
Abstract
Lipid bodies (LBs) have long been considered to be organelles merely for the storage of neutral lipids. However, recent studies have shown the significance of LBs in signal transduction, especially in glial cells, including microglia. Microglial cells are the resident mononuclear phagocytes in the central nervous system and have a close relationship with the aging process and neurodegenerative diseases. Evidence suggests that LBs accumulate and are remodeled during the aging process and the development of neuroinflammatory conditions. However, the mechanisms underlying the formation of LBs under these conditions and the mechanism by which LB remodeling influences the progression of neurodegeneration remain to be clarified. In this review, we have summarized the findings from recent studies with the aim of further elucidating these issues.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipid bodies (LBs), also referred to lipid droplets (LD), lipid particles and liposomes, are dynamic lipid-rich organelles of various sizes and compositions that contain neutral lipids, such as triacylglycerols (TAG), diacylglycerols (DAG) or steryl esters [1]. The LB surface is composed of a monolayer of amphipathic phospholipids, glycolipids and/or sterols [2] and is also decorated with various proteins, such as PAT family proteins (perilipin, adipose differentiation-related protein (ADRP), tail-interacting protein of 47 kDa (TIP-47) and caveolins [3]. Although the functions of these proteins on LBs were still not fully understood, the identities and functions of various proteins have been revealed gradually by proteomics analyses. These surface proteins play vital roles in the formation, maintenance, metabolism and degradation of LBs under the influence of various stimuli and physiological conditions. LBs were first thought to serve as a reservoir of lipids and to play a vital role in lipid metabolism. However, research suggests that LBs are closely related to atherosclerosis, metabolic disorders [4], cancer [5] and especially, aging and aging-related diseases. Evidence suggests that LB accumulation in the microglia during the aging process is associated with remodeling and altered interactions with other organelles [6]. Thus, we hypothesized that LBs play important roles in brain aging, especially in some neurodegenerative diseases.
Microglial cells are the resident macrophages and immune cells in the central nervous system (CNS) and play an important role in the aging process [7,8,9], neuroinflammation and as well as neuron circuit integrity and plasticity [10]. Most popular models of the aging process and other studies suggest that LBs originate in the endoplasmic reticulum (ER) [11]. ER stress and mitochondrial defects are characteristic of brain aging and result in the microglial cell activation. Another key consequence of mitochondrial dysfunction and reactive oxygen species (ROS) generation is the accumulation of LBs [12]. Activated microglia secretes proinflammatory cytokines, nitric oxide (NO) and ROS [13, 14]. These inflammatory conditions induce LBs to undergo a variety of changes via specific signaling pathways. All these reports imply that LBs play a pivotal role in brain aging.
Dysregulated microglia play a vital role in aging and related neurodegenerative conditions, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases as well as in schizophrenia [15]. The disruption of cholesterol metabolism and inflammation is closely linked to the initiation and progression of AD, which is modulated by liver X receptors (LXRs). APP/PS1 transgenic mice lacking either of the Lxrα or Lxrβ genes exhibit an increasing amyloid plaque load [16]. The disruption of LXRα or LXRβ also leads to excessive LB accumulation in mouse brain [17] We have summarized these studies in this review and proposed three potential mechanisms of LB formation. We also discuss reduction in LB numbers and remodeling as a therapeutic strategy in neurodegenerative diseases.
LB Formation in Microglia
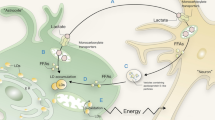
LBs exist in different cell types in various numbers and sizes, potentially reflecting lipid storage capacity. It is widely accepted that LBs originate in the ER [18,19,20]. LBs could be formed under two distinct but not mutually exclusive environmental conditions. First, LBs accumulate in cells in response to exogenous lipid availability [21]. Studies have shown that free fatty acids (FAs) and cholesterol from serum lipoproteins induce the formation of LBs, probably to meet the needs of the cell in terms of energy generation and biosynthesis of steroid hormones [22, 23]. Second, LB biogenesis is induced by a variety of cellular stress conditions, including starvation [24], inflammation and oxidative stress [21], the latter being closely associated with the aging process in microglia. Based on this emerging evidence, we propose three potential molecular mechanisms to account for the formation of LBs; these mechanisms are summarized in Fig. 1.

