
Abstract
Epidemiological studies indicate that a higher plasma level of uric acid (UA) associates with the reduced risk of Parkinson’s disease (PD). To confirm the role of UA as a biomarker for PD, we evaluated changes in the serum UA level in the 6-hydroxydopamine (6-OHDA)-induced hemiparkinsonism in rat. For this purpose, 6-OHDA was administered in the medial forebrain bundle by stereotaxic surgery. According to the apomorphine-induced rotational test, the increased intensity of behavioral symptoms as a function of time was associated with the further reduction of UA level. On the other hand, the level of UA increased in the midbrain of the injured hemisphere. The level of reduction in the serum UA level of rats with severe and moderate symptoms was significantly higher than that of rats with mild symptoms. The immunohistofluorescence and biochemical analyses showed that the serum UA level was also correlated with the death of tyrosine hydroxylase (TH)-positive neurons in the substantia nigra pars compacta (SNc), reduced level of striatal dopamine, and severity of oxidative stress in the midbrain. The rats with mild symptoms also showed a significant decrease in TH-positive neurons and striatal dopamine level. These findings suggest a positive correlation between the level of reduction in the serum urate level and severity of 6-OHDA-induced Parkinsonism. In addition, our findings indicated that UA had no marked neuroprotective effects, at least at concentrations obtained in this study. On the other hand, UA was introduced as a biomarker for PD, as a significant decline was observed in the serum UA level of rats with mild behavioral symptoms but with significant dopaminergic cell death in the SNc.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a chronic and progressive neurodegenerative disorder, affecting about 6 million people worldwide [1]. The prevalence of PD is expected to double within the next 20 years, considering the increasing age of the population [2]. The most important characteristic of this disease is the progressive degeneration of dopaminergic (DA) neurons within the substantia nigra pars compacta (SNc) of the midbrain. The pathophysiological mechanisms of DA degeneration remain unknown, although mitochondrial dysfunction and oxidative stress are believed to play important roles in the events leading to DA neuronal death. Levodopa treatment removes dopamine deficiency and effectively improves movement-related symptoms. However, it cannot halt or retard progressive neurodegeneration, and side effects, such as dyskinesia, are developed in the long term [3,4,5,6].
PD is currently examined via clinical evaluation of extrapyramidal signs, such as tremor, rigidity, slowness or absence of voluntary movements, postural instability, and freezing. These symptoms occur when more than 70% of DA neurons are lost [7, 8]. Therefore, early diagnosis of PD may help select and initiate the proper treatment to decelerate the neurodegenerative process and disease progression.
Identification of biomarkers for PD can be valuable in its early detection. The biomarkers also allow for a better follow-up of patients and facilitate objective measurements in clinical trials [9]. Nonetheless, research on the biomarkers for PD is still in its early stages, despite the urgent need for new treatment strategies [9]. Among the proposed biomarkers for PD, uric acid (UA) has attracted the greatest amount of attention. Urate, as the dissociated form of UA, is the final product of purine catabolism and has marked antioxidant properties [10]; it is also responsible for most of the antioxidant activities of the human plasma [11].
Involvement of urate in the pathophysiology of PD was first indicated by its low level in the SNc of patients with PD, as well as UA capacity to decrease dopamine oxidation [12]. A study on urate level and risk of PD among 7968 men, enrolled in the Honolulu Heart Program, was the first to introduce UA as a potential biomarker [13]. Two other prospective studies have also reported similar findings [14, 15]. However, studies comprising both males and females have reported conflicting results. Some claimed that the association of low serum urate level with the risk of PD is significant only for men [16,17,18], whereas others did not find any gender differences [14, 19, 20]. In addition, pharmacological therapy can increase this confliction, as additional oxidative stress may be induced by Levodopa treatment [21, 22]. Although the results of epidemiological studies are interesting, they do not clarify whether the inverse association between urate level and PD progression is merely related to a pre-existing high level of plasma UA or dynamic changes in the level of endogenous urate during DA nigrostriatal degeneration.
To confirm the role of UA as a biomarker for PD, we evaluated the serum UA changes in a well-established 6-hydroxydopamine (6-OHDA) animal model. 6-OHDA is an organic neurotoxic compound, which selectively destroys DA neurons in the SNc after intracranial injection into the medial forebrain bundle (MFB) or striatum [5, 23, 24]. A major advantage of this model is the quantifiable motor deficit, which was evaluated in a well-known rotational test. Following the systemic administration of dopamine agonists, 6-OHDA-treated rats show asymmetrical rotations. This behavior is particularly observed after toxin injection into the MFB, which causes major DA cell death in the SNc, besides marked depletion of dopamine in the striatum [23, 24]. Therefore, any correlation between the serum urate level and intensity of rotations indicates the association of urate level with DA cell loss in the SNc and striatal dopamine depletion.
