
Abstract
The aim of the study was to elucidate the therapeutic effects of Cytisine (CYT) on cerebral ischemia–reperfusion injury in mice. Male ICR mice were pretreated with reagents (drug), and then subjected to 2 h focal cerebral ischemia and 24 h reperfusion. Morphologically, the histopathological impairment were estimated by the TTC, HE and TUNEL staining. The expression of GluN2B-containing NMDA receptor, phosphorylation of extracellular regulated protein kinases, total ERK, phosphorylation of cAMP-response element binding protein and total CREB were determined by immunofluorescence and Western blot assay, respectively. The mRNA expression of NR2B, ERK and CREB were quantified by the real-time RT-PCR. CYT significantly diminished the infarct size and neuronal apoptosis. Additionally, it ameliorated histopathological lesion dramatically. CYT promoted the phosphorylation of ERK, CREB and their mRNA expression. In contrast, the expression of NR2B was suppressed in concomitant with the down-regulation of genes. The overall results thus far suggest that CYT confers the neuroprotection against cerebral I/R injury by regulating the NR2B-ERK/CREB signal pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past several decades, excitotoxicity, a type of neurotoxicity mediated by glutamate, has been regarded as the center stage in stroke research [1]. Cerebral ischemia leads a massive release of glutamate which over activates NMDA receptors (NMDARs) and leads an extensive calcium influx through ionotropic receptors. Then, the calcium-dependent activation of death-signaling proteins triggers a plethora of excitotoxic signaling cascades and induces neuronal death [2]. The NMDAR is a heteromeric glutamate receptor mainly composed of an obligatory NR1 subunit and modulatory subunits NR2A-D [3]. NR2B mostly distributed in extrasynaptic NMDAR sites tends to promote neuronal death [4]. It is noteworthy that the over-activated extrasynaptic NR2B mediating glutamate excitoxicity shows robust calcium permeability once excessive glutamate bind to ion channel-couple NR2B subunits [5]. CREB is the primary transcription factor involved in the transcriptional regulation of genes that are required for neuronal survival. ERK mediated the phosphorylation of CREB at serine-133 via the p90 ribosomal S6 kinase Rsk-2 (p90rsk) [1]. In particular, extrasynaptic NMDAR decreases the ERK, CREB and BDNF activation contributing to excitotoxicity [6]. Over decades more neuroprotective therapies were designed to disrupt extrasynaptic NMDARs-dependent death signal, such as NR2B subtype-specific antagonist ifenprodil and Ro 256981 [7, 8], the effectiveness of which is extremely limited in clinical therapeutics, owing to the clinical side effects such as headache, heart palpitations, rash, lethargy, loss of appetite, etc. In addition, an increasing body of researches indicated that administration of drug would have an opportunity to exert a pharmacological property to cerebral I/R injury [9]. In recent years, natural products used in traditional Chinese medicine have received increasing attention due to few side-effects, multi-targeted mechanisms of activity and no drug resistance. Therefore, we attempted to seek safe and effective neuroprotective nature compounds for cerebral I/R injury from Chinese herbal medicine.
CYT (Fig. 1) is a crucial herbal alkaloid extracted and purified from the seed of Sophora Secundiflora L. Traditionally, it was used for smoking cessation [10]. Studies have also demonstrated antianxiety effects in rodent models [11]. Surprisedly, recent research has shown that CYT protected cortical neurons by inhibiting the level of GluN2B-containing NMDA receptors [12]. However, no information is available on possible effects of CYT on cerebral I/R injury. Therefore, the study aims to evaluate the effects of CYT and elucidate the potential mechanism.

The chemical structure of Cytisine (CYT)
Materials and Methods
Animal Preparation and Drug Administration
All protocols of the experiments were performed in accordance with the National Institutes of Health guide for the care and use of Laboratory animals. All experimental procedures were approved by the Institutional Animal Ethics Committee of our University. A total of 144 healthy adult male ICR (Institute of Cancer Research) mice weighing 20 ± 2 g were obtained from animal center of our university. All mice were housed in individual cages in a 12-h light/dark cycle at 23 ± 1 °C, with food and water ad libitum. The CYT (C11H14N2O) was supplied from medical company (purity > 98.2%, HPLC), and dissolved in physiological saline. Nimodipine (Nim), as the positive control, was obtained from the Bayer Healthcare co., Ltd (30 mg active ingredient per tablet). Reagents were dissolved in the 0.9% (w/v) NaCl solution respectively. Mice were randomly divided into six groups (n = 24 in each group): (1) Sham group (0.9% NaCl, ip); (2) Vehicle treated cerebral I/R group (0.9% NaCl, ip); (3) CYT 0.25 mg/kg groups (ip); (4) CYT 0.5 mg/kg groups (ip); (5) CYT 1 mg/kg groups (ip); (6) Nim group (6 mg/kg, ip). All groups were administrated with drug or physiological saline under the same condition for seven consecutive days before cerebral ischemia. To raise the comparison ability of the result in our research by using the principle of randomize double-blind.
