
Abstract
Decades of research suggest that alterations in calcium are central to the pathophysiology of Alzheimer’s Disease (AD). Highly reproducible changes in calcium dynamics occur in cells from patients with both genetic and non-genetic forms of AD relative to controls. The most robust change is an exaggerated release of calcium from internal stores. Detailed analysis of these changes in animal and cell models of the AD-causing presenilin mutations reveal robust changes in ryanodine receptors, inositol tris–phosphate receptors, calcium leak channels and store activated calcium entry. Similar anomalies in calcium result when AD-like changes in mitochondrial enzymes or oxidative stress are induced experimentally. The calcium abnormalities can be directly linked to the altered tau phosphorylation, amyloid precursor protein processing and synaptic dysfunction that are defining features of AD. A better understanding of these changes is required before using calcium abnormalities as therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellular calcium regulation and metabolism/mitochondria are abnormal in Alzheimer’s Disease (AD). The understanding of the mechanism of these changes, their interactions and relationship to AD are limited. Both variables are dynamic and best measured in living cells. Since living neurons in AD brains cannot be studied, measures have to be made in “imperfect” model systems. Calcium homeostasis and mitochondria/metabolism are intimately linked. Most regulators of calcium homeostasis are modulated by metabolism including ATP, phosphorylation and reactive oxygen species (ROS). On the other hand, calcium directly effects mitochondrial metabolism and the production of oxidative stress [i.e., reactive oxygen species (ROS)]. AD-like changes in metabolism and oxidative stress can also lead to AD like change in calcium. An understanding of AD related changes in calcium and mitochondria/oxidative stress and their interactions will provide possibilities for new therapeutic intervention.
The changes in calcium and mitochondria/metabolism cause changes in protein processing that can lead to β-amyloid (Aβ) containing plaques and the hyperphosphorylated tau protein (i.e., tangles), the defining pathologies of AD. Calcium increases the formation of Aβ [1]. Oxidative stress is prominent in AD brains [2]. Enhanced ROS occurs in peripheral cells and is an early event that precedes plaque and tangle formation in transgenic mice [3–6]. The enzymes catalyzing Aβ formation are sensitive to oxidants so that any interruption of metabolism or abnormal ROS production increases Aβ production [7]. Reduced glucose metabolism always accompanies AD and has been directly linked to tau phosphorylation and the formation of tangles [8].
Cellular Calcium Regulation
Calcium regulates many cellular processes and a basic understanding of cellular calcium regulation is essential to understand the calcium abnormalities in AD. Calcium permeability channels including NMDA channels, AMPA receptors, nicotinic receptors and store operated calcium channels control calcium entry from outside the cell. An immense literature exists for each of these and they vary with each cell type. Calcium is also maintained by intracellular calcium signaling and the most replicable AD related changes are in intracellular calcium regulation. This review will focus on the aspects of calcium regulation that are well documented to be altered in AD and their interaction with metabolism.
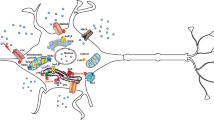
Endoplasmic Reticulum (ER) Calcium Store (Fig. 1)

Regulation of calcium in neurons. The details are provided in the text. The yellow circles indicate the process is regulated by ROS, ATP or phosphorylation and by presenilins. (Color figure online)
Endoplasmic reticulum (ER) is the main intracellular calcium store (Fig. 1) [9, 10]. ER calcium signaling maintains synaptic plasticity by regulating synaptic transmission and membrane excitability [11]. While cytosolic calcium is 100 nM, ER calcium may be about a 1000 times higher. The ER lumen contains high concentrations of Ca2+ binding proteins so the total amount of calcium in the lumen may be 1 mM. The concentration of free Ca2+ in the ER has been estimated to be between 100 and 700 nM [12]. High ER calcium is an important co-factor for ER chaperones [13]. ER calcium is maintained by very specific channels and transporters. The calcium is transported into the ER by a Ca2+-ATPase transporter. Calcium leaves the ER by two release channels on the outer membrane of the ER. One is activated by IP3 and one by ryanodine. In addition, a calcium leak channel has been proposed (Fig. 1).
Transport into the ER by the Sarco/Endoplasmic Reticulum Ca2+ATPase (SERCA)
Resting cytosolic Ca2+ is maintained at low levels by sarco ER Ca2+-ATPase (SERCA) pumps [14]. The SERCA pumps have the highest affinity for Ca2+ removal from the cytosol. In combination with plasma membrane Ca2+-ATPases and transporters, SERCA pumps determine the resting cytosolic Ca2+ concentration. Three differentially expressed genes encode at least five isoforms of the SERCA pump. SERCA2b is ubiquitously expressed in smooth muscle tissues and non-muscle tissues including neurons [15]. Activators of SERCA have been used to treat diseases associated with ER stress [16]. SERCA is regulated by phosphorylation (ATP), ROS as well as calcium [17].
The Inositol 1,4,5-Trisphosphate Receptor (IP3R) Activated Release Channel
The IP3Rs, a family of Ca2+ release channels, are concentrated in the soma and localized predominately in the endoplasmic reticulum. The IP3R channels are tetramers of IP3R molecules of multiple isoforms [12]. Activation of these channels release Ca2+ into the cytoplasm in response to IP3, which is produced by diverse stimuli such as bombesin or bradykinin. The channel gating is regulated not only by IP3, but by other ligands as well, in particular cytoplasmic Ca2+. Low concentrations of cytoplasmic calcium stimulate IP3R whereas high concentrations inhibit. Other ligands such as ATP, regulate channel activity mainly by potentiating Ca2+ activation. Together, cytoplasmic free ATP and IP3 act as allosteric regulators to tune the activation and inhibition. Phosphorylation by protein kinase A enhances single channel activity by approximately fivefold by increasing the apparent sensitivity to IP3 [18].
Ryanodine Receptors (RyR)
RyR are primarily localized in the distal processes, spines, dendrites and presynaptic terminals, but they are also present in soma [19–23]. RyRs are homotetramers that are comprised of a huge N-terminal cytoplasmic domain that serves as a scaffold for channel regulators while the remaining domain is in the ER lumen. Ca2+ activates RyRs at low nanomolar concentrations. Caffeine and ryanodine are pharmacological modulators of RyRs. Caffeine-evoked [Ca2+]i elevations are sensitive to pharmacological modulators interacting with RyRs or with SERCA pumps. These caffeine induced [Ca2+]i responses are blocked by ryanodine, ruthenium red, and procaine and disappear after inhibition of SERCA-dependent ER Ca2+ uptake with thapsigargin or cyclopiazonic acid. Caffeine-induced Ca2+ increases occur in individual spines of cultured hippocampal neurons, which are rich in RyR. Ryanodine locks the RyR channel to an “open state” at low concentrations [23]. RyRs are also directly or indirectly modulated by other channels and kinases. CaMKII associates with, phosphorylates, and regulates the activity of RyR. Mutations in RyR or alterations in the post-translational modifications of RyRs (i.e. hyper-phosphorylation, oxidation and nitrosylation) can shift RyRs from a finely regulated state to an unregulated Ca2+ leak channel [23]. ROS can also modify calcium regulation. For example, H2O2 can modify the redox state of RyR by increasing its S-glutathionylation, which potentiate Ca2+ release [24].