Experimental Procedures
Animals and Experimental Groups
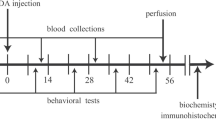
Adult male Wistar rats (Razi Institute, Karaj, Iran), weighing 250–300 g, were housed in large cages (38 × 59 × 20 cm) in a temperature-controlled colony room in a 12:12 h light/dark cycle with free access to tap water and standard feed. All procedures in this study were in accordance with the guidelines for animal experiments by the Research Council of Qazvin University of Medical Sciences. The rats were divided into three experimental groups with respect to the stereotaxic injections: (1) control group, receiving no treatment; (2) vehicle group, receiving the vehicle (normal saline containing ascorbic acid) intracerebrally; and (3) 6-OHDA group, receiving 6-OHDA intracerebrally. Each group consisted of 20 rats (Fig. 1).

Experiment design. Only rats showed less than 10 rotations per hour in presurgery apomorphine-induced rotational test were used for experiments. Rotational test was also carried out 3 and 6 weeks after the 6-OHDA injection. Blood sampling was carried out at three intervals: before the toxin and 3 and 6 weeks after the 6-OHDA injection. Six weeks after the surgery, six rats from each group were perfused and immunohistofluresance staining was carried out on the midbrain sections. For biochemical measurements, the brain of the other rats was freshly removed and separate homogenates were prepared from striatum and midbrain of both hemispheres. Dopamine concentration in striatum and urate concentration, superoxide dismutase (SOD), glutathione peroxidase (GPx) and malondialdehyde (MDA) concentrations in midbrain were measured using immunosorbent assay kits. Numbers show the days after the toxin injection. UA uric acid
Surgical Procedures
Except for the animals in the control group, all other rats were subjected to stereotaxic surgery. The vehicle and 6-OHDA were injected into the right MFB by stereotaxic surgery, using a 10-µL Hamilton syringe. Briefly, the rats were anesthetized with an intraperitoneal (i.p.) injection of a solution, containing ketamine (100 mg/kg) and xylazine (5 mg/kg). Subsequently, 4 µL of 6-OHDA (4 µg/µL; Sigma) was dissolved in isotonic sodium chloride (NaCl) solution (containing 0.2 mg/mL of ascorbic acid) and injected at 2 sites with the following coordinates: anterior-posterior (AP): − 4, lateral (L): − 1.8, dorsoventral (DV): 9; and AP: − 4.4, L: − 2, DV: 8.8. The AP and L coordinates were measured from the bregma, while DV was measured from the surface of the skull according to the Atlas by Paxinos and Watson [25]. The same volume of vehicle was injected at these sites in the vehicle group. At the end of each injection, the needle was left in place for 5 min and then withdrawn at a rate of 1 mm/min.
Apomorphine-Induced Rotational Test
The apomorphine-induced rotational test was carried out in all animals before the surgery and toxin injection, as well as 3 and 6 weeks after surgery according to the method described by Fujita et al. in [26]. The animals were initially allowed to acclimate to their environment for 5 min and then received an i.p. injection of apomorphine hydrochloride dissolved in saline (0.5 mg/kg). After 1 min, the number of full rotations was counted for 60 min in a cylindrical container (diameter = 28 cm; height = 38 cm). Contralateral (away from the lesion side) and ipsilateral (towards the lesion side) rotations were counted as positive and negative scores, respectively, and the net number of rotations was determined by subtracting the negative scores from the positive scores.
Immunohistofluorescence Staining
Immunohistofluorescence staining was carried out on tissues of 6 rats from each group. After anesthesia induction with ketamine and xylazine, the rats were perfused transcardially with phosphate-buffered saline (PBS), followed by 4% paraformaldehyde. Subsequently, their brains were removed and postfixed overnight in paraformaldehyde and transferred to 30% sucrose in PBS at 4 °C. The midbrain section was isolated and frozen in a cryostat embedding medium (Bio-Optica, Italy) at − 22 °C. The coronal sections (8-µm thickness) were prepared using a cryostat (Histo-Line Laboratories, Italy). One out of every serial three sections was permeabilized with 0.2% Triton X-100 and blocked with 10% normal goat serum for 1 h. The sections were incubated overnight at 4 °C with an anti-tyrosine hydroxylase (TH) antibody (1:250; Abcam) and incubated with proper fluorescently labeled rabbit secondary antibodies. Afterwards, the sections were coverslipped using a mounting medium (sc-24941; Santa Cruz), which contained 4′,6-diamidino-2-phenylindole (DAPI) as the nuclear stain. They were then visualized on an Olympus microscope at ×10 magnification, and sections including the SNc (AP, − 4.8 to − 5.2 relative to the bregma) were selected. Then five sections in each animal were selected from totally 50 sections which were divided into five series rostrocaudally. There were at least 5-section intervals between two selected sections. TH-positive cells were visualized and counted manually at ×100 magnification.
Blood Sampling and UA Measurement
The blood specimens were collected at three intervals: before the surgery and toxin injection, 3 weeks after the surgery, and 6 weeks after the surgery (Fig. 1). The first and second specimens were collected from the caudal vein, while the third sample was collected from the heart of animals under deep anesthesia. Blood was allowed to coagulate, and then, the serum was separated. To determine the urate level in the midbrain section, the brain was removed following deep anesthesia, and the midbrain portion was isolated. Then, it was sonicated in PBS (pH 7.4) and centrifuged, and the supernatant was collected. The sera and supernatants were stored at − 80 °C to urate measurements.