Cerebral I/R Model
The focal cerebral ischemia in mice was induced by the method as described previously [13]. After 2 h of middle cerebral artery occlusion, the suture was withdrawn. The Sham group was subjected to the same surgical procedure but the MCA was not occluded. Mice failed in the surgery over 15 min or died in the surgery were excluded from the study.
Infarction Evaluation
The evaluation of infarct was performed as previously described [14]. Mice were sacrificed at 24 h after reperfusion, six animals of each group were euthanized by cervical dislocation. Brains were coronally sectioned into 2 mm thick slices by using a brain matrix. Coronal sections were stained in 2% 2,3,5 triphenyl tetrazolium chloride (TTC) (Sigma, USA) for 30 min at 37 °C, then immersed in 4% formaldehyde solution at 4 °C overnight. The brain sections were photographed using a digital camera individually (Samsung, KOR). The size of infarct areas was calculated with microscope image-analysis software Image-Pro plus6.0 (Media Cybernetics, USA). The infarct volume was equal to the total infarct area integrating across each section multiplied by its thickness. Ratio of infarct volume to total brain volume represents the degree of cerebral infarction.
Histopathological Observation
After 2 h of ischemia followed by 24 h of reperfusion, the mice (n = 6 from each group) were anesthetized with 3.5% chloral hydrate and perfused transitorily with physiological saline until the liver appeared to be white, followed by 4% paraformaldehyde in the phosphate buffered saline (PBS) (0.1 M, PH 7.4). Each brain was rapidly removed and immersed in 10% formalin for 2 h, then was washed three times using the PBS. After being dehydrated and embedded with paraffin, the brain tissues were cut into 5-µm-thick coronal sections for the HE staining, TUNEL staining and immunohistochemistry study. The sections were counterstained with hematoxylin in nucleuses and differentiated by eosin in cytoplasm. Interested area in cortex was scanned under high-power magnification (Bar = 20 µm) for histopathological observation in a blinded manner.
TUNEL Staining
The protocol of the TUNEL staining was implemented by using the commercial kit as previously reported [15]. Briefly, sections were performed a dewaxing and rehydration process followed by a permeation with proteinase K. DNA fragmentation in apoptotic neurons bound to the terminal deoxynucleotidyl transferase (TdT) enzyme in a reaction buffer. Slides were mounted with DAPI (Sigma, USA) for nuclear staining after rinsing with PBS. Ultimately, the apoptotic neurons in three random cortical fields across each section from six groups were counted at the high-power magnification (Bar = 20 µm) by a blinded manner. Ratio of the apoptotic neurons to total neurons in each sample represents the level of cerebral apoptosis.
Immunofluorescence Assay
Immunofluorescence assay was used to detect the immune-reactivity as described previously [16]. Briefly, the antigen retrieval of paraffin sections was performed with the high pressure after dewaxing and dehydration. Brain sections were immersed in normal goat serum to block the non-specific antigen followed by incubation with rabbit polyclonal primary antibodies of NMDAR2B (1:500; Protientech, USA), Phospho-ERK (Thr202/yr204) (1:300; Cell Signaling Technology, USA), Phospho-CREB (Ser133) (87G3) (1:300; Cell Signaling Technology, USA) at 4 °C overnight. Finally, they were incubated with the TRITC-labeled secondary antibody. The positive fluorescence in the interested area of sections was photographed (Bar = 20 µm) by a single investigator who was blind to sample identity. The semi-quantitative analysis was calculated by the Image-Pro plus6.0.