Store Operated Calcium Entry (SOCE), Which is also Known as Capacitative Calcium Entry (CCE)
Depletion of ER calcium stores triggers influx of extracellular calcium into the cytosol that is rapidly used to refill the internal calcium stores. SOCE provides slow responses in calcium and are necessary to support the stability and maintenance of mushroom spines on neurons [25] and the proper function of calcium/calmodulin dependent processes including protein kinase 2 and calcium/calmodulin [25]. Stromal Interacting Molecules (STIM1 and STIM2) are required for SOCE (Fig. 1) [26, 27]. These proteins have a single transmembrane region with a putative Ca2+ binding domain in the lumen of the ER. Calcium store depletion causes a rapid translocation of STIM1 and STIM2 into puncta that accumulate near the plasma membrane. This leads to the subsequent activation of the plasma membrane ORAI Ca2+ channel to replenish Ca2+ stores in the ER [28].
STIM2-nSOC-CaMKII pathway plays an essential role in maintenance of mushroom spines in healthy neurons [29]. Postsynaptic spines contain multiple potential sources of Ca2+ influx, such as NMDAR, AMPAR, and voltage gated calcium channels [30], which provide rapid and massive Ca2+ influx during stimulation and remain silent at rest. Thus, these channels are poorly suited to support long-term maintenance mechanism. Steady-state CaMKII activity in the spines depends on continuous Ca2+ influx via STIM-regulated SOCE pathways [29].
Mitochondrial Associated Proteins and the ER
Multiple proteins in the cytosol serve as signaling proteins to and from the mitochondria. These proteins also interact with the ER. For example, the BCL-2 family of proteins, which interact with mitochondria in cell death pathways, also alter ER calcium homeostasis [31]. Cells lacking Bax/Bak have a decreased ER calcium [32]. Bcl-2 inhibits IP3 induced calcium release [33].
Mitochondria and ER calcium are linked functionally through energy dependent processes such as ATP and ROS, and morphologically through mitochondrial associated membranes (MAMS) (Fig. 1). Several proteins participate in stabilization of MAMS and alter Ca2+ transfer between ER and mitochondria. Glucose Regulated Protein 75 (GRP75) regulates the coupling of the voltage dependent anion channel of the mitochondria to the ER IP3R [34]. MAMS are enriched in mitofusin2 and dynamin related GTPases. ER mitofusin2 interacts with mitochondrial mitofusin1/2. Mitochondrial Fis1 and ER-located Bap31 are proposed as ER–mitochondria tethering proteins [35]. MAMS are also enriched with presenilins. Presenilins have been shown to affect ER–mitochondria associations and related functions as measured by cholesteryl ester and phospholipid synthesis. The interactions are increased significantly in presenilin-mutant cells and in fibroblasts from patients with AD [35, 36]. Similarly, alterations in the ER–mitochondrial associations and functions are seen in amyloid precursor protein (APP) transgenic mouse models, and treatment of neurons with Aβ alters ER–mitochondrial–ER contacts [37].
Mitochondrial and ER Interactions in the Regulation of Cellular Calcium (Fig. 1)
Mitochondria are also Important in the Regulation of Cellular Calcium
Voltage dependent anion channels (VDAC) regulate calcium across the outer mitochondrial membrane potential. Several proteins such as the BCL-2 family of proteins can regulate permeability by binding to VDACs [34] and these same proteins also bind to ER protein. The inner mitochondrial membrane potential is regulated by the calcium uniporter which removes calcium from the mitochondria. Calcium transport into the mitochondria is regulated by the mitochondrial membrane potential [38].
Role of ER in Regulating Mitochondrial Calcium, ROS Production and Respiration
Calcium released from the ER has profound effects on mitochondria [10]. Calcium that is released from the ER does not mix with cytosolic calcium but is rapidly taken up by mitochondria in close proximity [39]. In many cell types, agonist induced calcium signals appear in the mitochondria [39, 40]. Isoform specific IP3R are localized close to mitochondrial calcium uptake sites [39]. Suppression of the IP3R leads to suppression of calcium transfer to the mitochondria. At high concentrations calcium regulates the permeability transition which leads to release of calcium and many apoptosis inducing factors. The ER-originating calcium signals and the subsequent increase in mitochondrial calcium will activate the α-ketoglutarate dehydrogenase complex (KGDHC) and the pyruvate dehydrogenase complex (PDHC). Both PDHC and KGDHC produce ROS and NADH [41]. Calcium activation can increase mitochondrial NADH to exceed that needed to sustain oxidative phosphorylation, and lead to an enough electron-saturated respiratory chain that results in increased mitochondrial ROS production. The Ca2+ sensitive sites include several dehydrogenases and substrate transporters together with a post-translational modification of F1-FO-ATPase and cytochrome oxidase [42, 43].
Role of Mitochondria in Regulating ER Calcium
Mitochondria regulate ER calcium by regulating cytosolic calcium and by regulating all of the proteins that regulate calcium through ATP and ROS. Since the Ca2+-ATPase pumps and the IP3R of the ER are regulated by calcium, the local concentration of calcium around the mitochondria will determine the refilling of the ER and spatial temporal calcium signal. Both the SERCA pump, IP3R and RyR are regulated by ATP, which depends on the mitochondria. IP3 receptors are extensively phosphorylated and are controlled by phosphorylation [10]. Hyperphosphorylation of IP3R decreases ER calcium stores. Local NADH levels can also alter IP3 levels. Thus, mitochondria control the ER calcium through ATP, ROS and calcium.
AD-Related Changes in Calcium Homeostasis and Their Interaction with Mitochondrial Function
Changes in calcium with aging and AD have been hypothesized to be important in AD for more than 30 years [44–46]. Initial studies showed that calcium uptake is diminished in fibroblasts from AD patients [46] and lymphocytes [47]. A large number of studies in fibroblasts, animal models of AD [48], autopsy brain studies and genetically manipulated cells of many kinds support the suggestion that abnormal calcium regulation accompanies AD. Calcium uptake is also reduced in brains from aged mice [49]. Mouse models harboring presenilin mutations accumulate calcium in the ER before pathology is present [50–52]. Such changes can be inferred in patients [53]. However, the findings have not lead to better treatments. Thus, a more detailed understanding of the changes as well as the details in calcium regulation in the right cellular systems are required. Each of the calcium regulatory systems outlined above have been implicated in AD.
