience 111:19–34. https://doi.org/10.1016/S0306-4522(01)00619-4
Nagamatsu S, Sawa H, Kamada K, Nakamichi Y, Yoshimoto K, Hoshino T (1993) Neuron-specific glucose transporter (NSGT): CNS distribution of GLUT3 rat glucose transporter (RGT3) in rat central neurons. FEBS Lett 334:289–295. https://doi.org/10.1016/0014-5793(93)80697-S
Gómez O, Ballester-Lurbe B, Poch E, Mesonero JE, Terrado J (2010) Developmental regulation of glucose transporters GLUT3, GLUT4 and GLUT8 in the mouse cerebellar cortex. J Anat 217:616–623. https://doi.org/10.1111/j.1469-7580.2010.01291.x
Maher F, Davies-Hill TM, Lysko PG, Henneberry RC, Simpson IA (1991) Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: evidence for neuron-specific expression of GLUT3. Mol Cell Neurosci 2:351–360. https://doi.org/10.1016/1044-7431(91)90066-W
Simpson I, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27:1766–1791. https://doi.org/10.1038/sj.jcbfm.9600521
Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ (2008) The facilitative glucose transporter GLUT3: 20 years of distinction. AJP Endocrinol Metab 295:E242–E253. https://doi.org/10.1152/ajpendo.90388.2008
Leloup C, Arluison M, Lepetit N, Cartier N, Marfaing-Jallat P, Ferré P et al (1994) Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. Brain Res 638:221–226. https://doi.org/10.1016/0006-8993(94)90653-X
Penicaud L, Leloup C, Lorsignol A, Alquier T, Guillod E (2002) Brain glucose sensing mechanism and glucose homeostasis. Curr Opin Clin Nutr Metab Care 5:539–543
De los Angeles García M, Millán C, Balmaceda-Aguilera C, Castro T, Pastor P, Montecinos HH et al (2003) Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem 86:709–724. https://doi.org/10.1046/j.1471-4159.2003.01892.x
Koekkoek LL, Mul JD, la Fleur SE (2017) Glucose-sensing in the reward system. Front Neurosci. https://doi.org/10.3389/fnins.2017.00716
Kobayashi M, Nikami H, Morimatsu M, Saito M (1996) Expression and localization of insulin-regulatable glucose transporter (GLUT4) in rat brain. Neurosci Lett 213:103–106. https://doi.org/10.1016/0304-3940(96)12845-7
Duelli R, Kuschinsky W (2001) Brain glucose transporters: relationship to local energy demand. News Physiol Sci 16:0–5
Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci 91:10625–10629. https://doi.org/10.1073/pnas.91.22.10625
McCall AL (2004) Cerebral glucose metabolism in diabetes mellitus. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2004.02.052
Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. https://doi.org/10.1016/j.cmet.2011.08.016
Barros LF, Deitmer JW (2010) Glucose and lactate supply to the synapse. Brain Res Rev. https://doi.org/10.1016/j.brainresrev.2009.10.002
Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JDR et al (2015) Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat Commun. https://doi.org/10.1038/ncomms7807
Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. https://doi.org/10.1016/j.cmet.2017.06.021
Inoue N, Matsukado Y, Goto S, Miyamoto E (1988) Localization of glycogen synthase in brain. J Neurochem 50:400–405. https://doi.org/10.1111/j.1471-4159.1988.tb02926.x
Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF et al (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab 34:945–955. https://doi.org/10.1038/jcbfm.2014.33
Vilchez D, Ros S, Cifuentes D, Pujadas L, Vallès J, García-Fojeda B et al (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10:1407–1413. https://doi.org/10.1038/nn1998
Drulis-Fajdasz D, Gizak A, Wójtowicz T, Wiśniewski JR, Rakus D (2018) Aging-associated changes in hippocampal glycogen metabolism in mice. Evidence for and against astrocyte-to-neuron lactate shuttle. GLIA. https://doi.org/10.1002/glia.23319