Western Blot Analysis
After 24 h of reperfusion, the mice (n = 6 from each group) were selected randomly and decapitated, and brain tissue was taken from the ischemic parietal cortex. Western blot was employed according to the methods as described previously [12]. Briefly, the protein, extracted from supernatant of homogenate, was size fractionated on the Sodium dodecyl sulfate polyacrylamide gels. After electrotransfer, polyvinylidene fluoride membranes were blocked with the 5% Bovine Serum Albumin (BSA) or defatted milk, and then incubated with primary antibodies: NMDAR2B (1:1000, Protientech, USA); p44/42 MAPK (Erk1/2) (137F5) (1:1000); Phospho-ERK (p-ERK) (Thr202/yr204) (1:500); CREB (48H2) (1:500); Phospho-CREB (Ser133) (87G3) (1:500) (Cell Signaling Technology, USA). Secondary incubations were performed with the secondary antibody (1:3000, Protinetech, USA). The bands were visualized using Pierce® ECL Western Blotting Substrate (Thermo Fisher scientific, USA). The signals were quantified by the Western blotting detection system Quantity One (Bio-Rad, USA). The intensities of each bands were all normalized to β-actin.
qRT-PCR (Quantitative Real-Time PCR)
At 24 h after reperfusion, total RNA of ischemic parietal cortex (n = 6 from each group) was extracted strictly in accordance with the instructions of the commercial reagent (Axygen, China) [15]. Melting curve analysis was performed after the final amplification step to identify the PCR products. The relative expression of the target gene was normalized to the β-actin regarded as the endogenous control. Ultimately, the fold induction (\({2^ - }^{{\Delta \Delta {{\text{C}}_{\text{t}}}}}\)) was represented as the calculated results. Primers used are as follows:
NR2B (forward 5′-TCTGAGCATCGTTACCTTGG-3′/ reverse 5′-CTTCTGGCACGGGACTGTAT-3′),
CREB (forward 5′-TAGTCCCAGCAACCAAGT-3′/ reverse 5′-GGACGCCATAACAACTCCAG-3′),
ERK (forward 5′-CTCTCATTTGAGGACAGGCA-3′/ reverse 5′-ATGTGGCATGCAGTGTAGGT-3′) and β-actin (forward 5′-GAGACCTTCAACACCCCAGC-3′/ reverse5′-ATGTCACGCACGATTTCCC-3′).
Statistical Analysis
Statistical analysis was performed by the SPSS 19.0 statistical software (IBM, USA). All values were expressed as the means ± SEM. Statistical evaluation of the data was performed by the one-way analysis of variance (ANOVA). The significance between groups was determined by the Tukey’s post-hoc test. Values of p < 0.05 were considered to be statistically significant.
Results
Neuroprotection of CYT Against Cerebral Ischemia-Induced Infarct Volume and Neuron Apoptosis
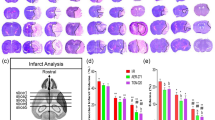
As shown in the images of the TTC-stained brain sections (Fig. 2a). No infarction was observed in the Sham group and an extensive infract area (37.04 ± 0.48%) was observed in the Vehicle treated cerebral I/R group. Total infarct volume was significantly reduced in the CYT groups (1 and 0.5 mg/kg, 30.74 ± 0.90% and 27.89 ± 0.85%) or Nim group (6 mg/kg, 25.91 ± 0.41%) compared with the Vehicle group (Fig. 2b, p < 0.01). As shown in Fig. 3a, there were virtually rare TUNEL-positive cells in the Sham group. Conversely, numerous apoptotic neurons were detected in the Vehicle treated cerebral I/R group (Fig. 3b, p < 0.01). However, in comparison with the Vehicle group, the neuronal apoptosis was substantially reduced in the groups pretreated with CYT (1 and 0.5 mg/kg) or Nim respectively (Fig. 3b, p < 0.01).

Protective effect of CYT against cerebral I/R injury in mice. a TTC staining of representative coronal sections at 24 h after reperfusion. b Quantitative analysis of infarct volumes at 24 h after reperfusion. Data are mean ± SEM (n = 10). ##p < 0.01 versus the Sham group; **p < 0.01 versus the Vehicle treated cerebral I/R group

The level of apoptosis in ischemic neurons was measured by TUNEL staining. a The representative fluorescence images of neurons apoptosis in the cortex at 24 h after reperfusion (Bar = 20 µM). b The quantitative data for the TUNEL-positive cells in above 6 groups from 3 distinct films of independent regions at 24 h after reperfusion. Data are mean ± SEM (n = 6). ##p < 0.01 versus the Sham group; **p < 0.01 versus the Vehicle treated cerebral I/R group
CYT Treatment on Ischemia-Induced Pathological Changes in the Ischemic Cortex
Neurons in Sham group remained intact and well-arranged, and the nuclei were centered with clear staining (Fig. 4a). However, Vehicle treated cerebral I/R group neurons showed nuclear pyknosis, interstitial oedema and swollen (Fig. 4b). Treatment with CYT (1 mg/kg) or Nim (6 mg/kg) was mostly confined to alleviate neuronal injury after ischemia (Fig. 4c–f).