Three postulated models of lipid body formation cell. LBs consist of a monolayer of amphipathic phospholipids decorated with various proteins (such as perilipin-2 and TIP-47 as shown in above). The figure shows three possible models for LB formation. (1) The first model is characterized by LPS-induced perilipin-2 expression. ILR4 phosphorylates TAK1 following LPS stimulation, and induces MKK phosphorylation. Phosphorylated MKKs activate the JNK and p38 pathways, leading to LB formation through expression of perilipin-2 by activating the downstream targets, AP-1 and ELK1. (2) In the second model, mitochondrial defects increase ROS production, which further activates the JNK and Akt-mTORC1-SREBP pathways in neurons, leading to accumulation of LBs in the surrounding microglia. (3) In the third model, the absence or dysfunction of CPT2 prevents the entry of TAG into the mitochondria for β-oxidation, leading to cytosolic accumulation and LB formation
The first two potential mechanisms are based on the observation that LB formation in the microglia is induced by ROS, generated either in the microglial cell itself or in neurons in the vicinity. This discovery was the result of intense investigations and continues to be a focus of research [25]. Although the relationship between ROS and LB formation has been known for some time, this association in the context of microglia has only recently been discovered. Lee et al. showed that ROS induced by hyperoxia/hypoxia resulted in loss of mitochondrial DNA (mtDNA) and also enhanced LB formation. This group also showed that LB formation is greatly influenced by heme oxygenase (HO)-1, an inducible stress protein [21]. Subsequently, Khatchadourian et al. reported that ROS produced by lipopolysaccharide (LPS)-stimulated microglia induce LB formation via the JNK and p38 MAPK stress signaling pathways, in which LD-associated protein perilipin-2 (also known as adipose differentiation-related protein, ADRP) and cytosolic phospholipase A2 (cPLA2-α) play vital roles [25]. The mechanism by which ROS induce LB formation and their relationship with neurodegeneration represent a current focus of research in this area. Recently, Liu et al. demonstrated that ROS induced by mitochondrial defects in microglia stimulated the formation of LBs and promoted neurodegeneration [12]. In fact, a number of seemingly autonomous observations indicate the close relationship between LBs and neurodegeneration. These observations include the discovery of mitochondrial mutants that are closely associated with neurodegenerative diseases and aging [26] as well as the correlation between LB accumulation and increased ROS, an effect that is ameliorated by a reduction in ROS. Furthermore, elevated ROS levels promote JNK/SREBP activation, which leads to LB accumulation and most importantly, to a reduction in LBs in microglia resulting in delayed neurodegeneration. Based on these observations, it was concluded that ROS and mitochondrial dysfunction stimulates neuronal c-Jun-N-terminal kinase (JNK) and sterol regulatory element binding protein (SREBP) activity, leading to LB accumulation in microglia, and probably resulting in neurodegeneration [12].
Thus, we postulate three possible mechanisms for LB formation. The first mechanism proposed for LB formation is based primarily on experiments involving LPS-induced ROS produced in microglia. LPS is an outer cell wall component of Gram-negative bacteria that is widely used to induce ROS generation both in vivo and in vitro [27]. LPS is also often used as a proinflammogen to stimulate the microglial activation that occurs as part of the aging process [28]. Normally, LPS binds to TLR4 to activate downstream signaling of mitogen-activated protein kinase (MAPK), JNK and p38 [29]. MAPK kinases (MKK) phosphorylate p38 and JNK, with resulting activation of the downstream targets activator protein-1 (AP-1) transcription complex (c-Jun/ATF2) and ELK1. This activation of AP-1 induces proinflammatory cytokine expression, which is the key characteristic of microglia in the aging process [30]. Activated AP-1 binds to the Ets/AP-1 binding site, increasing the expression of perilipin-2, a key regulator of LB formation [31]. This increased perilipin-2 expression stimulates LB formation not only in macrophages, but also in microglia [25, 31]. In addition to LPS, oleic acids or low-density lipoproteins have also been shown to induce perilipin-2 expression [32, 33]. Although the detailed mechanism by which perilipin-2 influences LB formation remains to be fully elucidated, a number of hypotheses have been proposed. One is based on the location of perilipin and other enzymes in the ER, where triglycerides accumulate at privileged sites to form nascent LD [11]. The binding of perilipin-2 to LBs does not require other LD proteins, which suggests that perilipin-2 is probably one of the first proteins to bind to LBs [34]. However, there is no direct evidence that perilipin-2 is located in the ER. Alternatively, binding of perilipin-2 to phospholipids at the surface of the nascent LD occurs via hydrophobic and electrostatic interactions. This binding results in accumulated binding of more lipoproteins and continuously increased size until the LD becomes large enough to bud from the ER, forming a new LB [35, 36]. This hypothesis is supported by several studies showing that perilipin-2 in the cytosol binds to LD in vitro without the assistance of other LD proteins, and also that perilipin-2 interacts directly with other LD proteins [34, 37,38,39].
