
Abstract
Neurons are long-lived post-mitotic cells that possess an elaborate system of endosomes and lysosomes (endolysosomes) for protein quality control. Relatively recently, endolysosomes were recognized to contain high concentrations (400–600 μM) of readily releasable calcium. The release of calcium from this acidic organelle store contributes to calcium-dependent processes of fundamental physiological importance to neurons including neurotransmitter release, membrane excitability, neurite outgrowth, synaptic remodeling, and cell viability. Pathologically, disturbances of endolysosome structure and/or function have been noted in a variety of neurodegenerative disorders including Alzheimer’s disease (AD) and HIV-1 associated neurocognitive disorder (HAND). And, dysregulation of intracellular calcium has been implicated in the neuropathogenesis of these same neurological disorders. Thus, it is important to better understand mechanisms by which calcium is released from endolysosomes as well as the consequences of such release to inter-organellar signaling, physiological functions of neurons, and possible pathological consequences. In doing so, a path forward towards new therapeutic modalities might be facilitated.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Endosomes
- Lysosomes
- Endolysosomes
- Calcium
- Store-operated calcium entry
- N-type calcium channels
- Neurodegenerative diseases
- HIV-1 associated neurocognitive disorder
- Alzheimer’s disease
- Neurons
27.1 An Evolutionary Perspective on Calcium, Intracellular Organelles and Endolysosomes
Intracellular calcium regulates many essential functions of neurons including neurotransmitter release, excitability, synaptic plasticity, and cell viability [1]. Levels of intraneuronal calcium are very tightly regulated both temporally and spatially by various mechanisms including calcium release from intracellular stores, calcium influx across plasma membranes, and its association with a whole host of calcium binding proteins. Because of its importance both physiologically and pathologically, we start our story about the presence and functional significance of readily releasable stores of calcium in neuronal endolysosomes with a brief evolutionary perspective about calcium and intracellular organelles.
Calcium is well-known to be important for signal transduction in most cells including neurons. Indeed, calcium has been referred to as a universal second messenger in eukaryotic cells. The approximate 10,000-fold gradient of extracellular to intracellular calcium originated evolutionarily because of the gradual rise in calcium levels from about 100 nM during the period when the basic building blocks of life developed in thermal ducts under the ocean floor to about 1 mM during the Pre-Cambrian period when multicellular life evolved [2, 3]. Due to the toxic nature of millimolar levels of calcium, evolutionary pressure was applied such that cellular survival dictated that semipermeable membranes appeared and a variety of mechanisms were formed to maintain appropriate calcium gradients across plasma membranes [3]. Simultaneously, embedded in the plasma membranes were newly developed calcium pumps and calcium binding proteins which helped with calcium homeostasis [3]. Together, in neurons, these evolutionary changes provide unique and complex spatial and temporal handling of calcium that is essential for not only proper cellular signaling but also neuronal cell life and death.
It was also during this billion-year evolutionary period that intracellular organelles began appearing including mitochondria resulting from symbiotic relationships with bacteria and the development of functional endocytic machinery [4]. Mitochondria are integral to the maintenance of cellular energetics and they are important ‘sinks’ for intracellular calcium [5]. However, when too much calcium is up-taken into mitochondria cellular energetics are compromised and the resulting calcium overload can lead to a cascade of events including increased oxidative stress and cell death. It has also become increasingly appreciated that organelles including endoplasmic reticulum, endosomes and lysosomes (hereafter referred to as endolysosomes) have readily releasable and functionally important pools of intracellular calcium. Although less well known, the approximate 500 μM levels of calcium in endolysosomes are similar to the calcium concentrations present in endoplasmic reticulum [6]. This is a very important concept because endoplasmic reticulum is commonly referred to as the principal intracellular store of readily releasable calcium. Furthermore, as the field of inter-organellar signaling as well as physical and chemical crosstalk between organelles has grown over the past decade it is prudent of us to now posit that this relatively new and highly complicated area of modern cell biology is key to our understanding of the regulation and dysregulation of calcium [7].
With this as a very quick trip across 1 billion years of evolutionary biology, here we embark on a brief but focused summary of findings that neuronal endolysosomes contain readily releasable stores of calcium and once released this calcium can lead to calcium influx into cells, calcium release from other organelles, and calcium dysregulation-induced neurotoxicity. The relevance of such an important upstream store of calcium to the regulation of physiological functions and pathophysiological events is obvious and will be discussed with particular relevance to the pathogenesis of two neurodegenerative disorders; Alzheimer’s disease (AD) and HIV-1 associated neurocognitive disorder (HAND).