The representative HE staining photomicrographs in the cortical pyramidal cell layer (Bar = 20 µm). a Sham group; b Vehicle treated cerebral I/R group; c–e CYT groups (0.25, 0.5 and 1 mg/kg); f Nim group (6 mg/kg)
All of these results demonstrated that most prominent protective effects were observed in CYT (1 mg/kg) groups and this dose was used in subsequent experiments.
Effects of CYT on NR2B, p-ERK and p-CREB Immune-Reactivity in the Ischemic Cortex
Immunofluorescence showed the fluorescence intensity with respect to the Rhodamine-labed positive cells (NR2B, p-ERK and p-CREB). NR2B positive cells were weakly stained in the Sham group (Fig. 5a), but ischemia induced an intense expression of NR2B in the Vehicle treated cerebral I/R group (Fig. 5b, p < 0.01). However, pretreatment with CYT (1 mg/kg) or Nim (6 mg/kg) significantly decreased NR2B immune-reactivity in the cortex (Fig. 5b, p < 0.01). It was worth to mentioning that NR2B positive signal was detected only in cytoplasm in the Sham group. We further examined the p-ERK and p-CREB. In the Sham group, p-ERK immunofluorescence intensity was feeble (Fig. 6a). Interestingly, the moderately increased p-ERK immune-reactivity was observed in the Vehicle treated cerebral I/R group as compared to the Sham group (Fig. 6b, p < 0.01). Likewise, the positive signal distributed in the cortex was augmented significantly with the administration of CYT (1 mg/kg) or Nim (6 mg/kg) in comparison with the Vehicle treated cerebral I/R group (Fig. 6b, p < 0.01). Similarly to the p-ERK, the p-CREB positive cells in the cortex (Fig. 7a) were slightly stained in the Sham group. However, the immunofluorescence of p-ERK was obviously (Fig. 7b, p < 0.01) strengthened in the Vehicle treated cerebral I/R group than the Sham group. Mice pretreated with CYT (1 mg/kg) or Nim (6 mg/kg) showed a significantly greater intensity versus the Vehicle treated cerebral I/R group (Fig. 7b, p < 0.01).

Effects of CYT on the immune-reactivity of NR2B. a The representative photomicrographs of the immune-reactivity of the NR2B in the cortex at 24 h after reperfusion in mice (Bar = 20 µM). b The quantification for the immunofluorescence intensity in above groups. Values were expressed as mean ± SEM (n = 6). ##p < 0.01 versus the Sham group; **p < 0.01 versus the Vehicle treated cerebral I/R group

Effects of CYT on the immune-reactivity of p-ERK. a The representative photomicrographs of the immune-reactivity of p-ERK in the cortex at 24 h after reperfusion in mice (Bar = 20 µm). b The quantification for the immunofluorescence intensity in above groups. Values were expressed as mean ± SEM (n = 6). ##p < 0.01 versus the Sham group; **p < 0.01 versus the Vehicle treated cerebral I/R group

Effects of CYT on the immune-reactivity of p-CREB. a The representative photomicrographs of the immune-reactivity of p-CREB in the cortex at 24 h after reperfusion in mice (Bar = 20 µm). b The quantification for the immunofluorescence intensity in above groups. Values were expressed as mean ± SEM (n = 6). ##p < 0.01 versus the Sham group; **p < 0.01 versus the Vehicle treated cerebral I/R group
Effects of CYT on the Proteins NR2B, p-ERK/t-ERK and p-CREB/t-CREB Expression in the Ischemic Cerebral Hemisphere
Under normal condition, the expression of NR2B protein remained a basal expression in the Sham group (Fig. 8a). However, its expression was intensively triggered (Fig. 8b, p < 0.01) in mice subjected to cerebral I/R. The ischemia-induced NR2B was remarkably suppressed by pretreatment with CYT (1 mg/kg) when compared to the Vehicle treated cerebral I/R group (Fig. 8b, p < 0.01). Compared with the Sham group, the phosphorylation of ERK1/2 (Fig. 9a) and CREB (Fig. 8c) protein was also potentiated in the Vehicle treated cerebral I/R group (Figs. 9b, 8d, p < 0.01). Both CYT (1 mg/kg) or Nim (6 mg/kg) significantly promoted the phosphorylation of ERK1/2 and CREB protein versus the Vehicle treated cerebral I/R group (Fig. 9b, p < 0.01 and Fig. 8d, p < 0.05). Whereas, there is no statistically significant difference about the expression of t-ERK or t-CREB in above groups (Fig. 9c, Fig. 8F).