Genetic engineering of cells and mice with presenilin mutations that cause AD is a powerful tool to study AD-related changes in ER and mitochondrial calcium. Mutations in presenilins 1(PS1) or 2 (PS2) are the cause of most common genetic forms of AD. Presenilins are multifunctional transmembrane proteins that are synthesized and localized in the ER membrane and endosomes [54]. Presenilins have many functions including as a γ-secretase that cleaves several transmembrane proteins, including amyloid precursor protein (APP) [43, 55, 56] (Fig. 2). γ-Secretase plays a central role in regulating intramembrane proteolysis, cleaving the transmembrane domains of hundreds of proteins, including Notch and APP. β-secretase (BACE) processing of the latter exposes a 99-amino-acid C-terminal fragment to γ-secretase. This C-99 retains the transmembrane domain, which γ-secretase first cuts internally between amino acids 48 and 49, or 49 and 50 (so-called epsilon cleavage). This releases into the cytosol either of two APP intracellular domains, AICD49-99 or AICD50-99. Then, γ-secretase proceeds in carboxypeptidase fashion to chop off C-terminal amino acids from the remaining transmembrane domain until Aβ peptides are finally released into the lumen. Most mutations in PS1 or PS2 that lead to AD cause abnormalities in calcium, whereas mutations that do not cause AD do not change calcium. This has made these mutations powerful tools for looking at mechanism.

Processing of amyloid precursor protein and its modification by presenilins
By contrast, few studies have been done to assess the effects of AD causing mutations in APP processing on neuronal calcium regulation. The one study in fibroblasts show the results to be quite different from those in cells from non-genetic forms or those bearing presenilin mutations [57].
IP3 Releasable Stores of Calcium
Exaggerated calcium signaling through IP3R is a disease specific and robust proximal mechanism of AD [43]. ER calcium stores in fibroblasts from AD patients have an exaggerated calcium signal in response to agonists such as bradykinin or bombesin that produce IP3. Early results suggest that the ER calcium pool is exaggerated [58]. Subsequent studies reveal that this exaggerated release is a very robust finding that is common and specific [19, 50, 59–61]. Studies on fibroblasts from hundreds of patients suggest the changes may be diagnostic for AD [43, 58, 62–64]. The abnormalities occur if the fibroblasts are taken before the patient developed AD, which suggests they are trait dependent markers, not state dependent markers [62]. The changes occur in fibroblasts from individuals that have presenilin mutations and those that do not. However, the changes do not occur in fibroblasts from patients bearing mutations in the processing of APP [57]. The increase in IP3 releasable stores in fibroblasts bearing presenilin mutations can be reproduced in mice that have human mutant presenilin. As in humans, the changes occur in fibroblasts. Importantly, the same change occur in neurons from the same mice. Together, the results suggest that neurons in patients with AD bearing presenilin mutations likely have exaggerated internal stores of calcium [48].
These well documented changes in human fibroblasts stimulated experiments in many in vitro models with other cell types. Presenilin mutations increase IP3 releasable calcium stores in multiple cell types including Sf9 cells, chicken DT40 B cells and human lymphoblasts from Familial AD (FAD) patients [20, 22, 43]. In Xenopus oocyte expression systems, photolysis of caged inositol IP3 reveals that the PS1 mutations that cause AD potentiate IP3-mediated release of Ca2+ from internal stores [59]. Single channel recordings of IP3R show that presenilin mutations cause an apparent sensitization of the IP3R channel to IP3, which produces an enhanced IP3R Ca2+ release channel gating [20]. Measures on the behavior of the IP3R measured at the single channel levels also reveal exaggerated IP3 calcium signals in lymphoblasts [43, 58] and other cell types [66, 67]. Further, genetic reduction of the type 1 IP3R by 50% normalized exaggerated Ca2+ signaling. In PS1M146V knock-in mice, reduced IP3R1 expression restores normal ryanodine receptor and cAMP response element-binding protein (CREB)-dependent gene expression and rescues aberrant hippocampal long-term potentiation (LTP) [67]. Presenilins interact with the gating of the IP3R [22]. The IP3R interacts with both wild type and mutant presenilin [20–22]. Electrophysiological recordings of IP3 receptor in FAD patient B cells, cortical neurons of PS1 AD mice and other cells suggest a higher occupancy in a high open probability mode resulting in enhanced calcium signaling [43].
Presenilin mutations alter APP processing and lead to excess Aβ but the changes in calcium are not likely to be secondary to Aβ. Functionally, mutant PS1 has a gain of function consequences when binding to the channels and this was associated with exaggerated [Ca2+]i signaling in intact cells [20, 22]. This was true even if the γ-secretase component of PS1 was inactive. Many FAD-linked PS mutations have been shown to alter Ca2+ homeostasis by Aβ independent mechanisms [43, 68–70]. The changes in calcium with mutated presenilins are also independent of the presenilin secretase activity [43]. Further evidence that Aβ is not involved is that in fibroblasts from patients bearing the APP mutation, the increase in IP3 mediated calcium release does not occur [57]. These observations provide molecular insights into the calcium dysregulation in AD pathogenesis and suggest novel targets for therapeutic intervention [20].
Ryanodine Receptors
The increased calcium release in AD models has also been attributed to enhanced release through ryanodine receptors [51, 60, 69]. Three RyR subtypes encoded by different genes were originally identified in skeletal muscle (type 1 RyR), heart (type 2 RyR), and brain (type 3 RyR) [71]. Ryanodine receptor activity is enhanced in dendrites and synaptic spines from pre-symptomatic mouse models of AD [72, 73]. Basal synaptic function and plasticity mechanisms appear similar between young non-transgenic and transgenic mice. However, when RyRs are manipulated, striking synaptic transmission and plasticity abnormalities are revealed in the transgenic mice. This is in large part due to calcium induced calcium release via the ryanodine receptor [51, 72, 73]. Electrophysiological recordings and two-photon calcium imaging in young (6–8 weeks old) 3xTg-AD and non-transgenic hippocampal slices reveal a marked increase in RyR-evoked calcium release within synapse-dense regions of CA1 pyramidal neurons in the AD mouse model. The RyR2 isoform is selectively increased more than fivefold in the hippocampus of 3xTg-AD mice. These novel findings demonstrate that 3xTg-AD CA1 neurons at pre-symptomatic ages operate under an aberrant calcium signaling and synaptic transmission system long before AD histopathology onset [51].