The Alpha Classic Autoanalyzer was used to determine the urate level. The urate level in the specimens was measured using uricase and a tribromophenol-aminoantipyrine chromogen. In this method, urate reacts with water and oxygen in the presence of uricase enzyme to produce allantoin and hydrogen peroxide. Enzymatic analysis of differential absorbance in these substances at 293 nm allows for the colorimetric assay of urate level.
Dopamine Measurement
Dopamine concentrations in the striatum were measured in 14 rats from each group, using the Dopamine Research ELISA™ Kit (BA E-5300; Nordhorn, Germany), according to the manufacturer’s instructions. Briefly, striatal tissues were homogenized in hydrogen chloride (0.01 N; 1 mL for 50 mg of tissue) with 1 mM EDTA and 4 mM sodium metabisulfite. Under these conditions, dopamine is charged positively, and its solubility reaches the optimized level. Afterwards, the homogenate was centrifuged at 15,000×g for 15 min (4 °C), and the supernatant was collected for the measurements. The measurements were performed in 20 µL of standard and diluted samples, using a microplate reader at 450 nm (reference wavelength 620–650 nm). The concentration of dopamine in the samples was calculated according to 6 standards from 0 to 90 ng. The ELISA kit provides a very sensitive approach (lower limit 0.7 ng/mL) for the measurement of dopamine.
Measurement of Oxidative Stress Markers
Three markers, including superoxide dismutase (SOD), glutathione peroxidase (GPx), and malondialdehyde (MDA), were measured in the midbrain homogenates (n = 14 per group), using specific research ELISA™ kits (EASTBIOPHARM, Hangzhou), according to the manufacturer’s instructions. The midbrain portion was isolated and sonicated in PBS (pH 7.4). After centrifugation, the supernatant was collected. The test wells were first filled with 40 µL of the sample, followed by both specific antibody and streptavidin-horseradish peroxidase. Subsequently, the sealing membrane was sealed, gently shaken, and incubated for 60 min at 37 °C. The absorbance was read at 450 nm for all markers.
Statistical Analysis
Data are expressed as mean ± standard error of mean (SE) in spite of the probable skewed distribution of scores. Data were initially analyzed by Kolmogorov–Smirnov test to determine their normal distribution. Then, analysis of variance (single-factor ANOVA), followed by Newman–Keuls test, was performed. Paired t test was also used for comparison of the number of rotations on two postsurgical rotational tests. The significance level was set at p ≤ 0.05.
Results
Apomorphine-Induced Rotational Test and Serum Urate Level
Before the stereotaxic injection of 6-OHDA, the mean ± SE of urate level was 3.54 ± 0.2 (range 1.2–9.8 mg/dL), the age of animals was 3–4 months, and their weight was 240–280 g (n = 112); these data are related to 7 different experimental groups of rats, which were intact and did not receive any treatments. However, we evaluated the urate level in three groups, including the control, vehicle, and toxin-treated (6-OHDA) rats (20 rats per group). Figure 2 quantifies the rotational activity, as well as the serum and midbrain urate levels.

Serum urate level significantly decreased in the toxin-treated rats. Upper plot: after the surgery, only 6-OHDA-treated rats showed significant rotations (n = 20 per group). Middle plots: urate level in 6-OHDA-treated group (PD) decreased remarkably in samples collected 3 and 6 weeks after the toxin injection (n = 20 per group). Left lower plot: urate level in right midbrain of toxin-treated rats was significantly higher than that in left midbrain and midbrains of both sides of control and vehicle groups (n = 8 per group). RM right midbrain, LM left midbrain. Data are expressed as means ± S.E. of animals in each group (n = 20). *p < 0.05, **p < 0.01 and ***p < 0.001 in compared to first test after the toxin in rotational activity(paired t test), in compared to serum urate level before the toxin injection (one way ANOVA) and in compared to midbrain urate level in control and vehicle groups(one way ANOVA). #p < 0.05 and ###p < 0.001 in compared to values in control and vehicle groups in the same sampling. One-way ANOVA followed by Newman–Keuls test. Right lower plot displays Pearson’s correlation coefficient between serum urate level and severity of rotational behavior. *p < 0.05 and **p < 0.01. See text for more information
Before toxin injection, no significant difference was observed in the rotational activity and serum urate level between the groups. After toxin injection, noticeable rotations were only observed in toxin-treated rats. The number of rotations increased as a function of time and was remarkably higher in the second postsurgical test, compared to the first postsurgical test (upper plot in Fig. 2). The biochemical data showed that serum urate level did not change in the control and vehicle groups during the experiments. On the other hand, in the toxin-treated group, the level of urate significantly decreased (3.6 ± 0.4 mg/dL before toxin injection, 2.7 ± 0.2 mg/dL at 3 weeks after toxin injection, and 1.84 ± 0.2 mg/dL at 6 weeks after toxin injection) (middle plot in Fig. 2). The statistical analysis showed a significant negative correlation between urate level and intensity of rotations after toxin injection (Pearson’s correlation coefficient − 0.541 and p < 0.05 for the third week after toxin injection; Pearson’s correlation coefficient − 0.741 and p < 0.01 for the sixth week after toxin injection). A significant positive correlation was found between urate level before toxin injection and intensity of rotations in the sixth week (correlation coefficient 0.481; p < 0.05) (right lower plot in Fig. 2).