Effects of CYT on NR2B and p-CREB/t-CREB proteins expression. a, c The representative Western blot bands of the NR2B, p-CREB/t-CREB proteins expression at 24 h after reperfusion in mice. b, d, e The quantification for the expression of NR2B, p-CREB/t-CREB. Values were expressed as mean ± SEM (n = 6). ##p < 0.01 versus the Sham group; *p < 0.05, **p < 0.01 versus the Vehicle treated cerebral I/R group

Effects of CYT on p-ERK/t-ERK proteins expression. a The representative Western blot bands of p-ERK/t-ERK proteins expression at 24 h after reperfusion in mice. b, c The quantification for the expression of p-ERK/t-ERK. Values were expressed as mean ± SEM (n = 6). ##p < 0.01 versus the Sham group; *p < 0.05, **p < 0.01 versus the Vehicle treated cerebral I/R group
Effects of CYT on the Levels of NR2B, ERK and CREB mRNA in the Ischemic Cerebral Hemisphere
As shown in Fig. 10a, the NR2B mRNA expression was significantly elevated in the Vehicle treated cerebral I/R group versus the Sham group (p < 0.01). However, administration with CYT (1 mg/kg) or Nim (6 mg/kg) prior to ischemia led to a marked decrease in the expression of NR2B mRNA (Fig. 10a, p < 0.01). Unexpectedly, the mRNA expression of ERK (Fig. 10b) and CREB (Fig. 10c) in the Vehicle treated cerebral I/R group actually showed a significant reduction when compared to the Sham group (p < 0.01). Nevertheless, CYT (1 mg/kg) or Nim (6 mg/kg) intervention robustly reversed the downregulation of ERK mRNA expression in comparison with the Vehicle treated cerebral I/R group in response to ischemia (Fig. 10b, p < 0.01). In the present experiment, the CREB mRNA in the CYT group showed an upregulation although there were no significances between the CYT group and the Vehicle treated cerebral I/R group (Fig. 10c). These results suggested that CYT-mediated regulation of NR2B, ERK and CREB mRNA expression may be closely associated with the activation of p-ERK and p-CREB following ischemia.

Relative quantitative mRNA levels of NR2B, ERK and CREB in the ischemic cerebral hemisphere. a The quantification for NR2B. b The quantification for ERK. c The quantification for CREB. Data represent the mean ± SEM (n = 6). ## p < 0.01 versus the Sham group; *p < 0.05, **p < 0.01 versus the Vehicle treated cerebral I/R group
Discussion
A complex series of biochemical and molecular mechanisms that include excitotoxicity, calcium overload, oxidative stress, inflammation and apoptosis are involved in cerebral focal ischemia-induced impairment, resulting in fatal neuronal death [17,18,19]. When cerebral ischemia occurs, the brain fails to generate sufficient ATP and maintain a normal Na+/H+ electrochemical gradient [20, 21]. It not only reverses the function of Na+-Ca2+ exchanger but also evokes the cellular membrane depolarization. In addition, the cytotoxic edema, as the result from the ionic gradients collapse in concomitant with influx water, facilitates the reversed glutamate transport and restrains the glutamate reuptake [22, 23]. Subsequently, these result in glutamate accumulation and irreversibly excitotoxicity injury [24]. The excitotoxicity plays an important role in the pathophysiology of several acute or chronic neurological disorders by mediating calcium overload [2]. The intracellular excess Ca2+ influx not only induces the activation of cPLA2 leading to production of neurotoxic metabolites and activation of the neutral protease, but also interrupts the electron transport chain generating the reactive oxygen species (ROS) and mitochondrial dysfunction [25,26,27]. Eventually, the derangement of cellular homeostasis contributes to neurodegeneration and neurons death [28]. Extrasynaptic NMDARs antagonize the prosurvival signal of synaptic NMDARs by disturbing the transduction of Ca2+ signal from the synapse to the nucleus [8]. Most of the extrasynaptic NMDARs are assembled with NR2B linked to the activation of cell-death pathways, especially calcium overload [29]. In view of the dual roles of NMDARs in mediating glutamate toxicity and promoting neuronal survival, novel approach has been utilized to design selective NR2B antagonists as potential therapeutic agents with fewer side effects [30]. A number of NR2B subunit antagonists, for instance, ifenprodil, dextromethorphan and dizocilpine have been developed. Nevertheless, ifenprodil was reported to have the modest neuroprotection in rodent models of stroke at the maximum tolerated dose. Dextromethorphan and dizocilpine reduced infarction volume in concomitant with common dose-related reversible adverse events or neuropsychiatric problems [31, 32].