Several studies suggest precise mechanism for these changes. Caffeine evokes a peak rise of [Ca2+]i in neurons from a 3xTg-AD that is significantly greater than those observed in non-transgenic neurons. This suggests that Ca2+ stores are greater in neurons from 3xTg-AD neurons or that the ryanodine receptor is altered. Protein levels of several Ca2+ binding proteins (SERCA2b, calbindin, calsenilin and calreticulin) implicated in the pathogenesis of AD did not change, whereas ryanodine receptor expression in both PS1 Knock In and 3xTg-AD cortex increases. RyR expression and protein levels also increase in AD brains [74]. The results suggest that the enhanced Ca2+ response to caffeine observed in 3xTg-AD neurons may be attributable to an increase in the steady-state levels of the ryanodine receptor [19].
Presenilins interact with the gating of the RyR [22]. The PS1 N-terminal fragment 1–82 allosterically potentiates RyRs, but the channel still requires Ca2+ for activation [21]. Application of the PS2 N-terminal fragment 1–87 to the cytoplasmic side of the RyR significantly increases single channel activity by favoring higher sublevel openings. The Ca2+ activation and desensitization ranges for RyRs are unchanged. The results demonstrate that PS2 N-terminal fragment 1–87 facilitates RyR gating [75].
Store Activated Calcium Entry (SACE) or Capacitative Calcium Entry (CCE)
SACE is activated in response to lowering calcium content in the ER. The ER calcium overload in mice bearing the M146V presenilin mutation down regulates SACE [25, 43, 48, 61, 65, 70]. Deficits in SACE were evident after agonist stimulation, but not if intracellular calcium stores were completely depleted with thapsigargin. Treatment with ionomycin and thapsigargin revealed that calcium levels within the ER were significantly increased in mutant PS1 knock-in cells. Thus, the overfilling of calcium stores may represent the fundamental cellular defects underlying the alterations in calcium signaling conferred by presenilin mutations [48]. In the presence of IP3, mitochondria buffer the local Ca2+ released from ER to serve as a negative feedback to the SACE [76].
SACE has been examined in considerable morphological detail. Cultured hippocampal neurons expressing mutant PS1 have attenuated SACE that is associated with destabilized dendritic spines, and SACE is rescued by either γ-secretase inhibition or over-expression of STIM1. γ-Secretase activity may physiologically regulate CCE by targeting STIM1 [28]. PS1-associated γ-secretase activity may be important in this process because knockout of PS1 and PS2 or expression of catalytically inactive PS1 mutants (D257A or D385A) is associated with enhanced SACE [28]. Forms of PS1 with FAD-associated mutations enhanced γ-secretase cleavage of the STIM1 transmembrane domain at a sequence that was similar to the gamma-secretase cleavage sequence of APP [28]. Results indicate that γ-secretase activity may physiologically regulate SACE by targeting STIM1 and that restoring STIM1 may be a therapeutic approach in AD [28].
STIM2 has also been implicated in the mutant presenilin related calcium deficits [29]. Specific downregulation of STIM2 occurs in PS1-FAD patient lymphoblasts [77]. In PS-FAD and aging neurons, an increase in ER Ca2+ levels causes compensatory downregulation of STIM2 expression, impaired SACE, reduced steady-state CaMKII activity in the spines, and eventual loss of mushroom spines. Loss of mushroom spines can be linked to memory impairment in aging and AD neurons [29].
STIM2 may also be involved in forms of AD not related to presenilin mutations. As emphasized elsewhere, elevated extracellular Aβ (perhaps due to oxidative stress) accompanies AD. Results are consistent with Aβ acting by down regulating STIM2. Extracellular Aβ may over activate mGluR5 receptor in vulnerable neurons, leading to elevated intracellular Ca2+ levels, compensatory downregulation of STIM2 expression, impaired synaptic SACE, and reduced CaMKII activity. Pharmacological inhibition of mGluR5 or overexpression of STIM2 rescues synaptic changes in internal calcium stores and prevents mushroom spine loss in hippocampal neurons [25].
Abnormalities in SERCA may also Underlie the Calcium Changes
Others have proposed that enhanced activity of the ER calcium pump [14] leads to enhanced loading of the ER lumen [19, 69]. SERCA pump activity is physiologically regulated by presenilin [14]. SERCA activity is diminished in fibroblasts lacking both PS1 and PS2 genes, despite elevated SERCA2b steady state levels. Presenilins and SERCA physically interact. Ca2+ homeostasis, overexpression of wild-type PS1 or PS2 in oocytes causes enhanced IP3-mediated Ca2+ release, an effect that is exacerbated by mutations in both genes [59]. Enhancing presenilin levels in Xenopus oocytes accelerates clearance of cytosolic Ca2+, whereas higher levels of SERCA2b phenocopy PS1 overexpression, accelerating Ca2+ clearance and exaggerating IP3—mediated Ca2+ liberation. The critical role that SERCA2b plays in the pathogenesis of AD is supported by findings that show modulating SERCA activity alters amyloid production. Considerable data suggests a physiological role for the presenilins in Ca2+ signaling via regulation of the SERCA pump.
Calcium Leak Channel
An alternative hypothesis is that presenilins form low conductance ER Ca2+ leak channels and that the mutations in presenilin that cause AD block the leak channels [78–81]. Although not without controversy [82], considerable data supports that conclusion. Some but not all genetic mutations in PS disrupt this function leading to overfilling of ER with calcium. In response, the cell upregulates the function of the ER calcium releasing channels such as IP3 and ryanodine receptors. This link channel function is independent of the gamma-secretase activity [78]. Crystal structures show that presenilin has a large hole that transverses the entire protein that is large enough to allow passage of small molecules [83]. The results are in agreement with other studies utilizing mutagenesis [80]. The crystal structure of presenilin reveals a conspicuous pore in a bundle of nine α-helices, which was originally thought to adopt a novel protein fold. However, the presenilin fold is a variant of the ClC chloride channel/transporter fold. Several mutations that affect calcium leak activity line the presenilin “hole”. The fundamental architectural similarities between presenilin and ClCs may therefore have important implications for understanding the presenilin function, including its possible physiological role as a calcium channel [84].
The suggestion of a role for the calcium leak channel in the cells bearing presenilin mutations was verified by analysis of 250 small interfering RNAs on Ca2+ signals in single cells. The AD–linked protein presenilin-2 and the channel protein ORAI2 prevented overload of ER Ca2+ and that feedback from Ca2+ to membrane lipids [85].