The second proposed mechanism of LB formation involves defects in neuronal mitochondria that lead to elevated levels of ROS, resulting in activation of the JNK signaling pathway and also SREBP. This important regulator of LB biogenesis is a highly conserved, membrane-bound, basic helix-loop-helix leucine zipper transcription factor that is crucial for lipid homeostasis [12, 40]. This activation of JNK and SREBP in neurons induces accumulation of LBs in glial cells, including microglia. However, the specific mechanism of LB formation in response to this stimulation by surrounding neurons remains unknown and it is noteworthy that mitochondrial defects in neurons, but not in glial cells, induce downstream LB accumulation in microglia [12].
In addition to non-cell-autonomous LB accumulation, LBs can also accumulate in microglia in a cell-autonomous manner. Thus, a third explanation for LB accumulation in microglia is raised. Triglycerides (TAGs) accumulate to form abundant LBs in glial cells, including microglia, which lack the mitochondrial enzyme carnitine palmitoyltransferase 2 (CPT2) that is necessary for β-oxidation of long-chain FAs [41, 42]. This accumulation of LBs has also been observed in vitro [43]. CPT2 catalyzes the replacement of carnitine by CoA, which is catabolized by β-oxidation and generates ATP in brain tissues [44, 45]. In the absence of CPT2 in microglia, TAGs do not undergo β-oxidation in mitochondria, resulting in LB accumulation in the microglial cytoplasm and a reduction in the energy supply to the surrounding neurons. This suggests a possible relationship between LB accumulation and neurodegenerative diseases via a mechanism that does not involve SREBP, which is an important molecule in non-cell-autonomous LB accumulation [41].
Lipid Bodies and Neurodegenerative Diseases
LBs are associated with neurodegenerative diseases, although LB accumulation has not been reported in patients with neurodegenerative diseases such as Leigh Syndrome or in corresponding animal models resulting from LBs’ transient accumulation and intensive occurrence during the presymptomatic stages [12]. Nevertheless, an association between LB accumulation and neurodegenerative diseases is indicated based primarily on evidence that LBs release proinflammatory molecules. Some studies have shown an increasing number of LBs and alterations in lipid metabolism in autosomal dominant neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), although the specific mechanisms remain unknown.
Alzheimer’s Disease
AD, which is the most common neurodegenerative disease, is closely associated with aging, microglia and the metabolism of lipids, such as cholesterol, resulting in amyloid β accumulation. However, the association of LBs with AD has recently been revealed with the discovery of protein-O-linked N-acetyl-β-d-glucosaminidase (O-GlcNAcase, OGA). Evidence has shown that OGA accumulates on the surface of the nascent LD along with perilipin-2. Furthermore, OGA knockout causes increased levels of perilipin-2 and perilipin-3, suggesting that OGA is a regulator of LD formation. OGA has also been linked with AD via the hexosamine signaling pathway. Therefore, these findings indicate that LB assembly and mobilization are associated with AD [46].
The three isoforms of apolipoprotein E (ApoE) encoded by the ε2, ε3 and ε4 alleles play important roles in lipid metabolism and transport from one tissue or cell to another in the brain. The ε4 allele of the apolipoprotein E gene (ApoE4) is a major risk factor for later onset AD. Studies have revealed that the ApoE4-lipoproteins bind to Aβ with lower affinity than that of the ApoE3-lipoproteins, which may lead to less efficient Aβ clearance by microglial cells [47, 48]. Furthermore, ATP-binding cassette sub-family A member 1 (ABCA1)-deficiency is associated with reduced efflux of cholesterol to exogenous ApoE in the microglia. Expression of both ABCA1 and ApoE is induced following LXR stimulation, which is closely related to the amyloid plaque load [49]. Taken together, these observations indicate the close relationship between lipid metabolism in microglial cells and AD.
Parkinson’s Disease
PD is the second most common neurodegenerative disorder associated with microglia and aging. Studies have shown that activated microglia accumulate in damaged areas of the brain in patients with PD [50, 51]. The release of proinflammatory cytokines, such as IL-1β, by activated microglia has been demonstrated in animal models of PD induced by MPTP, 6-OHDA and LPS [52]. LBs play a role in this process as a site of proinflammatory cytokine storage. In addition, LBs interact with α-synuclein, a key player in PD and triglyceride turnover. Normally, α-synuclein exists in the soluble state in the cytosol and adopts an α-helical structure when it interacts with membranes containing acidic phospholipids [53]. The A53T mutant of synuclein, which causes PD [54], is redistributed to the lipid droplet surface, where it forms oligomers. This is an initial step in the formation of α-synuclein patches, which are a characteristic of PD [55].