Choi IY, Seaquist ER, Gruetter R (2003) Effect of hypoglycemia on brain glycogen metabolism in vivo. J Neurosci Res 72:25–32. https://doi.org/10.1002/jnr.10574
Suh SW, Bergher JP, Anderson CM, Treadway JL, Fosgerau K, Swanson RA (2007) Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J Pharmacol Exp Ther 321:45–50. https://doi.org/10.1124/jpet.106.115550
Matsui T, Soya S, Okamoto M, Ichitani Y, Kawanaka K, Soya H (2011) Brain glycogen decreases during prolonged exercise. J Physiol 589:3383–3393. https://doi.org/10.1113/jphysiol.2010.203570
DiNuzzo M, Mangia S, Maraviglia B, Giove F (2012) The role of astrocytic glycogen in supporting the energetics of neuronal activity. Neurochem Res. https://doi.org/10.1007/s11064-012-0802-5
Dakic TB, Jevdjovic TV, Peric MI, Bjelobaba IM, Markelic MB, Milutinovic BS et al (2017) Short-term fasting promotes insulin expression in rat hypothalamus. Eur J Neurosci 46:1730–1737. https://doi.org/10.1111/ejn.13607
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin–Phenol reagent. J Biol Chem 193:265–275. https://doi.org/10.1016/0304-3894(92)87011-4
Rasouli M, Shokri-Afra H, Ostovar-Ravari A (2015) A new protocol for separation of acid soluble and insoluble fractions from total glycogen and simultaneous measurements. Eur Rev Med Pharmacol Sci 19:1785–1789
Rasouli M, Ostovar-Ravari A, Shokri-Afra H (2014) Characterization and improvement of phenol-sulfuric acid microassay for glucose-based glycogen. Eur Rev Med Pharmacol Sci 18:2020–2024
Mitrakou A, Ryan C, Veneman T, Mokan M, Jenssen T, Kiss I et al (1991) Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am J Physiol 260:E67–E74. https://doi.org/10.1152/ajpendo.1991.260.1.E67
Giudice J, Barcos LS, Guaimas FF, Penas-Steinhardt A, Giordano L, Jares-Erijman E et al (2013) Insulin and insulin like growth factor II endocytosis and signaling via insulin receptor B. Cell Commun Signal 11:18. https://doi.org/10.1186/1478-811X-11-18
Marks JL, Porte D, Stahl WL, Baskin DG (1990) Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 127:3234–3236. https://doi.org/10.1210/endo-127-6-3234
Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM et al (1987) Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 121:1562–1570. https://doi.org/10.1210/endo-121-4-1562
Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L (2002) Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 5:566–572. https://doi.org/10.1038/nn861
Unger J, McNeill TH, Moxley RT, White M, Moss A, Livingston JN (1989) Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 31:143–157. https://doi.org/10.1016/0306-4522(89)90036-5
Gray SM, Meijer RI, Barrett EJ (2014) Insulin regulates brain function, but how does it get there? Diabetes. https://doi.org/10.2337/db14-0340
Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A (2013) Insulin in the brain: sources, localization and functions. Mol Neurobiol. https://doi.org/10.1007/s12035-012-8339-9
Furtado LM, Somwar R, Sweeney G, Niu W, Klip A (2002) Activation of the glucose transporter GLUT4 by insulin. Biochem Cell Biol 80:569–578. https://doi.org/10.1139/o02-156
Richter E, Hargreaves M, Exercise (2013) GLUT4, and skeletal muscle glucose uptake. Physiol Rev 93:993–1017. https://doi.org/10.1152/physrev.00038.2012
Clarke DW, Boyd FT, Kappy MS, Raizada K (1984) Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J Biol Chem 259:11672–11675
McAllister MS, Krizanac-Bengez L, Macchia F, Naftalin RJ, Pedley KC, Mayberg MR et al (2001) Mechanisms of glucose transport at the blood-brain barrier: an in vitro study. Brain Res 904:20–30. https://doi.org/10.1016/S0006-8993(01)02418-0