Recently, the Stroke Therapy Academic Industry Roundtable (STAIR) is focusing on the assessment of neurology deficits extent and cognitive limitations for stroke therapies [33, 34]. Some studies showed that the relevant correlation coefficients predicted from the infarction volume to functional evaluation reached 91%. Progressive oedema and tissue swelling dominantly contribute to the neuronal death in malignant infarction after stroke [35]. In the present study, our results clearly demonstrated that pretreatment with CYT (1 or 0.5 mg/kg) apparently reduced the infarct volumes and alleviated histopathology injury in mice after cerebral ischemia. It suggests that the intervention of CYT may exert a neuroprotection in mouse which are undergoing cerebral ischemia injury.
Furthermore, pretreatment with CYT inhibited the NR2B expression. NR2B occurred a nuclear translocation in immunofluorescent assay in all groups except the Sham group. Interestingly, it is consistent with the finding that CYT blocked the upregulation of NR2B upon NMDA stimuli in cultured cortical neurons [12]. ERK, a member of the MAPK family, was thought to be involved in the mechanism of ischemic tolerance and neuroprotection [36]. Synaptic and extrasynaptic NMDAR are mutually antagonistic with regard to Ras-mediated ERK signal. Extrasynaptic NMDARs strongly inactivate Ras, and then promote ERK dephosphorylation in cortical neurons. Activated ERK is translocated into the nucleus where it regulates gene expression via the upregulation of specific transcription factors [8, 37, 38]. In present study, p-ERK positive neurons was barely observed in cortex. Noticeably, the potentiation of p-ERK immune-reactivity in concomitant with nuclear translocation in above regions after ischemia may be associated with the intervention of CYT. We have also observed more ERK phosphorylation in the DG neurons than ischemia-vulnerable CA1 neurons (data not shown), suggesting a protective role of ERK phosphorylation, it is consistent with previous reports [35]. In our experiment, phosphorylation of ERK protein is mainly distributed in the penumbra regions indicating that p-ERK inhibits apoptosis and expansion of infarction by phosphorylating the downstream target such as CREB.
CREB phosphorylation is critical in regulating the CRE-mediated genes in brain [39]. Activation of the extrasynaptic NR2B shuts off the robust phosphorylation of CREB induced by synaptic NR2B [35, 40, 41]. In our results, the activation of p-CREB was potentiated significantly with the administration of CYT (1 mg/kg) contrast to the Vehicle group. Correspondingly, the neurons apoptosis was decreased in the cortex compared to the Vehicle group by using the TUNEL staining. It infers that CYT may exert neurons pro-survival effect by meditating the activation of ERK/CREB. What noteworthy is the inconsonant variation tendency between mRNA expression and protein phosphorylation for ERK, CREB in the Vehicle group contrast to the Sham group. The decrease of the CREB and ERK mRNA in the Vehicle group may be related to the massive necrosis and apoptosis in this region at the outset after ischemia [42]. Nevertheless, it is well known that protein expression is modulated in turn by transcription, translation and histone modification process.
Conclusion
In summary, the present research has preliminary demonstrated that CYT might antagonize NR2B, and then activate CREB-mediated pro-survival signal pathway via inducing ERK phosphorylation subsequently. Compared with other NR2B antagonists and currently available stroke medications, CYT should receive more attention and are being increasingly investigated due to the advantage in terms of abundant resources, few side effects, lower cost and no drug resistance. Besides, further observation is deserved to elucidate whether other potential and interrelated mechanisms underlying the effects of CYT.