Hereditary Sensory Neuropathy Type 1(HSN-1)
Hereditary sensory neuropathy type 1(HSN-1), an autosomal dominant neurodegenerative disease, is caused by SPTLC1 gene missense mutations [56] which encodes the serine palmitoyltransferase long-chain subunit of serine palmitoyltransferase (SPT). This ubiquitously expressed, and highly regulated ER-bound membrane enzyme maintains sphingolipid concentrations and is involved in lipid metabolism and signaling [57,58,59]. Studies have shown increased LB accumulation in HSN-1 patient-derived lymphoblasts, which is consistent with earlier reports that HSN-1 is caused by the accumulation of two neurotoxic sphingolipids [57]. Although co-localization with mitochondria was not detected [56], this evidence is controversial because it was based on immunoblotting of markers of these two organelles in fractions rather than observation in unbroken cells in vivo. This method does not exclude the possibility that these interactions are weak and may be broken during the purification of the fractions.
No research into LBs has yet been conducted in HSN-1 patient-derived microglia. However, since microglia and lymphoblasts are both immune cells, we assume that the results obtained by studying lymphoblasts can be applied in microglia. Thus, we propose the hypothesis that mutations in the SPTLC1 gene result in the production of toxic lipids by the mutated SPT enzyme. These toxic lipids accumulate in LBs, which interact with mitochondria to promote neurodegeneration and induce the death of peripheral neurons to cause HSN-1.
Remodeling of Lipid Bodies in Microglia
So far, we have discussed LB accumulation during the aging process and the potential mechanisms. However, this process involves the active formation of large LBs in preparation for storage of various inflammatory mediators [60]. This remodeling represents the morphological and functional changes in organelles as a result of several types of stimulation, including docosahexaenoic acid (DHA). Furthermore, emerging evidence suggests that the LB remodeling is an early biomarker of neuroinflammation and modulator of neurodegenerative diseases [6]. In this review, we describe the morphological changes and transforming interactions between LBs and other organelles during LB remodeling.
Morphological Changes
LPS is used to stimulate microglia to mimic the state occurring in aging and neuroinflammatory conditions. Using confocal microscopy and transmission electric microscopy (TEM), Tremblay showed that LBs accumulate in various sizes in N9 microglial cells exposed to LPS. Under LPS stimulation, the newly formed LBs were in larger in size than those in the control cells. The electron density was also found to vary, with electron-lucent content observed in LBs in LPS-stimulated microglia. However, following the addition of DHA, LBs reorganized and aggregated, with the formation of medium-sized LBs and electron-dense bodies. This remodeling could be induced by the activation and phosphorylation of p38 [12, 25, 61]. This result was observed in both human and mouse microglia [6].
Interactions with Other Organelles
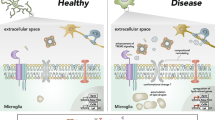
LB remodeling also influences their spatial relationship with other organelles, as shown in Fig. 2.

Remodeling of lipid bodies and interactions with other organelles. The left-hand schematic shows LPS-stimulated mimicry of the aging state in microglia, while the right-hand schematic shows DHA-induced remodeling of microglia. Striking differences occur: stretches decrease obviously through this remodeling; the number and size of LB reduces significantly; mitochondrial function is greatly rescued with increasing cristae and membrane integrity through remodeling; interaction of LB with mitochondria and ER increases. Possible regulators are shown in the box (perilipin-2 and Rab18; perilipin-2, perilipin-5 and SNAP23, α-SNAP, Erg6 and Pet10)
Mitochondria
In LPS-stimulated microglia, LBs and mitochondria are in close proximity, but with few direct contacts and no fusion. DHA induces numerous direct contacts with mitochondria and LB remodeling. Interestingly, LPS stimulation results in partial or even complete loss of mitochondrial cristae and the double membrane, leading to decreased mitochondrial membrane potential and ROS generation. ROS not only stimulates the formation of LBs and release of TNFα, IL-1β and NO, leading to inflammation and neurodegeneration, but also causes damage to mtDNA, proteins and lipids, and influences phagocytosis. Dysregulated phagocytosis promotes cytokine production, leading to mitochondrial modification and LB remodeling [61,62,63]. DHA rescues mitochondrial integrity and ameliorates the defects associated with mitochondrial damage, probably through LB remodeling [61].