27.2 Endolysosomes Contain Readily Releasable Pools of Calcium
Neurons are long-lived post-mitotic cells that possess an elaborate endolysosome system for quality control especially for proteins. Endolysosomes are well known to be acidic organelles that contain high levels of cations including calcium, iron, zinc and copper. However, for the cation calcium it was not until fairly recently that these organelles were described as being ‘acidic calcium stores’ because the luminal pH of endolysosomes is acidic and endolysosomes contain high (400–600 μM) levels of readily releasable calcium [8, 9].
Neuronal calcium signals display spectacular spatiotemporal complexity and understanding how calcium signals are generated spatially and temporally is necessary to understand calcium-dependent cellular processes. Endolysosome calcium levels are maintained by a variety of uptake and efflux mechanisms. Essential for uptake of calcium into endolysosomes, proton gradients are established mainly by vacuolar H+-ATPase (v-ATPase) that pumps H+ into the lumen and this helps regulate Ca2+ levels [9,10,11]. Four main mechanisms for calcium release from endolysosomes have been described including: (1) Calcium release through two-pore channels (TPCs) triggered by nicotinic acid adenine dinucleotide phosphate (NAADP) [12,13,14,15,16,17]; (2) Elevation of endolysosome pH with, for example, the selective v-ATPase inhibitor bafilomycin (BAF) or the alkaline lysosomotropic agents NH4Cl and chloroquine [8, 18, 19]; (3) Involvement of TRPML1 mucolipin-type channels and P2X4 receptors [20,21,22]; and (4) Selective disruption of endolysosome membranes with Gly-Phe-β-naphtylamide (GPN) [23, 24]. Of physiological significance, calcium released from endolysosomes has been shown to contribute to a variety of calcium-dependent neuronal processes including neurotransmitter release, neuronal excitability, neurite outgrowth, synaptic remodeling, and cell viability [25,26,27].
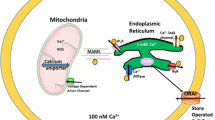
Endolysosomes can release calcium transiently and in a highly localized and distinct fashion [17, 28, 29]. Endolysosome calcium can affect the release of calcium from organellar stores as well as through plasma membrane-based calcium influx mechanisms. The inter-organellar signaling and signaling with the plasma membrane is explained at least in part by findings that endolysosomes are highly mobile in cells, are highly dynamic metabolically, have high rates of biogenesis, and can interact physically and functionally with other intracellular organelles (Fig. 27.1).

HIV-1 proteins and other neurotoxic insults can cause deacidification of endolysosomes. Increasing endolysosome pH can release calcium and other cations from endolysosomes. Calcium released from readily releasable stores in endolysosomes can increase the release of calcium from other intracellular stores and can increase the influx of extracellular calcium. Such increases in pH and calcium levels can cause endoplasmic reticulum (ER) and mitochondrial dysfunction, Alzheimer’s disease (AD)-like pathology, and synaptodendritic damage
At least three models of acidic store-induced calcium signaling mechanisms have been described [9]. (1) Acidic stores of calcium might communicate with endoplasmic reticulum calcium stores such that calcium released from endolysosomes can enhance endoplasmic reticulum calcium loading [30] and calcium-induced calcium release [13, 15]. (2) Changes in endolysosome pH may release calcium from a subgroup of acidic calcium stores and the released calcium may affect other subgroups of acidic stores through mechanisms such as vesicular fusion of late endosomes and lysosomes [9, 15, 31]. (3) Calcium released from acidic calcium stores might depolarize plasma membranes, evoke calcium-dependent currents, and stimulate calcium influx across plasma membranes [12].
27.3 Acidic Store-Operated Calcium Entry in Neurons
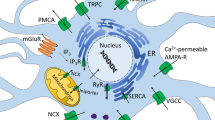
Acidic store-operated calcium entry (aSOCE) is a unique mechanism that links readily releasable calcium in endolysosomes with influx of extracellular calcium into neurons. This is a novel means by which intraneuronal stores of calcium can contribute to spatial and temporal integration of calcium signaling. In support of this novel mechanism, we found that calcium could be released from endolysosomes following stimulation of a number of different mechanisms, that the calcium release could be independent of other organellar stores of calcium, that release of calcium from endolysosomes triggered calcium influx, and that the calcium influx was regulated by N-type calcium channels and lysosome exocytosis (Fig. 27.2).