What Causes the Change in Calcium in AD Fibroblasts Without Genetic Mutations?
The increase in releasable stores of calcium have been found in fibroblasts from the small percent (<4%) of patients bearing presenilin mutations as well as in fibroblasts from patients that do not have any known genetic cause who reflect the vast majority of patient population. The previous sections describe efforts made to understand the changes in greater functional and morphological detail in cells bearing presenilin mutations. The experiments described here were designed to determine what might cause the change in non-genetic forms of AD.
Oxidative stress is common in AD and oxidants can lead to changes in calcium that resemble those in AD. Multiple oxidants can induce the change in IP3-releasable calcium stores (Fig. 3). The most effective oxidant in producing AD like changes in calcium is t-butryrl-hydroperoxide [86]. Enhanced H2O2 production in cells expressing mutant PS1 requires the expression of the IP3R. The agonist induced changes in ROS were also totally blocked by using BAPTA to buffer calcium [43]. This suggests the elevated calcium was stimulating ROS production by the mitochondria. Interestingly, SERCA2b is also the most sensitive of the SERCA pumps to oxidative stress [87, 88].

Diverse oxidants induce select changes in releasable calcium stores. The indicated oxidants were added to cells. Internal calcium stores were measured exactly as in cell from AD patients. Data are from [86]
Thus, the findings support the hypothesis that the calcium changes are related to ROS in both cells bearing presenilin mutations and in cells with non-genetic forms of AD.
Lowering the activity of a key mitochondrial enzyme complex, the alpha-ketoglutarate dehydrogenase complex (KGDHC) also increases IP3 releasable stores of calcium (Fig. 4). Neurons cultured from either embryonic or mature mice deficient in KGDHC have an exaggerated IP3-inducible calcium change that resembles the change in AD [89]. Reducing KGDHC in N2a cells by shRNA also increases IP3 releasable calcium [89].

Bradykinin or caffeine releasable calcium stores are exaggerated in response to reduced KGDHC. In the top panel, KGDHC was reduced with shRNA in either N2a cells or cultured neurons. In the lower panel, neurons were cultured from mice with diminished KGDHC [89]
Relation of the IP3 Releasable Calcium to Mitochondria and Mitochondrial Calcium Buffering
The many connections of the ER and mitochondria make it difficult to distinguish cause and effect relationships. Stable expression of PS1 or PS1 mutants does not alter H2O2 production in DT40 cells. However, upon activation both manipulations increase H2O2 production and the increase in cells bearing the PS1 mutation is greater. In the absence of IP3R, the change is not apparent [43]. Qualitatively similar results occur in PC12 cells wherein the increase in ROS is blocked when calcium is buffered [43]. The excess release of calcium results in enhanced generation of ROS. However, in response to excess Ca2+ release, the Ca2+-induced increase in mitochondrial NADH can exceed that needed to sustain oxidative phosphorylation, and lead to an electron-saturated respiratory chain that results in increased mitochondrial ROS production [43]. The reduction in KGDHC leads to an exaggeration in the response to an oxidant challenge [90].
The Changes in Calcium can be Directly Linked to AD like Pathophysiological Changes
The increase in ER calcium release results in altered pre-synaptic and post-synaptic transmission mechanisms [51, 91, 92]. Studies of neurons in hippocampal slices from AD transgenic mice and appropriate controls reveal that upon theta burst stimulation or high-frequency stimulation, input-specific LTP in AD mice have a larger initial amplitude and are more persistent than that in controls. These data suggest that the AD mutations lead to higher degree of LTP induction [91]. Additional studies of either presynaptic (CA3) or postsynaptic (CA1) neurons of the hippocampal Schaeffer-collateral pathway reveal that long-term potentiation induced by theta-burst stimulation decreased after presynaptic, but not postsynaptic deletion of presenilins. Moreover, presynaptic but not postsynaptic inactivation of presenilins alters short-term plasticity and synaptic facilitation. Depletion of endoplasmic reticulum Ca2+ stores by thapsigargin, or blockade of Ca2+ release from these stores by ryanodine receptor inhibitors, mimics and occludes the effects of presynaptic presenilin inactivation. Collectively, these results indicate a selective role for presenilins in the activity-dependent regulation of neurotransmitter release and long-term potentiation induction by modulation of intracellular Ca2+ release in presynaptic terminals, and further suggest that presynaptic dysfunction might be an early pathogenic event leading to dementia and neurodegeneration in AD [92].
Two-photon calcium imaging, flash photolysis of caged glutamate, and patch-clamp electrophysiology in cortical brain slices reveal increased ryanodine receptor-evoked calcium signals within dendritic spine heads, dendritic processes, and the soma of pyramidal neurons from AD transgenic mice. In addition, synaptically evoked postsynaptic calcium responses and calcium signals generated from NMDAR activation are larger in the AD strains. Concurrent activation of RyRs with either synaptic or NMDAR stimulation generate a supra-additive calcium response in the AD strains, suggesting an aberrant calcium-induced calcium release effect within spines and dendrites. The results suggest that presenilin-linked disruptions in RyR signaling and subsequent calcium-induced calcium release via NMDAR-mediated calcium influx alters synaptic function and serves as an early pathogenic factor in AD [73].
The exaggerated calcium store is likely coupled to the pathology. Elevating calcium increases Aβ production [1]. In 3xTg mice, reduced IP3R1 expression profoundly attenuated Aβ accumulation and tau hyperphosphorylation and rescued hippocampal LTP and memory deficits. The critical role that SERCA2b plays in the pathogenesis of AD is underscored by findings that modulating SERCA activity alters Aβ production [14]. Thus, reduction of the calcium change ameliorates the pathology in animal models [67].
Another possible way that the calcium changes contribute to AD is by altering ROS production [43]. Oxidative damage due to ROS is very common in brains of patients that died with AD. Enhanced generation of ROS is believed to be an important component in AD pathogenesis [93–96]. Exaggerated Ca2+ signaling results in enhanced generation of ROS. The increased ROS production in PS1 mutants depends entirely on expression of the IP3R [43]. The reduction in KGDHC that accompanies AD leads to an exaggeration in the response to an oxidant challenge [90]. The calcium changes are present from birth in mice, and perhaps in humans, but the brain compensates. The compensatory process then exaggerates the pathophysiology. The initial response is increased post-synaptic small conductance potassium activated calcium currents and increased presynaptic vesicle release which lead to synaptic depression and increased long term depression [11].