The molecules involved in the LB-mitochondrial interaction in microglia have not yet been fully clarified, although this interaction has been elucidated in other cells. LBs play an important role in cells under conditions of starvation stress by functioning as storage and transportation compartments for FAs. Furthermore, the LB-mitochondria interaction is beneficial in starved cells, which use LBs as a conduit for β-oxidation of FAs by mitochondria [24]. However, the mechanisms underlying the responses of microglia under nutrient stress conditions and the proteins involved in the fusion of LBs and mitochondria remain to be clarified. Perilipin-2 (ADFP) is found in significant levels in the mitochondrial fraction, suggesting a potential interaction with mitochondria. Synaptosome-associated protein 23 (SNAP23) and the soluble NSF adaptor protein receptor (α-SNAP) are also found in the LB-mitochondria interactional area. Ablation of SNAP23 reduces perilipin-2 levels, indicating a decrease in LB-mitochondria complex formation [64]. Perilipin-5, also known as LB-associated protein, was first detected in isolated mitochondria and recruits mitochondria to the LB surface. Moreover, perilipin-5 inhibits LB hydrolysis and prevents excessive fatty acid (FA) oxidation in the mitochondria by storing FAs transiently in the LBs [65]. Erg6 and Pet10 are two proteins located in LBs that have been shown to interact with a series of mitochondrial proteins, including Mcr1, Mdv1, Om14 and Cox7, using bimolecular fluorescence complementation (BiFC) assays [66], but further underling mechanisms still need to be elucidated.
Endoplasmic Reticulum
In contrast to the interaction between LBs and other organelles, the LB–ER interaction has been regarded as the result of budding of LBs from the ER [67,68,69]. Excess FAs and cholesterol are stored in the ER as neutral lipids, such as triglycerides and cholesterol esters. The terminal enzymes involved in neutral lipid synthesis, including acyl-CoA: cholesterol acyltransferase (ACAT) and acyl-CoA: diacylglycerol acyltransferase (DGAT), are also localized in the ER [70]. Acyl-CoA synthetase 3 (ACSL3) is another enzyme that is redistributed from the ER to LBs and is necessary for the formation of LBs in the early stages [11]. The hydrophobic nature of neutral lipids is also important for the budding of lipid droplets from the ER; this widely accepted hypothesis for LBs formation is reviewed elsewhere [20]. There is, however, evidence also showing that LB–ER interactions are independent of LB formation [71, 72]. There is a lack of direct evidence to support both of the hypotheses that have been proposed previously. Although the detailed molecular mechanism of the LB–ER interaction in microglia remains to be elucidated, molecules involved in this process have been identified in other non-adipocyte cells. Proteomic analyses have revealed a number of Rab proteins in LBs. Among them, Rab18 plays a central role in the LB–ER interaction and Rab18 overexpression significantly reduces perilipin-2 levels. However, in contrast to the effects in the LB–mitochondrial interaction, reduced perilipin-2 induces membrane apposition between LBs and the ER, as well as the ER-derived membrane [73]. Furthermore, regulation of the LB–ER interactions is dependent on the GTP/GDP state of Rab18 [74].
Few focal contacts between LBs and stretches of ER occur in response to LPS stimulation. However, following the addition of DHA, electron microscopy showed stretches of ER running parallel to the LBs and their intermingling over long distances, suggesting the occurrence of specifically orientated functional interactions induced by LB remodeling [6]. Studies have shown that LBs are related to, and probably derive from the ER, which contains enzymes involved in LB formation and accumulation. Budding from the ER occurs once the LBs reach a certain size [18, 75, 76]. Furthermore, this remodeling is critical for neurodegeneration. DHA treatment increases the direct contacts between these two organelles as well as the levels of phosphatidyl serine (PS), which is the most abundant negatively charged phospholipid in the eukaryotic membrane [77]. PS is critically involved in signal transduction, suggesting that DHA-induced remodeling plays a role in releasing proinflammatory cytokines in the microglia [78].
Lipofuscin Granules
Lipofuscin granules, which are formed in microglia and neurons during the aging process in animals as a result of oxidative stress [79], interfere with several metabolic processes [80] and are implicated in the development of neurodegenerative diseases. After remodeling, LBs are juxtaposed to very large lipofuscin granules, with some regions of fusion [6]. However, the significance of this interaction in neurodegeneration remains to be clarified.
Perspective
In the main, we have discussed LB formation and remodeling in relation to neurodegenerative diseases. Studies have shown that LB accumulation results in the release of proinflammatory molecules and neuroinflammation, which serves as a signaling platform and indicates the potential of LBs as therapeutic targets in neurodegenerative diseases [81, 82]. Two aspects of LB function are implicated as targets for the treatment of aging-related neurodegenerative diseases; LB remodeling and reduction in LB numbers, especially large LBs. As we have discussed earlier in this review, LB remodeling induced by DHA restores normal LB function in aging microglia by increasing contacts with other organelles that are required for normal function of activated microglia [57]. DHA treatment has also been shown to reduce the number of large LBs [6, 83], thus, indicating the marked potential of ω-3 polyunsaturated fatty acids (PUFAs) for the treatment of neurodegenerative diseases.