HIV-1 Tat de-acidifies endolysosomes, increases amyloidogenesis, and releases calcium from readily releasable stores in endolysosomes. Calcium released from endolysosomes can affect mitochondrial and endoplasmic reticulum (ER) calcium stores, and increase store operated calcium (SOCE) mechanisms. Mechanistically, following de-acidification endolysosome calcium is released through TRPML1 and two pore channels (TPCs). The calcium signals can be amplified by releasing calcium from other organelles including mitochondria and ER, and by activating ER-based SOCE involving STIM1 and Orai channels as well as acidic store operated calcium entry involving N-type calcium channels (NTCCs)
Capacitative influx of calcium into cells was described over 30 years ago [32]. Such calcium influx mechanisms, that are now commonly referred to as store-operated calcium entry (SOCE), are principally initiated by a reduction in endoplasmic reticulum calcium stores followed by influx of extracellular calcium in a variety of cells including neurons in order to refill the depleted stores of calcium. Mechanistically, depleting endoplasmic reticulum calcium stores drives the oligomerization and translocation of stromal interaction molecule 1 (STIM1) proteins to endoplasmic reticulum junctions close to the plasma membrane. Such STIM1 translocation induces the clustering of calcium release-activated calcium modulator 1 (Orai1) channels and/or transient receptor potential (TRP) cation channels into plasma membranes thereby enabling extracellular calcium entry [33].
Conceptually, but not mechanistically, we observed similar store-operated calcium entry involving endolysosomes in neurons. Using primary cultures of rat cortical neurons, we found that calcium was released from endolysosomes following treatment with the two-pore channel agonist NAADP-AM, the v-ATPase inhibitor BAF, and the lysosomotropic agent GPN; all of which de-acidify endolysosomes [34]. However, when these experiments were conducted in the absence of extracellular calcium, de-acidification of endolysosomes with NAADP-AM, BAF and GPN increased only slightly levels of free cytosolic calcium. When these same experiments were conducted in the presence of extracellular calcium, NAADP-AM, BAF and GPN all increased significantly the levels of free cytosolic calcium. Although it is not well understood currently, the relatively small release of calcium from endolysosomes causes a much larger influx of extracellular calcium and this might be due to plasma membrane depolarization as is accompanied by NAADP-induced endolysosome calcium release [34, 35]. Besides neurons, phenomena similar to aSOCE have been described in other cell types where NAADP has been found to induce endolysosome calcium release and large influxes of calcium across plasma membranes [12, 36,37,38,39]. These observations suggested to us that endolysosome de-acidification by three completely different mechanisms led directly or indirectly to an enhanced influx of calcium into neurons. Accordingly, we next tested more specifically the extent to which a store-operated mechanism might control the observed calcium influx across the plasma membrane.
Using approaches similar to those used by others and us, we began studying store-operated calcium mechanisms including the classical endoplasmic reticulum-based capacitative SOCE. Indeed, we confirmed that in the absence of extracellular calcium and following depletion of endoplasmic reticulum calcium with the SERCA pump inhibitor thapsigargin (TG) there was a significant increase in levels of free intracellular calcium only when calcium was re-introduced to the extracellular medium. With this positive control for the functional presence of endoplasmic reticulum-based SOCE in our cultured neurons, we conducted similar experiments with agents that de-acidify endolysosomes and release calcium from endolysosome stores. Even after depleting ER pools of calcium with TG, application of NAADP-AM, BAF and GPN still caused increased influx of extracellular calcium and still induced increased levels of intracellular calcium. Thus, in these neurons there appeared to be at least two separate and functional store-operated calcium mechanisms; one governed by endoplasmic reticulum and the other by endolysosomes.