The lower plot in Fig. 2 illustrates the urate level in the midbrain portion of the brain. In both hemispheres of the control and vehicle groups, as well as the left midbrain of toxin-treated rats (8 rats per group), the urate level was roughly close to the serum level of the control group. However, in the right midbrain of toxin-treated rats, the urate level was remarkably higher than that of the control group.
The intensity of rotational behaviors was not similar in toxin-treated rats. To further evaluate the association between Parkinsonism and serum urate level, we divided toxin-treated rats into three groups: severe group (n = 8) with more than 300 rotations per hour in one of the postsurgical tests; moderate group (n = 6) with 60–300 rotations; and mild group (n = 6) with less than 30 rotations. Figure 3 quantifies the rotational activity and urate level in these groups. As can be seen in the upper plot, the intensity of rotational activities increased remarkably in the second postsurgical test in the severe and moderate groups, but not the mild group.

Toxin-treated rats showed different degree of rotational behavior and urate profile. Left upper plot: based on the number of rotations, toxin-treated rats were divided into three groups of severe (more than 300 rotations/h, n = 8), moderate (between 60 and 300 rotations/h, n = 6) and mild (mild, less than 30 rotations/h, n = 6). Right upper plot and left lower plot: before the toxin injection = serum urate level in severe and moderate groups was significantly further than that in mild group. After the toxin injection, urate level in severe and moderate groups decreased remarkably in the both postsurgical sampling. Such decrease in mild group was only observed 6 weeks after toxin injection. Data are expressed as means ± S.E. of animals in each group (n = 8, 6 and 6 for severe, moderate and mild groups, respectively). *p < 0.05, **p < 0.01 and ***p < 0.001 in compared to first test after the toxin injection in rotational test (paired t test) and in compared to urate level before the toxin injection (paired t test). #p < 0.05, ##p < 0.01 and ###p < 0.001 in compared to values in severe and mild group in rotational test and severe and moderate groups in urate level in the same sampling. One-way ANOVA followed by Newman–Keuls test. Right lower plot displays Pearson’s correlation coefficient between serum urate level and severity of rotational behavior in severe group. *p < 0.05 and **p < 0.01. See text for more information
Before toxin injection, the serum urate level in the mild group was significantly lower than that of the severe group. After toxin injection, all groups showed a significant reduction in the urate level; however, the rate of this decline in the severe group was much higher than that of the mild group (lower diagram in Fig. 3). Also, statistical analysis showed a significant negative correlation between urate level after toxin injection and intensity of rotations in the severe group (correlation coefficient − 0.691 and p < 0.01 for the third week after toxin injection; correlation coefficient − 0.785 and p < 0.01 for the sixth week after toxin injection). In this group, there was a positive correlation between urate level before toxin injection and intensity of rotations in the sixth week (correlation coefficient 0.561; p < 0.01) (right lower plot in Fig. 3).
Evaluation of TH-Positive Neurons in the SNc and Assessment of Striatal Dopamine Level
To evaluate the association between urate level and 6-OHDA-induced DA cell death, we traced TH-positive neurons in the SNc, using immunohistofluorescence staining. We also measured the striatal dopamine level to evaluate the association with toxin-induced loss of DA terminals in the striatum. The photomicrographs in Fig. 4 present TH-positive neurons, and the upper plot represents the counts. Expectedly, in the severe group (n = 2), massive DA cell loss was observed in the right injured SNc (B); in fact, the number of TH-positive neurons in the right SNc was > 70% lower than that of the left SNc (upper plot in Fig. 4). Nevertheless, no significant difference was observed in the number of TH-positive neurons between the severe and moderate (n = 2) groups. On the other hand, in the mild group (n = 2), although a major lesion was identified in the SNc, the number of TH-positive neurons was remarkably higher than that of the severe and moderate groups. The statistical analysis revealed that the number of TH-positive neurons was positively correlated with urate level (correlation coefficient 0.588; p < 0.05 in the sixth week) (lower plot in Fig. 4).