Even with these exciting advances in our understanding of the role of microglia in aging and neurodegeneration, significant challenges remain. First, relevant animal models of neurodegenerative disease have not yet been established. Moreover, reports of LBs in humans with neurodegenerative diseases are scarce, probably because LB formation and degradation is a highly dynamic process, which is therefore difficult to monitor. However, overcoming the difficulties associated with investigations of neurodegenerative diseases will enable researchers to gain a further understanding of the role of LBs in these diseases.
Abbreviations
- ADRP:
-
Adipose differentiation-related protein
- AD:
-
Alzheimer’s disease
- CNS:
-
Central nervous system
- JNK:
-
c-Jun-N-terminal kinase
- DAG:
-
Diacylglycerols
- DHA:
-
Docosahexaenoic acid
- ER:
-
Endoplasmic reticulum
- HSN-1:
-
Hereditary sensory neuropathy type 1
- LBs:
-
Lipid bodies
- LPS:
-
Lipopolysaccharide
- LXRs:
-
Liver X receptors
- OGA:
-
O-GlcNAcase
- SPT:
-
Palmitoyltransferase
- SNAP23:
-
Synaptosome-associated protein 23
- CPT2:
-
Palmitoyltransferase 2
- PD:
-
Parkinson’s disease
- PS:
-
Phosphatidyl serine
- SREBP:
-
Sterol regulatory element binding protein
- TEM:
-
Transmission electric microscopy
- TAG:
-
Triacylglycerols
References
Murphy DJ (2001) The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40:325–438
Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T (2002) The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J Biol Chem 277:44507–44512
Bickel PE, Tansey JT, Welte MA (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta 1791:419–440
Bell M, Wang H, Chen H, McLenithan JC, Gong DW, Yang RZ, Yu D, Fried SK, Quon MJ, Londos C, Sztalryd C (2008) Consequences of lipid droplet coat protein downregulation in liver cells: abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes 57:2037–2045
Bozza PT, Viola JP (2010) Lipid droplets in inflammation and cancer. PLEFA. doi:10.1016/j.plefa.2010.02.005
Tremblay ME, Zhang I, Bisht K, Savage JC, Lecours C, Parent M, Titorenko V, Maysinger D (2016) Remodeling of lipid bodies by docosahexaenoic acid in activated microglial cells. J Neuroinflammation 13:116
McGeer PL, McGeer EG (2001) Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging 22:799–809
von Bernhardi R (2007) Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox Res 12:215–232
von Bernhardi R (2010) Immunotherapy in Alzheimer’s disease: where do we stand? Where should we go? J Alzheimers Dis 19:405–421
Sierra A, Abiega O, Shahraz A, Neumann H (2013) Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 7:6
Kassan A, Herms A, Fernandez-Vidal A, Bosch M, Schieber NL, Reddy BJ, Fajardo A, Gelabert-Baldrich M, Tebar F, Enrich C, Gross SP, Parton RG, Pol A (2013) Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol 203:985–1001
Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, Bellen HJ (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160:177–190
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Gavgiotaki E, Filippidis G, Kalognomou M, Tsouko AA, Skordos I, Fotakis C, Athanassakis I (2015) Third Harmonic Generation microscopy as a reliable diagnostic tool for evaluating lipid body modification during cell activation: the example of BV-2 microglia cells. J Struct Biol 189:105–113
Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P (2007) Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci USA 104:10601–10606
Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA (2002) Liver X receptors in the central nervous system: From lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci USA 99:13878–13883
Martin S, Parton RG (2006) Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol 7:373–378
Wang CW 2016 Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta 1861:793–805
Hashemi HF, Goodman JM (2015) The life cycle of lipid droplets. Curr Opin Cell Biol 33:119–124
Lee SJ, Zhang J, Choi AM, Kim HP (2013) Mitochondrial dysfunction induces formation of lipid droplets as a generalized response to stress. Oxid Med Cell Longev 2013:327167
Gubern A, Casas J, Barcelo-Torns M, Barneda D, de la Rosa X, Masgrau R, Picatoste F, Balsinde J, Balboa MA, Claro E (2008) Group IVA phospholipase A2 is necessary for the biogenesis of lipid droplets. J Biol Chem 283:27369–27382
Gubern A, Barcelo-Torns M, Barneda D, Lopez JM, Masgrau R, Picatoste F, Chalfant CE, Balsinde J, Balboa MA, Claro E (2009) JNK and ceramide kinase govern the biogenesis of lipid droplets through activation of group IVA phospholipase A2. J Biol Chem 284:32359–32369