In testing the distinctive nature of the two store-operated calcium mechanisms governed by endoplasmic reticulum or endolysosomes, we used pharmacological and molecular/genetic strategies. Using siRNA to knock-down protein expression levels of STIM1, a protein that is central to SOCE, and the SOCE blockers SKF-96365 and 2-APB we were able to block significantly TG-induced release of calcium from endoplasmic reticulum, but we were unable to block significantly NAADP-AM-, BAF- and GPN-induced calcium influx. However, we were able to block significantly NAADP-AM-, BAF- and GPN-induced calcium influx with the selective N-type calcium channel (NTCC) blocker (ω-conotoxin). The selective and specific nature of this inhibition by ω-conotoxin was confirmed further by showing that NAADP-AM-, BAF- and GPN-induced calcium influx was not blocked by inhibitors of L-type (nimodipine, verapamil) and P/Q-type (ω-agatoxin) calcium channels. Moreover, we confirmed these pharmacological findings by showing that siRNA knockdown of NTCCs attenuated significantly NAADP-AM-, BAF- and GPN-induced calcium influx, but did not affect TG-induced SOCE. Together, the above results demonstrated that calcium released from endolysosomes can be distinct from calcium released from endoplasmic reticulum through SOCE mechanisms and that the calcium released from endolysosomes is capable of activating cell surface calcium channels to stimulate calcium influx. These findings support and extend earlier findings that calcium released from endolysosomes did not stimulate endoplasmic reticulum-dependent SOCE in MDCK epithelial cells [23]. Accordingly, this new mechanism was termed by us as “acidic store-operated calcium entry” (aSOCE) [34].
27.4 Role of Lysosome Exocytosis in Acidic Store-Operated Calcium Entry (aSOCE)
Multiple mechanisms might control aSOCE involving NTCCs. One such mechanism might involve lysosome exocytosis because we have shown using a quantitative biotinylation of surface proteins assay that NAADP-AM, BAF and GPN all increased cell surface protein expression levels of NTCCs and lysosome-associated glycoprotein 1 (LAMP1). Next, we addressed the possibility that lysosome exocytosis and NTCCs were linked directly by conducting co-immunoprecipitation studies and found a physical interaction between NTCCs and LAMP1. Because LAMP1 is critical for lysosome exocytosis [40], those observations suggested to us that lysosome exocytosis might be a functional partner in aSOCE especially because aSOCE was inhibited following siRNA knockdown of protein expression levels of LAMP1. Thus, de-acidification of endolysosomes might be of central importance because NAADP-AM, BAF and GPN through very different initial mechanisms all appeared to enhance lysosome exocytosis and the recycling of NTCCs to the plasma membrane where they participated in calcium influx generally and aSOCE more specifically. Physically, this makes sense as well because of findings that de-acidification of endolysosomes changes cellular distribution patterns of these organelles from a mostly peri-nuclear pattern to one where the endolysosomes migrate close to the plasma membrane [41]. Thus, functionally and physically there is evidence favoring endolysosomes and endolysosome exocytosis in calcium entry.
27.5 Physical Interactions and Functional Relevance of Inter-organellar Signaling
In addition to physical interactions between endolysosomes and plasma membranes, it is becoming increasingly clear that endolysosomes physically and functionally interact as well with other intracellular organelles including mitochondria and endoplasmic reticulum. Such recognition has led to an appreciation for dynamic physical and chemical communications between intracellular organelles including those regulated by pH and calcium.
Physical interactions between mitochondria and endoplasmic reticulum were first described about 60 years ago and the functional significance of mitochondria-associated membranes was first characterized about 30 years ago [42]. Even today, there continues to be work focused on the physical and functional interactions between organelles [43] as well as the role that organellar interactions plays in the pathogenesis of neurodegenerative diseases [44, 45]. As it relates to endolysosomes, it is now known that there are extensive physical interactions between endolysosomes and mitochondria and that these inter-organellar communications participate in lipid and metabolite exchange as well as mitochondrial quality control [46]. Conversely, mitochondrial dysfunction has been found to negatively affect lysosome structure and function through reactive oxygen species-dependent mechanisms [47]. Extensive membrane contact sites have been described between lysosomes and endoplasmic reticulum, that these contact sites were evolutionarily conserved, and that calcium released from lysosomes was sufficient to stimulate endoplasmic reticulum-dependent calcium-induced calcium release [48, 49, 50]. However, only recently was it shown that endolysosomes maintain their 1000-fold calcium concentration gradient in cells in part by refilling endolysosome stores of calcium from IP3-regulated stores of calcium in endoplasmic reticulum [51]. Some of the differences in findings as to calcium movements between organelles might be because of cell-specific mechanisms. In addition, the difficult nature of understanding inter-organellar calcium dynamics is highlighted by work showing that STIM1 and STIM2 are expressed in endolysosomes, at least in platelets, and that depletion of acidic organellar stores of calcium can increase protein-protein interactions between STIM proteins with Orai1 and TRPC channels to induce SOCE [52]. It is further complicated by findings that calcium released through endolysosome-resident TRML1 channels can cause calcium release from endoplasmic reticulum and calcium influx [53] and that NAADP has been implicated in this “cross-talk” [54].