Toxin-treated rats showed different degree of both DA cell loss in SNc and decrease in striatal dopamine level. Photomicrographs present tyrosine hydroxylase (TH)-positive neurons in SNc (AP: − 5 relative to bregma) of veh (a), severe (b), moderate (c) and mild (d) groups. Note to massive DA cell loss in severe and moderate groups. Scale bar 100 µm. Upper plot quantifies number of remaining TH-positive neurons in injured hemisphere. The number of TH-positive neurons was counted and then calculated as a ratio of the injured side to intact side of SNc (n = 6 for control and veh groups and n = 2 for other groups). Middle plot quantifies striatal dopamine level. Level of dopamine in the both hemisphere was measured using immunosorbent assay kit and then calculated as a ratio of the injured side to intact side. Data are expressed as means ± S.E. of animals in each group (n = 14 for control and veh groups and n = 6, 4 and 4 for severe, moderate and mild groups, respectively). **p < 0.01 and ***p < 0.001 in compared to veh group. #p < 0.05 and ##p < 0.01 in compared to severe group. One-way ANOVA followed by Newman–Keuls test. Lower plot displays Pearson’s correlation coefficient between serum urate level and number of TH-positive neurons and striatal dopamine level. *p < 0.05 and **p < 0.01. See text for more information
The striatal dopamine level could mostly discriminate toxin-treated rats (Middle plot in Fig. 4). Relative to the intact side, 70, 55, and 30% reductions were observed in the dopamine level of the injured striatum in the severe (n = 6), moderate (n = 4), and mild (n = 4) groups, respectively. A significant positive correlation was found between dopamine and urate levels in the sixth week (correlation coefficient 0.686; p < 0.01) (lower plot in Fig. 4).
Evaluation of Oxidative Stress in the Midbrain
Three conventional markers of oxidative stress, including SOD, GPx, and MDA, were measured in the midbrain of rats. Plots in Fig. 5 quantify the activity and level of these markers. In all toxin-treated rats (n = 10), substantial oxidative stress was found in the midbrain of the injured hemisphere. However, no significant difference was observed between the severe (n = 6) and moderate (n = 4) groups. On the other hand, the level of oxidative stress in the mild group (n = 4) was significantly lower than that of the severe group.

Toxin-treated rats showed different degree of oxidative stress in SNc. Plots display concentration of superoxide dismutase (SOD), glutathione peroxidase (GPx) and malondialdehyde (MDA) in the injured midbrain of control, vehicle and different groups of toxin treated rats. Note oxidative stress was almost the same in severe and moderate groups but in mild group it was remarkably less severe. Data are expressed as means ± S.E. of animals in each group (n = 14 for con and veh groups and n = 6, 4 and 4 for severe, moderate and mild groups, respectively). *p < 0.05, **p < 0.01 and ***p < 0.001 and in compared to con and veh group. ##p < 0.01 in compared to severe group. One-way ANOVA followed by Newman–Keuls test
Discussion
According to several prospective and cohort studies, a higher plasma urate level associates with the reduced risk of PD and can predict the slower progression of this disease [14,15,16,17,18,19,20,21,22]. Urate is an important natural antioxidant, iron chelator, and free radical scavenger in the blood and brain tissues [10, 11]. Moreover, urate has been shown to reduce the rate of DA autoxidation in homogenates of the caudate and SNc of patients with PD [12].
In the present study, to further investigate the association between PD and urate level, serum urate changes were evaluated in an experimental rat model of PD, developed by unilateral intracranial injection of 6-OHDA. Overall, 6-OHDA causes DA neuronal death in the SNc through major oxidative stress [23, 24, 27]. Oxidative stress is also the most common mechanism, which has been suggested for DA cell death in the SNc among patients with PD [3, 5, 6]. Therefore, a common pathophysiological mechanism underlies DA cell death in PD among humans and 6-OHDA-induced Parkinsonism in rats. Our findings showed that the serum urate level decreased in 6-OHDA-treated rats. The level of reduction was directly correlated with the severity of behavioral symptoms. First, increased severity of rotational behaviors as a function of time was associated with further reduction in urate level. Second, the level of reduction in rats with more rotations (severe group) was significantly higher than that of rats with fewer rotations (mild group). These findings are in agreement with human studies, showing that UA level is inversely correlated with disease severity and duration in patients with PD. In fact, patients at the stages of third and upper have significantly less plasma UA levels relative to patients at the first two stages [21, 22].
Rotational activity is the behavioral outcome of unilateral DA degeneration in the SNc and striatal DA depletion [5, 23, 24, 28]. Our histological and biochemical findings confirmed this result, and an inverse relationship was observed between the intensity of rotational activities, survival of DA neurons in the SNc, and striatal DA level. Therefore, further reduction in the serum urate level was associated with the greater severity of DA neuronal loss and further reduction in the striatal DA level.
We also measured three markers of oxidative stress, including SOD, MDA, and GPx. Our findings showed that the intensity of oxidative stress in rats with severe Parkinsonism was significantly higher than that of the mild group. Based on the findings, severity of oxidative stress was directly correlated with the severity of DA neuronal loss, reduction in striatal DA level, and severity of rotational behaviors. This finding confirms a large body of evidence, indicating oxidative stress as the main pathophysiological mechanism of 6-OHDA-induced Parkinsonism [3,4,5,6, 23, 24, 27, 28]. Therefore, in 6-OHDA-treated rats, an increase in oxidative stress was accompanied by a decline in serum urate level; in fact, urate was used as an antioxidant against 6-OHDA-induced oxidative stress. Similarly, Andreadou et al. [22] suggested the possibility of increased consumption of UA as a scavenger of oxidants in PD. Our findings are also in agreement with studies, which revealed the protective effects of urate on DA neurons against 6-OHDA neurotoxicity [29,30,31]. The Akt/GSK3β and Nrf2 signaling pathways underlie the neuroprotection produced by UA against 6-OHDA [29, 31].