Rambold AS, Cohen S, Lippincott-Schwartz J (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32:678–692
Khatchadourian A., Bourque SD, Richard VR, Titorenko VI, Maysinger D. (2012) Dynamics and regulation of lipid droplet formation in lipopolysaccharide (LPS)-stimulated microglia. Biochim Biophys Acta 1821:607–617
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553
Buchanan MM, Hutchinson M, Watkins LR, Yin H (2010) Toll-like receptor 4 in CNS pathologies. J Neurochem 114:13–27
Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9:537–549
Gu JQ, Ikuyama S, Wei P, Fan B, Oyama J, Inoguchi T, Nishimura J (2008) Pycnogenol, an extract from French maritime pine, suppresses Toll-like receptor 4-mediated expression of adipose differentiation-related protein in macrophages. Am J Physiol Endocrinol Metab 295:E1390–E1400
Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, Kimmel AR, Londos C (2005) Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J Biol Chem 280:42841–42847
Wang X, Reape TJ, Li X, Rayner K, Webb CL, Burnand KG, Lysko PG (1999) Induced expression of adipophilin mRNA in human macrophages stimulated with oxidized low-density lipoprotein and in atherosclerotic lesions. FEBS Lett 462:145–150
Sletten A, Seline A, Rudd A, Logsdon M, Listenberger LL (2014) Surface features of the lipid droplet mediate perilipin 2 localization. Biochem Biophys Res Commun 452:422–427
Guo Y, Cordes KR, Farese Jr RV, Walther TC (2009) Lipid droplets at a glance. J Cell Sci 122:749–752
Thiam AR, Farese Jr RV, Walther TC (2013) The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 14:775–786
Yang L, Ding Y, Chen Y, Zhang S, Huo C, Wang Y, Yu J, Zhang P, Na H, Zhang H, Ma Y, Liu P (2012) The proteomics of lipid droplets: structure, dynamics, and functions of the organelle conserved from bacteria to humans. J Lipid Res 53:1245–1253
Nakamura N, Akashi T, Taneda T, Kogo H, Kikuchi A, Fujimoto T (2004) ADRP is dissociated from lipid droplets by ARF1-dependent mechanism. Biochem Biophys Res Commun 322:957–965
Yamaguchi T, Omatsu N, Matsushita S, Osumi T (2004) CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem 279:30490–30497
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Schulz JG, Laranjeira A, Van Huffel L, Gartner A, Vilain S, Bastianen J, Van Veldhoven PP, Dotti CG (2015) Glial beta-oxidation regulates Drosophila energy metabolism. Sci Rep 5:7805
Welte MA (2015) Expanding roles for lipid droplets. Curr Biol 25:R470–R481
Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, Chu CT, Kagan VE, Clark RS (2009) Starving neurons show sex difference in autophagy. J Biol Chem 284:2383–2396
Bonnefont JP, Demaugre F, Prip-Buus C, Saudubray JM, Brivet M, Abadi N, Thuillier L (1999) Carnitine palmitoyltransferase deficiencies. Mol Genet Metab 68:424–440
Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto S, Cha SH, Shimokawa T, Lane MD (2006) The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc Natl Acad Sci USA 103:7282–7287
Keembiyehetty CN, Krzeslak A, Love DC, Hanover JA (2011) A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J Cell Sci 124:2851–2860
LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem 269:23403–23406
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV (2008) apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest 118:4002–4013
Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL (2004) Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem 279:41197–41207
Sawada M, Imamura K, Nagatsu T (2006) Role of cytokines in inflammatory process in Parkinson’s disease. J Neural Transm 70:373–381
Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2003) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol (Berl) 106:518–526
Long-Smith CM, Sullivan AM, Nolan YM (2009) The influence of microglia on the pathogenesis of Parkinson’s disease. Prog Neurobiol 89:277–287
McPhee JC, Dang YL, Davidson N, Lester HA (1998) Evidence for a functional interaction between integrins and G protein-activated inward rectifier K+ channels. J Biol Chem 273:34696–34702
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL (2002) Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J Biol Chem 277:6344–6352
Marshall LL, Stimpson SE, Hyland R, Coorssen JR, Myers SJ (2014) Increased lipid droplet accumulation associated with a peripheral sensory neuropathy. J Chem Biol 7:67–76
Penno A, Reilly MM, Houlden H, Laurá M, Rentsch K, Niederkofler V, Stoeckli ET, Nicholson G, Eichler F, Brown RH Jr., von Eckardstein A, Hornemann T (2010) Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285:11178–11187