27.6 Possible Role of Endolysosomes and aSOCE in Pathogenesis of Alzheimer’s Disease and HIV-1 Associated Neurocognitive Disorder (HAND)
Disturbances in endolysosome structure and/or function have been noted in a variety of neurodegenerative disorders including Alzheimer’s disease (AD) and HIV-1 neurocognitive disorder (HAND) [55,56,57,58,59]. AD is a devastating age-related neurodegenerative disease that is the commonest cause of dementia in people over the age of 65. People with HAND, on the other hand, exhibit neurological complications ranging from mild (mild cognitive impairment) to severe (dementia). In the current era of anti-retroviral therapeutics HIV-1 infected individuals are living almost full life-spans, but are now experiencing a prevalence rate of over 50% for HAND [60, 61]. Clinically and pathologically people living with neuroHIV-1 are exhibiting AD-like symptoms including learning and memory deficits as well as increased amyloidogenesis. Although the pathogenesis of HAND is not fully understood, HIV-1 proteins including the HIV-1 transactivator of transcription protein Tat have been implicated by others and us to be causative virotoxins in HAND [62,63,64,65,66,67,68,69]. Among the HIV-1 viral proteins, HIV-1 Tat is present in brains of HIV-1 infected individuals and its levels stay elevated in CSF even when HIV-1 viral levels are immeasurable [70]. Others and we have shown that HIV-1 Tat directly excites neurons [65, 71, 72], disturbs neuronal calcium homeostasis [64, 73], disrupts synaptic integrity [74, 75], and induces neurotoxicity [68, 76].
Endolysosome dysfunction has been implicated in the development of at least two pathological hallmarks of AD and HAND; Aβ accumulation and neurofibrillary tangle formation. Endolysosomes are very important for amyloidogenic processing of AβPP to Aβ because amyloid β precursor protein (AβPP) is first endocytosed, the amyloidogenic enzymes BACE-1 and γ-secretase are almost exclusively located in endosomes and lysosomes, the acidic environment of endolysosomes is favorable for amyloidogenic metabolism of AβPP, and Aβ can be either accumulated in or released by exocytosis from endolysosomes [77,78,79,80,81,82,83]. Tau is a microtubule-associated protein, and when hyperphosphorylated it aggregates and contributes to the formation of neurofibrillary tangles. Tau aggregates can be degraded by cathepsin D in autophagosomes-lysosomes [84, 85], and endolysosome dysfunction contributes to tau aggregation and neurofibrillary tangle formation [86, 87]. On the other hand, transcriptional activation of lysosome biogenesis can clear aggregated tau [88]. Thus, endolysosomes are important sites for development of these neurological disorders.
Dysregulation of intracellular calcium has also been implicated in the neuropathogenesis of these same neurological disorders. And it is clear (see above) that de-acidification of endolysosomes releases calcium from these acidic stores [28, 89, 90]. We found that HIV-1 Tat protein elevated endolysosome pH and disturbed the structure and function of endolysosomes [74], a prominent and early pathological feature of HAND [57, 58]. Clearly, endolysosome calcium stores contribute to neuronal calcium signaling and function [91,92,93] and calcium release from endolysosomes triggers calcium release from endoplasmic reticulum [11, 17] and through plasma membranes via aSOCE (see above).
HIV-1 proteins including HIV-1 Tat, and anti-retroviral therapeutic drugs contribute to the development of AD-like pathology including increases in Aβ levels [94,95,96,97,98,99]. HIV-1 Tat enters neurons via receptor-mediated endocytosis [100,101,102]. The Tat-induced de-acidification of endolysosomes and resulting effects on calcium dyshomeostasis likely results from the ability of HIV-1 Tat to decrease the levels and activity of vacuolar-ATPase as well as compensatory increases in cathepsin D and LAMP-1 [103]. The consequences of such alterations in calcium dynamics and homeostasis are synaptic disruption and neurotoxicity [104,105,106].