In contrast to serum UA, the level of UA increased in the right midbrain of 6-OHDA-treated rats. This finding is in agreement with a study by De Luca et al. [32], which reported that striatal urate level increases in the injured hemisphere of 6-OHDA-treated rats. On the other hand, human studies have shown that urate level in the midbrain of patients with PD is lower than that of age-matched controls [12, 20, 33]. The observed disagreement may be due to different modes of DA degeneration in humans and 6-OHDA-treated rats. DA degeneration in humans is chronic and progressive, while 6-OHDA stimulates fast and severe degeneration after injection into the MFB. Similarly, De Luca et al. [32] showed that partial DA degeneration, induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), only triggered a trend towards a significant increase in the striatal urate level of mice. Another difference between humans and rats can further explain this disagreement. In humans, a tenfold gradient from the blood to cerebrospinal fluid has been suggested for urate [18]. On the other hand, our data showed that UA concentration in the midbrain and serum of rats is roughly equal. This finding is in agreement with a study by Gong et al. [31], which reported a major correlation in the UA levels of blood and brain in male Sprague–Dawley rats. Furthermore, UA level in the brain is associated with the integrity of the blood–brain barrier (BBB), and its impairment increases the urate level in the cerebrospinal fluid [34]. Several studies have shown that intracranial injection of 6-OHDA causes major damage to BBB [35, 36]. The higher level of urate in the rat midbrain may be due to the in situ production of urate as a compensative mechanism [34]. Our data showed that 6-OHDA increases the cerebral urate level, which could be a compensatory mechanism to attenuate 6-OHDA-induced oxidative stress and DA neurodegeneration in the SNc.
However, urate was not highly effective against 6-OHDA-induced neurotoxicity, as further reduction in serum urate level was associated with more severe DA cell loss and Parkinsonism. Also, before the surgery and 6-OHDA injection, rats with severe Parkinsonism had higher serum urate levels, compared to those with mild Parkinsonism. This finding indicates that rats with higher urate levels are more susceptible to the development of 6-OHDA-induced Parkinsonism. This finding is in contrast with epidemiological studies, which suggested high urate level as a predictor of a lower risk of PD [13,14,15,16,17,18,19,20,21,22]. There is a marked difference in urate metabolism between humans and rodents. In human body fluids, urate is present in an anionic form of UA (2,6,8-trioxy-purine) and accounts for most of the antioxidant capacity of the plasma [10, 11]. On the other hand, in rodents, urate is further metabolized to allantoin by urate oxidase [37]. Therefore, the serum urate concentration in the blood of rats might not be adequate to ameliorate 6-OHDA-induced Parkinsonism. In fact, before toxin injection, urate concentration in our control specimens (3.54 ± 0.2 mg/dL) and even rats with severe Parkinsonism (4.46 ± 0.7 mg/dL) was lower than that of humans (about 6 mg/dL in men) [38]. Similarly, Gong et al. [31] increased the plasma urate level in rats after i.p. injection of urate at doses higher than 200 mg/kg for 10 days (twice daily). This treatment ameliorated the behavioral deficits, DA neuronal loss, and dopamine depletion in the nigrostriatal system of 6-OHDA-treated rats. They induced Parkinsonism in rats through injection of 6-OHDA into the striatum [31], which causes moderate and progressive Parkinsonism [39]. Also, the neurodegeneration produced by 6-OHDA injection into MFB may be so fast and severe that it counteracts the antioxidant potential of urate.
Remarkable DA degeneration and significant decline in the level of striatal dopamine were also observed in 6-OHDA-treated rats with a mild number of rotations. Rotational test is a well-established and common approach for the assessment of the intensity of Parkinsonism in 6-OHDA rat models [40, 41]. However, evaluation of histological, behavioral, and neurochemical outcomes of 6-OHDA neurotoxicity showed that it can only distinguish partial lesions from near-complete lesions (> 90%) of DA neurons in the SNc and cannot discriminate a lesion size of 50–80% [39]. Therefore, in the mild group, DA degeneration was so slight that it could not produce significant behavioral deficits. On the other hand, serum urate level in this group of rats decreased. In human being, clinical symptoms of PD are recognized when more than 60–70% of DA neurons are lost in the SNc [7, 8]. Therefore, the mild group of rats was similar to individuals in early stages of PD with remarkable DA cell death, but without any clinical symptoms. Therefore, serum urate changes can predict mild and subclinical DA degeneration and can predict development of PD.
In conclusion, our findings showed that in 6-OHDA-induced Parkinsonism, there is a significant correlation between the level of reduction in serum urate level and intensity of behavioral deficits, degree of DA neuronal cell death in the SNc, level of reduction in the striatal dopamine level, and severity of oxidative stress in the midbrain. In contrast, the midbrain urate level increased in the injured hemisphere. These findings support previous human studies, describing an important role for urate in the development and progression of PD. However, since further reduction in serum urate level is correlated with more severe behavioral deficits and DA neuronal death, it is difficult to consider a marked neuroprotective role for urate, at least at concentrations obtained in this study. However, higher concentrations of urate may produce significant neuroprotective effects. Our findings introduced serum urate level as a biomarker for PD, since a significant decline was observed in the urate level of 6-OHDA-treated rats, which had major DA cell death in the SNc, but did not show marked behavioral symptoms.