Hornemann T, Richard S, Rutti MF, Wei Y, von Eckardstein A (2006) Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem 281:37275–37281
Yard BA, Carter LG, Johnson KA, Overton IM, Dorward M, Liu H, McMahon SA, Oke M, Puech D, Barton GJ, Naismith JH, Campopiano DJ (2007) The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J Mol Biol 370:870–886
Melo RC, D’Avila H, Wan HC, Bozza PT, Dvorak AM, Weller PF (2011) Lipid bodies in inflammatory cells: structure, function, and current imaging techniques. J Histochem Cytochem 59:540–556
Chang PK, Khatchadourian A, McKinney RA, Maysinger D (2015) Docosahexaenoic acid (DHA): a modulator of microglia activity and dendritic spine morphology. J Neuroinflammation 12:34
Chen S, Zhang H, Pu H, Wang G, Li W, Leak RK, Chen J, Liou AK, Hu X (2014) n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci Rep 4:7458
Lu X, Ma L, Ruan L, Kong Y, Mou H, Zhang Z, Wang Z, Wang JM, Le Y (2010) Resveratrol differentially modulates inflammatory responses of microglia and astrocytes. J Neuroinflammation 7:46
Jagerstrom S, Polesie S, Wickstrom Y, Johansson BR, Schroder HD, Hojlund K, Bostrom P (2009) Lipid droplets interact with mitochondria using SNAP23. Cell Biol Int 33:934–940
Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52:2159–2168
Pu J, Ha CW, Zhang S, Jung JP, Huh WK, Liu P (2011) Interactomic study on interaction between lipid droplets and mitochondria. Protein cell 2:487–496
Novikoff AB, Novikoff PM, Rosen OM, Rubin CS (1980) Organelle relationships in cultured 3T3-L1 preadipocytes. J Cell Biol 87:180–196
Stemberger BH, Walsh RM, Patton S (1984) Morphometric evaluation of lipid droplet associations with secretory vesicles, mitochondria and other components in the lactating cell. Cell Tissue Res 236:471–475
Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RG, Goodman JM (2007) The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci USA 104:20890–20895
Buhman KK, Chen HC, Farese RV (2001) The enzymes of neutral lipid synthesis. J Biol Chem 276:40369–40372
Zehmer JK, Huang Y, Peng G, Pu J, Anderson RG, Liu P (2009) A role for lipid droplets in inter-membrane lipid traffic. Proteomics 9:914–921
Robenek H, Hofnagel O, Buers I, Robenek MJ, Troyer D, Severs NJ (2006) Adipophilin-enriched domains in the ER membrane are sites of lipid droplet biogenesis. J Cell Sci 119:4215–4224
Ozeki S, Cheng J, Tauchi-Sato K, Hatano N, Taniguchi H, Fujimoto T (2005) Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J Cell Sci 118:2601–2611
Martin S, Driessen K, Nixon SJ, Zerial M, Parton RG (2005) Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J Biol Chem 280:42325–42335
Murphy DJ (2001) The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40:325–348
Robenek MJ, Severs NJ, Schlattmann K, Plenz G, Zimmer KP, Troyer D, Robenek H (2004) Lipids partition caveolin-1 from ER membranes into lipid droplets: updating the model of lipid droplet biogenesis. FASEB J 18:866–868
Herms A, Bosch M, Ariotti N, Reddy BJ, Fajardo A, Fernandez-Vidal A, Alvarez-Guaita A, Fernandez-Rojo MA, Rentero C, Tebar F, Enrich C, Geli MI, Parton RG, Gross SP, Pol A (2013) Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Curr Biol 23:1489–1496
Schonthal AH (2012) Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica 2012:857516
Tremblay ME, Zettel ML, Ison JR, Allen PD, Majewska AK (2012) Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60:541–558
Kennedy CJ, Rakoczy PE, Constable IJ (1995) Lipofuscin of the retinal pigment epithelium: a review. Eye 9(Pt 6):763–771
Murphy S, Martin S, Parton RG (2009) Lipid droplet-organelle interactions; sharing the fats. Biochim Biophys Acta 1791:441–447
Lecchi C, Invernizzi G, Agazzi A, Modina S, Sartorelli P, Savoini G, Ceciliani F (2013) Effects of EPA and DHA on lipid droplet accumulation and mRNA abundance of PAT proteins in caprine monocytes. Res Vet Sci 94:246–251
Yurko-Mauro K, Alexander DD, Van Elswyk ME (2015) Docosahexaenoic acid and adult memory: a systematic review and meta-analysis. PLoS ONE 10:e0120391
Acknowledgements
This work was supported by the National Science Foundation of China (81373150 to Wei Ge and Benhong Xu).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Hu, X., Xu, B. & Ge, W. The Role of Lipid Bodies in the Microglial Aging Process and Related Diseases. Neurochem Res 42, 3140–3148 (2017). https://doi.org/10.1007/s11064-017-2351-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2351-4