Possible Therapeutic Approaches to Study the Calcium Change
Since the AD related changes in calcium are accompanied by an increase in IP3R and RyRs, blocking these receptors is a possible therapy. However, these channels are involved in many cellular pathways so this approach will likely have many side effects. Nevertheless, blocking RyR with dantrolene has been studied extensively [60, 74, 97, 98]. The stabilizing effect of dantrolene inhibits aberrant activation of the channel and prevents excessive Ca2+ release from intracellular stores. Pharmacological inhibitors of SACE (dantrolene), of calcium-activated potassium channels (apamin), and of calcium-dependent phosphatase calcineurin (FK506) are able to rescue structural plasticity defects in neurons from AD mouse models. Incubation with dantrolene or apamin also rescues LTP defects in hippocampal slices from AD mouse models [99]. Dantrolene treatment fully normalizes ER Ca2+ signaling within somatic and dendritic compartments in early and late stage AD mice in hippocampal slices. The elevated RyR2 levels in AD mice are restored to control levels with dantrolene treatment, as are synaptic transmission, and synaptic plasticity. Aβ deposition within the cortex and hippocampus is also reduced in dantrolene-treated AD mice [100].
Preventing the changes in calcium can also alter the defining pathologies in AD. Dantrolene-induced lowering of RyR-mediated Ca2+ release leads to the reduction of both intracellular and extracellular Aβ load in neuroblastoma cells as well as in primary cultured neurons derived from Tg2576 mice [98]. There are several studies showing that dantrolene treatment is effective in vivo. Aβ deposition within the cortex and hippocampus is also reduced in dantrolene-treated AD mice [100]. Dantrolene significantly improves cognition in a murine model of AD and is associated with a reduction in amyloid plaque burden [98, 100, 101]. However, other studies show that there may be undesirable effects. Long term feeding increases plaque formation and leads to loss of synaptic markers [52]. The difficulty is that dantrolene lacks specificity and may be more directed to muscle ryanodine receptors [102].
A genetic approach has also been used to evaluate the importance of RyR3 in AD pathology. The deletion of RyR3 increased hippocampal neuronal network excitability and accelerated AD pathology, leading to mushroom spine loss and increased amyloid accumulation. In contrast, deletion of RyR3 in older APPPS1 mice (≥6 mo) rescued network excitability and mushroom spine loss, reduced amyloid plaque load and reduced the number of spontaneous seizures. The results suggest that in young AD neurons, RyR3 protects AD neurons from synaptic and network dysfunction, while in older AD neurons, increased RyR3 activity contributes to pathology. These results imply that blockade of RyR3 may be beneficial for those in the later stages of the disease, but RyR activators may be beneficial when used prior to disease onset or in its initial stages [103].
An alternative approach is based on manipulating the STIM2 that is so critical in regulation of internal calcium stores. The Transient Receptor Potential Canonical 6 (TRPC6) and ORAI2 channels form a STIM2-regulated SACE Ca2+ channel complex in hippocampal mushroom spines. A known TRPC6 activator, hyperforin, and a novel SACE positive modulator, NSN21778, can stimulate activity of SACE in the spines and rescue mushroom spine loss in both presenilin and APP knock-in mouse models of AD. Further, the NSN21778 rescues hippocampal long-term potentiation impairment in APP knock-in mouse model. Thus, the STIM2-regulated TRPC6/ORAI2 SACE channel complex in dendritic mushroom spines is a potential new therapeutic target for the treatment of memory loss in AD and that NSN21778 is a potential candidate molecule for therapeutic intervention [104].
Conclusions
Abnormalities in calcium regulation are central to AD pathophysiology—a calcinist view of AD. Although the changes have been known for decades, the targets have not translated to the clinic. Calcium regulation including the interaction of mitochondria to ER and to SACE is very cell-type specific. The initial AD-related changes were all discovered in fibroblasts. Much of the remainder of the modeling has been done in cell lines or mouse or rat neurons. The relation of any of these results to what happens in human neurons or other brain cell types is unknown. This question can now be addressed with the use of neurons derived from fibroblasts. Similar methods could be used to explore these dynamics in other cell types. Stem cell derived neurons provide a better model for exploring detailed mechanistic studies which will be required to determine the clinical implications of our findings.
Abbreviations
- Aβ:
-
Amyloid β peptide
- AD:
-
Alzheimer’s disease
- CCE:
-
Capacitative calcium entry
- CREB:
-
cAMP Response element-binding
- ER:
-
Endoplasmic reticulum
- FAD:
-
Familial Alzheimer’s disease
- IP3 :
-
Inositol 1,4,5-triphosphate
- IP3R:
-
Inositol 1,4,5-triphosphate receptor
- KGDHC:
-
α-Ketoglutarate dehydrogenase complex
- LTP:
-
Long term potentiation
- MAMS:
-
Mitochondrial associated membranes
- PS-1:
-
Presenilin-1
- PS-2:
-
Presenilin-2
- PDHC:
-
Pyruvate dehydrogenase complex
- ROS:
-
Reactive oxygen species
- RyR:
-
Ryanodine receptor
- SACE:
-
Store activated calcium entry
- VDAC:
-
Voltage dependent anion channels
References
Querfurth HW, Selkoe DJ (1994) Calcium Ionophore Increases amyloid beta. Peptide production by cultured cells. BioChemistry 33:4550–4561
Calingasan NY, Uchida K, Gibson GE (1999) Protein-bound acrolein. J Neurochem 72:751–756
Castellani RJ, Lee H-G, Zhu X, Nunomura A, Perry G, Smith MA (2006) Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol 111:503–509
Cecchi C, Fiorillo C, Sorbi S, Latorraca S, Nacmias B, Bagnoli S, Nassi P, Liguri G (2002) Oxidative stress and reduced antioxidant defenses in peripheral cells from familial Alzheimer’s patients. Free Radical Biol Med 33:1372–1379
Cecchi C, Latorraca S, Sorbi S, Iantomasi T, Favilli F, Vincenzini MT, Liguri G (1999) Gluthatione level is altered in lymphoblasts from patients with familial Alzheimer’s disease. Neurosci Lett 275:152–154
Eckert A, Schindowski K, Leutner S, Luckhaus C, Touchet N, Czech C, Müller WE (2001) Alzheimer’s disease-like alterations in peripheral cells from presenilin-1 transgenic mice. Neurobiol Dis 8:331–342
O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R (2008) Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 60:988–1009
Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong C-X (2009) Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132:1820–1832
Decuypere J-P, Monaco G, Missiaen L, De Smedt H, Parys JB, Bultynck G (2011) IP3 receptors, mitochondria, and Ca2+ signaling: implications for aging. J Aging Res 2011:20
Sammels E, Parys JB, Missiaen L, De Smedt H, Bultynck G (2010) Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium 47:297–314
Chakroborty S, Kim J, Schneider C, Jacobson C, Molgó J, Stutzmann GE (2012) Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J Neurosci 32:8341–8353