References
Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188
Wang Y, Qin ZH (2010) Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 15(11):1382–1402
Knox R, Zhao C, Miguel-Perez D, Wang S, Yuan J, Ferriero D, Jiang X (2013) Enhanced NMDA receptor tyrosine phosphorylation and increased brain injury following neonatal hypoxia-ischemia in mice with neuronal Fyn Overexpression. Neurobiol Dis 51:113–119
Köhr G (2006) NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res 326(2):439–446
Zhang Z, Wang CZ, Wen XD, Shoyama Y, Yuan CS (2013) Role of saffron and its constituents on cancer Chemoprevention. Pharm Biol 51(7):920–924
Léveillé F, Gaamouch E, Gouix F, Lecocq E, Lobner M, Nicole D, Buisson O (2008) A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J 22(12):4258–4271
Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH, Xiong ZQ (2008) Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39(11):3042–3048
Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11(10):682–696
Manning NW, Campbell BC, Oxley TJ, Chapot R (2014) Acute ischemic stroke: time, penumbra, and reperfusion. Stroke 45(2):640–644
Thompson-Evans TP, Glover MP, Walker N (2011) Cytisine’s potential to be used as a traditional healing method to help indigenous people stop smoking: a qualitative study with Māori. Nicotine Tob Res 13(5):353–360
Igari M, Alexander JC, Ji Y, Qi X, Papke RL, Bruijnzeel AW (2014) Varenicline and cytisine diminish the dysphoric-like state associated with spontaneous nicotine withdrawal in rats. Neuropsychopharmacology 39(2):455–465
Li YJ, Yang Q, Zhang K, Guo YY, Li XB, Yang L, Zhao MG, Wu YM (2013) Cytisine confers neuronal protection against excitotoxic injury by down-regulating GluN2B-containing NMDA receptors. Neurotoxicology 34:219–225
McCullough LD, Tarabishy S, Liu L, Benashski S, Xu Y, Ribar T, Means A, Li J (2013) Inhibition of calcium/calmodulin-dependent protein kinase kinase β and calcium/calmodulin-dependent protein kinase IV is detrimental in cerebral ischemia. Stroke 44(9):2559–2566
Vakili A, Sharifat S, Akhavan MM3, Bandegi AR (2014) Effect of lavender oil (Lavandula angustifolia) on cerebral edema and its possible mechanisms in an experimental model of stroke. Brain Res 1548:56–62
Luo Y, Yang YP, Liu J, Li WH, Yang J, Sui X, Yuan X, Nie ZY, Liu YQ, Chen D, Lin SH, Wang YA (2014) Neuroprotective effects of madecassoside against focal cerebral ischemia reperfusion injury in rats. Brain Res 1565:37–47
Wang TF, Lei Z, Li YX, Wang YS, Wang J, Wang SJ, Hao YJ, Zhou R, Jin SJ, Du J, Li J, Sun T, Yu JQ (2013) Oxysophoridine protects against focal cerebral ischemic injury by inhibiting oxidative stress and apoptosis in Mice. Neurochem Res 38(11):2408–2417
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion—from mechanism to translation. Nat Med 17(11):1391–1401
Lee RHC, Lee MHH, Wu CYC, Couto E, Silva A, Possoit HE, Hsieh TH, Minagar A, Lin HW (2018) Cerebral ischemia and neuroregeneration. Neural Regen Res 13(3):373–385
Perez-Alvarez MJ, Villa Gonzalez M, Benito-Cuesta I, Wandosell FG (2018) Role of mTORC1 controlling proteostasis after brain ischemia. Front Neurosci 12:60
Szeto V, Chen NH, Sun HS, Feng ZP (2018) The role of KATP channels in cerebral ischemic stroke and diabetes. Acta Pharmacol Sin 9(5):683–694
Ma Z, Xin Z, Di W, Yan X, Li X, Reiter RJ, Yang Y (2017) Melatonin and mitochondrial function during ischemia/reperfusion injury. Cell Mol Life Sci 74(21):3989–3998
Sopjani M, Thaçi S, Krasniqi B, Selmonaj M, Rinnerthaler M, Dërmaku-Sopjani M (2017) Regulation of ion channels, cellular carriers and Na(+)/K(+)/ATPase by Janus Kinase 3. Curr Med Chem 24(21):2251–2260
Larsen BR, Stoica A, MacAulay N (2016) Managing brain extracellular K(+) during neuronal activity: the physiological role of the Na(+)/K(+)-ATPase subunit isoforms. Front Physiol 7:141
Kulbe JR, Mulcahy Levy JM, Coultrap SJ, Thorburn A, Bayer KU (2014) Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res 1542:12–19
Puyal J, Ginet V, Clarke PG (2013) Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 105:24–48