References
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373(9680):2055–2066. https://doi.org/10.1016/S0140-6736(09)60492-X
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68(5):384–386
Moon HE, Paek SH (2015) Mitochondrial dysfunction in Parkinson’s disease. Exp Neurobiol 24:103–116. https://doi.org/10.5607/en.2015.24.2.103
Tsang AHK, Chung KKK (2009) Oxidative and nitrosative stress in Parkinson’s disease. Biochem Biophys Acta 1792:643–650. https://doi.org/10.1016/j.bbadis.2008.12.006
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39(6):889–909. https://doi.org/10.1016/S0896-6273(03)00568-3
Jenner P, Olanow CW (1996) Oxidative stress and the pathogenesis of Parkinson’s disease. Neurol 47:S161–S170
Shulman JM, De Jager PL, Feany MB (2011) Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol 6:193–222. https://doi.org/10.1146/annurev-pathol-011110-130242
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79(4):368–376. https://doi.org/10.1136/jnnp.2007.131045
Frasier M, Chowdhury S, Eberling J, Sherer T (2010) Biomarkers in Parkinson’s disease: a funder’s perspective. Biomark Med 4(5):723–729. https://doi.org/10.2217/bmm.10.89
Ames BN, Cathcart R, Schwiers E, Hochstein P (1981) Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA 78(11):6858–6862
Yeum KJ, Russell RM, Krinsky NI, Aldini G (2004) Biomarkers of antioxidant capacity in the hydrophilic and lipophilic compartments of human plasma. Arch Biochem Biophys 430(1):97–103. https://doi.org/10.1016/j.abb.2004.03.006
Church WH, Ward VL (1994) Uric acid is reduced in the substantia nigra in Parkinson’s disease: effect on dopamine oxidation. Brain Res Bull 33(4):419–425
Davis JW, Grandinetti A, Waslien CI, Ross GW, White LR, Morens DM (1996) Observations on serum uric acid levels and the risk of idiopathic Parkinson’s disease. Am J Epidemiol 144(5):480–484
de Lau LM, Koudstaal PJ, Hofman A, Breteler MM (2005) Serum uric acid levels and the risk of Parkinson disease. Ann Neurol 58(5):797–800. https://doi.org/10.1002/ana.20663
Weisskopf MG, O’Reilly E, Chen H, Schwarzschild MA, Ascherio A (2007) Plasma urate and risk of Parkinson’s disease. Am J Epidemiol 166(5):561–567. https://doi.org/10.1093/aje/kwm127
Alonso A, Rodríguez LA, Logroscino G, Hernán MA (2007) Gout and risk of Parkinson disease: a prospective study. Neurology 69(17):1696–1700. https://doi.org/10.1212/01.wnl.0000279518.10072.df
Chen H, Mosley TH, Alonso A, Huang X (2009) Plasma urate and Parkinson’s disease in the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol 169(9):1064–1069. https://doi.org/10.1093/aje/kwp033
Schwarzschild MA, Macklin EA, Ascherio A (2014) Parkinson Study Group SURE-PD Investigators: urate and neuroprotection trials. Lancet Neurol 13(8):758. https://doi.org/10.1016/S1474-4422(14)70138-3
De Vera M, Rahman MM, Rankin J, Kopec J, Gao X, Choi H (2008) Gout and the risk of Parkinson’s disease: a cohort study. Arthritis Rheum 59(11):1549–1554. https://doi.org/10.1002/art.24193
Annanmaki T, Muuronen A, Murros K (2007) Low plasma uric acid level in Parkinson’s disease. Mov Disord 15(8):1133–1137
Vieru E, Köksal A, Mutluay B, Dirican AC, Altunkaynak Y, Baybas S (2016) The relation of serum uric acid levels with L-Dopa treatment and progression in patients with Parkinson’s disease. Neurol Sci 37(5):743–747. https://doi.org/10.1007/s10072-015-2471-z
Andreadou E, Nikolaou C, Gournaras F, Rentzos M, Boufidou F, Tsoutsou A, Zournas C, Zissimopoulos V, Vassilopoulos D (2009) Serum uric acid levels in patients with Parkinson’s disease: their relationship to treatment and disease duration. Clin Neurol Neurosurg 111(9):724–728. https://doi.org/10.1016/j.clineuro.2009.06.012
Bové J, Perier C (2012) Neurotoxin-based models of Parkinson’s disease. Neuroscience 211:51–76. https://doi.org/10.1016/j.neuroscience.2011.10.057
Blandini F, Armentero MT, Martignoni E (2008) The 6-ydroxydopamine model: news from the past. Parkinsonism Relat Disord 14(Suppl 2):S124–S129. https://doi.org/10.1016/j.parkreldis.2008.04.015
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates, 6th edn. Academic Press, San Diego
Fujita M, Nishino H, Kumazaki M, Shimada S, Tohyama M, Nishimura T (1996) Expression of dopamine transporter mRNA and its binding site in fetal nigral cells transplanted into the striatum of 6-OHDA lesioned rat. Mol Brain Res 39:127–136