Foskett JK, White C, Cheung K-H, Mak D-OD (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87:593–658
Corbett EF, Michalak M (2000) Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem Sci 25:307–311
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J Cell Biol 181:1107–1116
Baba-Aissa F, Raeymaekers L, Wuytack F, Dode L, Casteels R (1998) Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol Chem Neuropathol 33:199–208
Kang S, Dahl R, Hsieh W, Shin A, Zsebo KM, Buettner C, Hajjar RJ, Lebeche D (2016) Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J Biol Chem 291:5185–5198
Adachi T (2010) Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. In: Paul MV (ed) Advances in pharmacology. Academic Press, New York, pp 165–195
Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang G-X, Allescher H-D, Korth M, Wilm M, Hofmann F, Ruth P (2000) Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase I[beta]. Nature 404:197–201
Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM (2005) Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J Neurochem 94:1711–1718
Cheung K-H, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VMY, Foskett JK (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58:871–883
Rybalchenko V, Hwang S-Y, Rybalchenko N, Koulen P (2008) The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol 40:84–97
Cheung K-H, Mei L, Daniel Mak D-O, Hayashi I, Iwatsubo T, Kang DE, Foskett JK (2010) Gain-of-function enhancement of InsP(3) receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signaling 3:ra22
Del Prete D, Checler F, Chami M (2014) Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol Neurodegener 9:1–15
Kemmerling U, Muñoz P, Müller M, Sánchez G, Aylwin ML, Klann E, Carrasco MA, Hidalgo C (2007) Calcium release by ryanodine receptors mediates hydrogen peroxide-induced activation of ERK and CREB phosphorylation in N2a cells and hippocampal neurons. Cell Calcium 41:491–502
Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, Bezprozvanny I (2015) Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer’s disease. J Neurosci 35:13275–13286
Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem 281:20661–20665
Liou J, Kim ML, Do Heo W, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15:1235–1241
Tong BC-K, Lee CS-K, Cheng W-H, Lai K-O, Kevin Foskett J, Cheung K-H (2016) Familial Alzheimer’s disease–associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+. Sci Signaling 9:ra89
Sun S, Zhang H, Liu J, Popugaeva E, Xu N-J, Feske S, White Iii Charles L, Bezprozvanny I (2014) Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82:79–93
Murakoshi H, Yasuda R (2012) Postsynaptic signaling during plasticity of dendritic spines. Trends Neurosci 35:135–143
Lam M, Dubyak G, Chen L, Nuñez G, Miesfeld RL, Distelhorst CW (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci 91:6569–6573
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA 102:105–110
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166:193–203
Tsujimoto Y, Shimizu S (2000) VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ 7:1174
Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T, Miller CCJ (2016) There’s something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci 39:146–157
Area-Gomez E, del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJC, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31:4106–4123
Hedskog L, Pinho CM, Filadi R, Rönnbäck A, Hertwig L, Wiehager B, Larssen P, Gellhaar S, Sandebring A, Westerlund M, Graff C, Winblad B, Galter D, Behbahani H, Pizzo P, Glaser E, Ankarcrona M (2013) Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci 110:7916–7921
Hurst S, Hoek J, Sheu S-S (2016) Mitochondrial Ca2+ and regulation of the permeability transition pore. J Bioenerg Biomembr 1–21. doi:10.1007/s10863-016-9672-x
Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262:744–747
Hajnóczky G, Csordás G, Yi M (2002) Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32:363–377
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta (BBA) 1787:1309–1316
Balaban RS (2009) The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta (BBA) 1787:1334–1341
Müller M, Cheung K-H, Foskett JK (2010) Enhanced ROS generation mediated by Alzheimer’s disease presenilin regulation of InsP3R Ca2+ signaling. Antioxidants Redox Signaling 14:1225–1235
Khachaturian ZS (1994) Calcium hypothesis of Alzheimer’s disease and brain aginga. Ann N Y Acad Sci 747:1–11
Gibson GE, Peterson C (1987) Calcium and the aging nervous system. Neurobiol Aging 8:329–343
Peterson C, Gibson GE, Blass JP (1985) Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer’s disease. N Engl J Med 312:1063–1065
Gibson GE, Nielsen P, Sherman KA, Blass JP (1987) Diminished mitogen-induced calcium uptake by lymphocytes from alzheimer patients. Biol Psychiatry 22:1079–1086
Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM (2000) Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol 149:793–798
Gibson G, Perrino P, Dienel GA (1986) In vivo brain calcium homeostasis during aging. Mech Ageing Dev 37:1–12
Stutzmann GE, Caccamo A, LaFerla FM, Parker I (2004) Dysregulated IP3 signaling in cortical neurons of knock-In mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci 24:508–513
Chakroborty S, Goussakov I, Miller MB, Stutzmann GE (2009) Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 29:9458–9470
Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I (2010) Role of presenilins in neuronal calcium homeostasis. J Neurosci 30:8566–8580
Emilsson L, Saetre P, Jazin E (2006) Alzheimer’s disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol Dis 21:618–625
Annaert W, De Strooper B (1999) Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci 22:439–443
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391:387–390
Li H, Wolfe MS, Selkoe DJ (2009) Toward structural elucidation of the γ-secretase complex. Structure 17:326–334
Gibson GE, Vestling M, Zhang H, Szolosi S, Alkon D, Lannfelt L, Gandy S, Cowburn RF (1997) Abnormalities in Alzheimer’s disease fibroblasts bearing the APP670/671 mutation. Neurobiol Aging 18:573–580
Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL (1994) Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci 91:534–538
Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM (1999) Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus. J Neurochem 72:1061–1068
Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ Disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 26:5180–5189
Herms J, Schneider I, Dewachter I, Caluwaerts N, Kretzschmar H, Van Leuven F (2003) Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J Biol Chem 278:2484–2489
Etcheberrigaray R, Hirashima N, Nee L, Prince J, Govoni S, Racchi M, Tanzi RE, Alkon DL (1998) Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol Dis 5:37–45
Leissring MA, LaFerla FM, Callamaras N, Parker I (2001) Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis 8:469–478
Hirashima N, Etcheberrigaray R, Bergamaschi S, Racchi M, Battaini F, Binetti G, Govoni S, Alkon DL (1996) Calcium responses in human fibroblasts: a diagnostic molecular profile for Alzheimer’s disease. Neurobiol Aging 17:549–555