Ngarmukos C, Baur EL, Kumagai AK (2001) Co-localization of GLUT1 and GLUT4 in the blood-brain barrier of the rat ventromedial hypothalamus. Brain Res 900:1–8. https://doi.org/10.1016/S0006-8993(01)02184-9
Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F et al (1999) Blood-brain barrier glucose transporter: effects of hypo- and hyper-glycemia revisited. J Neurochem 72:238–247. https://doi.org/10.1046/j.1471-4159.1999.0720238.x
Silva-Alvarez C, Carrasco M, Balmaceda-Aguilera C, Pastor P, García MDLA, Reinicke K et al (2005) Ependymal cell differentiation and GLUT1 expression is a synchronous process in the ventricular wall. Neurochem Res 30:1227–1236. https://doi.org/10.1007/s11064-005-8794-z
Garcia MA, Carrasco M, Godoy A, Reinicke K, Montecinos VP, Aguayo LG et al (2001) Elevated expression of glucose transporter-1 in hypothalamic ependymal cells not involved in the formation of the brain-cerebrospinal fluid barrier. J Cell Biochem 80:491–503
Benarroch EE (2014) Brain glucose transporters: Implications for neurologic disease. Neurology 82:1374–1379. https://doi.org/10.1212/WNL.0000000000000328
Hara M, Matsuda Y, Hirai K, Okumura N, Nakagawa H (1989) Effect of glucose starvation on glucose transport in neuronal cells in primary culture from rat brain. J Neurochem 52:909–912
Liu XH, Morris R, Spiller D, White M, Williams G (2001) Orexin a preferentially excites glucose-sensitive neurons in the lateral hypothalamus of the rat in vitro. Diabetes. https://doi.org/10.2337/diabetes.50.11.2431
Brown AM, Ransom BR (2007) Astrocyte glycogen and brain energy metabolism. GLIA. https://doi.org/10.1002/glia.20557
Rui L (2014) Energy metabolism in the liver. Compr Physiol 4:177–197. https://doi.org/10.1002/cphy.c130024
Cohen P, Nimmo HG, Proud CG (1978) How does insulin stimulate glycogen synthesis? Biochem Soc Symp 43:69–95
Vujovic P, Lakic I, Laketa D, Jasnic N, Djurasevic SF, Cvijic G et al (2011) Time-dependent effects of starvation on serum, pituitary and hypothalamic leptin levels in rats. Physiol Res 60(Suppl 1):S165–S170
Garriga J, Cussó R (1992) Effect of starvation on glycogen and glucose metabolism in different areas of the rat brain. Brain Res 591:277–282
Acknowledgements
The authors wish to thank Michael A. Chirillo MD/PhD (Department of Internal Medicine, The University of Utah School of Medicine, Salt Lake City, Utah, USA) for his help with editing and proofreading and Marko Miler PhD (Department of Cytology, Institute for Biological Research “Sinisa Stankovic”, Belgrade, Serbia) for his assistance with histological staining. This study was supported by Ministry of Education, Science and Technological Development, Republic of Serbia (173023).
Funding
This study was supported by Ministry of Education, Science and Technological Development, Republic of Serbia (173023).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in study involving animals were in accordance with the rules of the Federation of European Laboratory Animal Science and the ethical standard of the Faculty of Biology, University of Belgrade and Serbian Ministry of Agriculture, Forestry and Water Management, Veterinary Directorate.
Rights and permissions
About this article

Cite this article
Dakic, T., Jevdjovic, T., Lakic, I. et al. Food For Thought: Short-Term Fasting Upregulates Glucose Transporters in Neurons and Endothelial Cells, But Not in Astrocytes. Neurochem Res 44, 388–399 (2019). https://doi.org/10.1007/s11064-018-2685-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2685-6