Sarookhani MR, Haghdoost-Yazdi H, Sarbazi-Golezari A, Babayan-Tazehkand A, Rastgoo N (2017) Involvement of adenosine triphosphate -sensitive potassium channels in the neuroprotective activity of hydrogen sulfide in the 6-hydroxydopamine- induced animal model of Parkinson’s disease. Behav Pharmacol. https://doi.org/10.1097/FBP.0000000000000358
Sarukhani M, Haghdoost-Yazdi H, Sarbazi A, Babayan-Tazehkand A, Dargahi T, Rastgoo N (2018) Evaluation of the antiparkinsonism and neuroprotective effects of hydrogen sulfide in acute 6-hydroxydopamine- induced animal model of Parkinson’s disease: behavioral, histological and biochemical studies. Neurol Res (unpublished data)
Zhang N, Shu HY, Huang T, Zhang QL, Li D, Zhang GQ, Peng XY, Liu CF, Luo WF, Hu LF (2014) Nrf2 signaling contributes to the neuroprotective effects of urate against 6-OHDA toxicity. PLoS ONE 9(6):e100286. https://doi.org/10.1371/journal.pone.0100286
Zhu TG, Wang XX, Luo WF, Zhang QL, Huang TT, Xu XS, Liu CF (2012) Protective effects of urate against 6-OHDA-induced cell injury in PC12 cells through antioxidant action. Neurosci Lett 506(2):175–179. https://doi.org/10.1016/j.neulet.2011.10.075
Gong L, Zhang QL, Zhang N, Hua WY, Huang YX, Di PW, Huang T, Xu XS, Liu CF, Hu LF, Luo WF (2012) Neuroprotection by urate on 6-OHDA-lesioned rat model of Parkinson’s disease: linking to Akt/GSK3β signaling pathway. J Neurochem 123(5):876–885. https://doi.org/10.1111/jnc.12038
De Luca MA, Cauli O, Morelli M, Simola N (2014) Elevation of striatal urate in experimental models of Parkinson’s disease: a compensatory mechanism triggered by dopaminergic nigrostriatal degeneration? J Neurochem 131(3):284–289. https://doi.org/10.1111/jnc.12809
Fitzmaurice PS, Ang L, Guttman M, Rajput AH, Furukawa Y, Kish SJ (2003) Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov Disord 18(9):969–976. https://doi.org/10.1002/mds.10486
Bowman GL, Shannon J, Frei B, Kaye JA, Quinn JF (2010) Uric acid as a CNS antioxidant. J Alzheimers Dis 19(4):1331–1336. https://doi.org/10.3233/JAD-2010-1330
Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, Snyder J, Zhu YG, Ling ZD (2005) 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci 22(5):1158–1168. https://doi.org/10.1111/j.1460-9568.2005.04281.x
Cooper PH, Novin D, Butcher LL (1982) Intracerebral 6-hydroxydopamine produces extensive damage to the blood-brain barrier in rats. Neurosci Lett 30(1):13–18
Paganoni S, Schwarzschild MA (2017) Urate as a marker of risk and progression of neurodegenerative disease. Neurotherapeutics 14(1):148–153. https://doi.org/10.1007/s13311-016-0497-4
Wen M, Zhou B, Chen YH, Ma ZL, Gou Y, Zhang CL, Yu WF, Jiao L (2017) Serum uric acid levels in patients with Parkinson’s disease: a meta-analysis. PLoS ONE 12(3):e0173731. https://doi.org/10.1371/journal.pone.0173731
Yuan H, Sarre S, Ebinge G, Michotte Y (2005) Histological, behavioural and neurochemical evaluation of medial forebrain bundle and striatal 6-OHDA lesions as rat models of Parkinson’s disease. J Neurosci Methods 144:35–45. https://doi.org/10.1016/j.jneumeth.2004.10.004
Abrous DN, Rodriguez JJ, Montaron MF, Aurousseau C, Le Moal M, Barneoud P (1998). Behavioural recovery after unilateral lesion of the dopaminergic mesotelencephalic pathway: effect of repeated testing. Neurosci 84:213–221
Iancu R, Mohapel P, Brundin P, Paul G (2005) Behavioral characterization of a unilateral 6-OHDA-lesion model of Parkinson’s disease in mice. Behav Brain Res 162:1–10. https://doi.org/10.1016/j.bbr.2005.02.023
Acknowledgements
We would like to thank Mrs. Ayda Faraji for her assistance in stereotaxic surgeries and Ms. Zare for her contribution to immunohistofluorescence experiments. This study was supported by a Grant-in-aid for scientific research from the Research Council of Qazvin University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Sarukhani, M.R., Haghdoost-Yazdi, H. & Khandan-Chelarci, G. Changes in the Serum Urate Level Can Predict the Development of Parkinsonism in the 6-Hydroxydopamine Animal Model. Neurochem Res 43, 1086–1095 (2018). https://doi.org/10.1007/s11064-018-2522-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2522-y