Stutzmann GE (2005) Calcium dysregulation, IP3 signaling, and Alzheimer’s disease. Neuroscientist 11:110–115
Stutzmann GE (2007) The pathogenesis of Alzheimers disease: is it a lifelong “calciumopathy”? Neuroscientist 13:546–559
Shilling D, Müller M, Takano H, Daniel Mak D-O, Abel T, Coulter DA, Foskett JK (2014) Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J Neurosci 34:6910–6923
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat Rev Neurosci 3:862–872
Smith IF, Green KN, LaFerla FM (2005) Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium 38:427–437
Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim T-W (2000) Presenilin-mediated modulation of capacitative calcium entry. Neuron 27:561–572
Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP (2000) Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem 275:18195–18200
Goussakov I, Chakroborty S, Stutzmann GE (2011) Generation of dendritic Ca2+ oscillations as a consequence of altered ryanodine receptor function in AD neurons. Channels (Austin) 5:9–13
Goussakov I, Miller MB, Stutzmann GE (2010) NMDA-mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J Neurosci 30:12128–12137
Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE (2012) Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 33(1001):e1001–e1006
Hayrapetyan V, Rybalchenko V, Rybalchenko N, Koulen P (2008) The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein–protein interaction. Cell Calcium 44:507–518
Huang H-M, Chen H-L, Gibson GE (2014) Interactions of endoplasmic reticulum and mitochondria Ca2+ stores with capacitative calcium entry. Metab Brain Dis 29:1083–1093
Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska-Kijek A, Gruszczynska-Biegala J, Kuznicki J (2009) Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim Biophys Acta (BBA) 1793:1050–1057
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G, Bezprozvanny I (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126:981–993
Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I (2007) Familial Alzheimer disease–linked mutations specifically disrupt Ca(2+) leak function of presenilin 1. J Clin Invest 117:1230–1239
Nelson O, Supnet C, Tolia A, Horré K, De Strooper B, Bezprozvanny I (2011) Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J Biol Chem 286:22339–22347
Bezprozvanny I (2013) Presenilins and calcium signaling: systems biology to the rescue. Sci Signaling 6:pe24
Shilling D, Mak D-OD, Kang DE, Foskett JK (2012) Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J Biol Chem 287:10933–10944
Li X, Dang S, Yan C, Gong X, Wang J, Shi Y (2013) Structure of a presenilin family intramembrane aspartate protease. Nature 493:56–61
Theobald DL (2016) Presenilin adopts the ClC channel fold. Protein Sci 25:1363–1365
Bandara S, Malmersjö S, Meyer T (2013) Regulators of calcium homeostasis identified by inference of kinetic model parameters from live single cells perturbed by siRNA. Sci Signaling 6:ra56
Huang H-M, Chen H-L, Xu H, Gibson GE (2005) Modification of endoplasmic reticulum Ca2+ stores by select oxidants produces changes reminiscent of those in cells from patients with Alzheimer disease. Free Radical Biol Med 39:979–989
Grover AK, Samson SE, Misquitta CM (1997) Sarco(endo)plasmic reticulum Ca2+ pump isoform SERCA3 is more resistant than SERCA2b to peroxide. Am J Physiol 273:C420–C425
Grover AK, Kwan C-Y, Samson SE (2003) Effects of peroxynitrite on sarco/endoplasmic reticulum Ca2+ pump isoforms SERCA2b and SERCA3a. Am J Physiol 285:C1537–C1543
Gibson GE, Chen H-L, Xu H, Qiu L, Xu Z, Denton TT, Shi Q (2012) Deficits in the mitochondrial enzyme α-ketoglutarate dehydrogenase lead to Alzheimer’s disease-like calcium dysregulation. Neurobiol Aging 33(1121):e1113–e1121, e1124
Chen H, Denton TT, Xu H, Calingasan N, Beal MF, Gibson GE (2016) Reductions in the mitochondrial enzyme α-ketoglutarate dehydrogenase complex in neurodegenerative disease: beneficial or detrimental? J Neurochem 139:823–838
Parent A, Linden DJ, Sisodia SS, Borchelt DR (1999) Synaptic transmission and hippocampal long-term potentiation in transgenic mice expressing FAD-linked presenilin 1. Neurobiol Dis 6:56–62
Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J (2009) Presenilins are essential for regulating neurotransmitter release. Nature 460:632–636
Gibson G, Huang H-M (2004) Mitochondrial enzymes and endoplasmic reticulum calcium stores as targets of oxidative stress in neurodegenerative diseases. J Bioenerg Biomembr 36:335–340
Mattson MP, Guo Q (1997) Cell and molecular neurobiology of presenilins: a role for the endoplasmic reticulum in the pathogenesis of Alzheimer’s disease? J Neurosci Res 50:505–513
Reddy PH, Beal MF (2005) Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res Rev 49:618–632
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G (2000) Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta (BBA) 1502:139–144
Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O’Neill C (1999) Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and β-amyloid pathologies. Neuroscience 92:499–513
Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Trebak PP, Checler F. BF, Chami M (2012) Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci 32(34):11820–11834
Zhang H, Liu J, Sun S, Pchitskaya E, Popugaeva E, Bezprozvanny I (2015) Calcium signaling, excitability and synaptic plasticity defects in mouse model of Alzheimer’s disease. J Alzheimer’s Dis 45:561–580
Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J (2012) Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PLoS One 7:274–279
Peng J, Liang G, Inan S, Wu Z, Joseph DJ, Meng Q, Peng Y, Eckenhoff MF, Wei H (2012) Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci Lett 516:274–279
Popugaeva E, Bezprozvanny I (2013) Role of endoplasmic reticulum calcium signaling in the pathogenesis of Alzheimer disease. Front Mol Neurosci 6:29
Liu J, Supnet C, Sun S, Zhang H, Good L, Popugaeva E, Bezprozvanny I (2014) The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels (Austin) 8:230–242
Zhang H, Sun S, Wu L, Pchitskaya E, Zakharova O, Fon Tacer K, Bezprozvanny I (2016) Store-operated calcium channel complex in postsynaptic spines: a new therapeutic target for Alzheimer’s disease treatment. J Neurosci 36:11837–11850
Acknowledgements
Supported by 2P01AG014930-15A1 (NIH) and the Burke Medical Research Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is dedicated to Dr. Ursula Sonnewald. Her pioneering, unique and creative application of MR spectroscopy to neuroscience made it possible for us and others to understand fundamental changes in metabolism that are critical to our understanding of neurodegenerative diseases. The metabolic changes she helped us delineate underlie many of the calcium changes described in this review.
Rights and permissions
About this article

Cite this article
Gibson, G.E., Thakkar, A. Interactions of Mitochondria/Metabolism and Calcium Regulation in Alzheimer’s Disease: A Calcinist Point of View. Neurochem Res 42, 1636–1648 (2017). https://doi.org/10.1007/s11064-017-2182-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2182-3