Rostas JAP, Spratt NJ, Dickson PW, Skelding KA (2017) The role of Ca2+-calmodulin stimulated protein kinase II in ischaemic stroke—a potential target for neuroprotective therapies. Neurochem Int 107:33–42
Sun Y, Cheng X, Hu J, Gao Z (2018) The role of GluN2A in cerebral ischemia: promoting neuron death and survival in the early stage and thereafter. Mol Neurobiol 55(2):1208–1216
Blackstone NW (2015) The impact of mitochondrial endosymbiosis on the evolution of calcium signaling. Cell Calcium 57(3):133–139
Yurkewicz L, Weaver J, Bullock MR, Marshall LF (2005) The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J Neurotrauma 22(12):1428–1443
Li Y, Yu M, Zhao B, Wang Y, Zha Y, Li Z, Yu L, Yan L, Chen Z, Zhang W, Zeng X, He Z (2018) Clonidine preconditioning improved cerebral ischemia-induced learning and memory deficits in rats via ERK1/2-CREB/ NF-κB-NR2B pathway. Eur J Pharmacol 818:167–173
Lee S, Yoon S, Kim DH, Gynecol Onco (2007) A high nuclear basal level of ERK2 phosphorylation contributes to the resistance of cisplatin-resistant human ovarian cancer cells. Gynecol Oncol 104(2):338–344
Lee YJ, Choi SY, Yang JH (2014) NMDA receptor-mediated ERK 1/2 pathway is involved in PFHxS-induced apoptosis of PC12 cells. Sci Total Environ 491(49):227–234
López-Valdés HE, Clarkson AN, Ao Y, Charles AC, Carmichael ST, Sofroniew MV, Brennan KC (2014) Memantine enhances recovery from stroke. Stroke 45(7):2093–2100
Saver JL, Albers GW, Dunn B, Johnston KC, Fisher M, STAIR VI Consortium (2009) Stroke Therapy Academic Industry Roundtable (STAIR) recommendations for extended window acute stroke therapy trials. Stroke 40(7):2594–2600
Kahle KT, Simard JM, Staley KJ, Nahed BV, Jones PS, Sun D (2009) Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology 24:257–265
Chang C, Zhao Y, Song G, She K (2018) Resveratrol protects hippocampal neurons against cerebral ischemia-reperfusion injury via modulating JAK/ERK/STAT signaling pathway in rats. J Neuroimmunol 315:9–14
Huang B, Chen P, Huang L, Li S, Zhu R, Sheng T, Yu W, Chen Z, Wang T (2017) Geniposide attenuates post-ischaemic neurovascular damage via GluN2A/AKT/ ERK-dependent mechanism. Cell Physiol Biochem 43(2):705–716
Shioda N, Han F, Fukunaga K (2009) Role of Akt and ERK signaling in the neurogenesis following bra brain ischemia. Int Rev Neurobiol 85:375–387
Kitagawa K (2007) CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J 274(13):3210–3217
Chen S, Yin W, Bi K, Lu B (2018) MicroRNA497 attenuates cerebral infarction in patients via the TLR4 and CREB signaling pathways. Int J Mol Med. https://doi.org/10.3892/ijmm.2018.3611
Zhang W, Song JK, Yan R, Li L, Xiao ZY, Zhou WX, Wang ZZ, Xiao W, Du GH (2018) Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol Sin. https://doi.org/10.1038/aps.2017.149
Zhang ZH, Fang XB, Xi GM, Li WC, Ling HY, Qu P (2010) Calcitonin gene-related peptide enhances CREB phosphorylation and attenuates tau protein phosphorylation in rat brain during focal cerebral ischemia/reperfusion. Biomed Pharmacother 64(6):430–436
Acknowledgements
This work was supported by the Colleges and Universities of Science and Technology Research Projects (Grant No. NGY2013078).
Author information
Authors and Affiliations
Contributions
Jian-Qiang Yu contributed to the conception of the study and approved the final version. Peng Zhao and Yue Liu significantly performed the experiments and drafted the manuscript preparation. Yin-Ju Hao and Yu-Xiang Li performed the data analyses. Nan Li and Jing Wang contributed reagents and materials. Yang Niu and Tao Sun revised the manuscript. We thank Drs. Lu-Ning Cui for his critical review and subsequent editing of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Zhao, P., Yang, JM., Wang, YS. et al. Neuroprotection of Cytisine Against Cerebral Ischemia–Reperfusion Injury in Mice by Regulating NR2B-ERK/CREB Signal Pathway. Neurochem Res 43, 1575–1586 (2018). https://doi.org/10.1007/s11064-018-2572-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2572-1