Endolysosomes contain physiologically important levels of calcium that is readily releasable by a number of stimuli and insults. The calcium can exit through a number of channels including TRPML and two pore channels. Once released the calcium can signal other organelles to release calcium and for greater influx of calcium through plasma membrane-resident calcium channels especially N-type calcium channels. These effects on endolysosome structure and function have clear implications to the pathogenesis of AD and HAND; neurological disorders that show overlap in terms of clinical and pathological features. We are excited to be part of this emerging area of cell biology focused on inter-organellar signaling and look forward to further studies elucidating physiological and pathological consequences of calcium release from endolysosome stores.
References
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529
Jaiswal JK (2001) Calcium – how and why? J Biosci 26(3):357–363
Case RM, Eisner D, Gurney A, Jones O, Muallem S, Verkhratsky A (2007) Evolution of calcium homeostasis: from birth of the first cell to an omnipresent signalling system. Cell Calcium 42(4–5):345–350
Wideman JG, Leung KF, Field MC, Dacks JB (2014) The cell biology of the Endocytic system from an evolutionary perspective. Cold Spring Harb Perspect Biol [Internet] 6(4):a016998. Apr 1 [cited 2018 July 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24478384
Carafoli E (2010) The fateful encounter of mitochondria with calcium: how did it happen? Biochim Biophys Acta Bioenerg [Internet] 1797(6–7):595–606. June 1 [cited 2018 July 16]. Available from: https://www.sciencedirect.com/science/article/pii/S0005272810001301#fig1
Patel S, Cai X (2015) Evolution of acidic Ca2+stores and their resident Ca2+−permeable channels [internet]. Cell Calcium 57:222–230. [cited 2018 July 14]. Available from: https://www.sciencedirect.com/science/article/pii/S0143416014002012
Raffaello A, Mammucari C, Gherardi G, Rizzuto R (2016) Calcium at the Center of Cell Signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 41:1035–1049. [cited 2017 May 17]. Available from: http://www.cell.com/trends/biochemical-sciences/pdf/S0968-0004(16)30147-5.pdf
Christensen KA, Myers JT, JA S (2002) pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 115(Pt 3):599–607
Morgan AJ, Platt FM, Lloyd-Evans E, Galione A (2011) Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J [Internet] 439(3):349–374. Available from: http://www.biochemj.org/content/439/3/349.abstract
Moreno SNJ, Docampo R (2009) The role of acidocalcisomes in parasitic protists. J Eukaryot Microbiol 56:208–213
Patel S, Docampo R (2010) Acidic calcium stores open for business: expanding the potential for intracellular Ca2+signaling. Trends Cell Biol 20:277–286
Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X et al (2009) Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling (a). J Cell Biol 186(2):201–209
Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X et al (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459(7246):596–600
Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rötzer K et al (2009) The two-pore channel TPCN2 mediates NAADP-dependent Ca2+−release from lysosomal stores. Pflugers Arch Eur J Physiol [Internet] 458(5):891–899. Sept 26 [cited 2018 July 14]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19557428
Ruas M, Rietdorf K, Arredouani A, Davis LC, Lloyd-Evans E, Koegel H et al (2010) Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+signaling and Endolysosomal trafficking. Curr Biol [Internet] 8(20):703–709. Apr 27 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20346675
Schieder M, Rötzer K, Brüggemann A, Biel M, Wahl-Schott CA (2010) Characterization of two-pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J Biol Chem [Internet] 285(28):21219–21222. July 9 [cited 2018 July 14]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20495006
Zhu MX, Ma J, Parrington J, Calcraft PJ, Galione A, Evans AM (2010) Calcium signaling via two-pore channels: local or global, that is the question. AJP Cell Physiol [Internet] 298(3):C430–C441. Available from: http://ajpcell.physiology.org/cgi/doi/10.1152/ajpcell.00475.2009
Camacho M, Machado JD, Alvarez J, Borges R (2008) Intravesicular calcium release mediates the motion and exocytosis of secretory organelles: a study with adrenal chromaffin cells. J Biol Chem 283(33):22383–22389
Machado JD, Camacho M, Alvarez J, Borges R (2009) On the role of intravesicular calcium in the motion and exocytosis of secretory organelles. Commun Integr Biol 2(2):71–73
Starkus JG, Fleig A, Penner R (2010) The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J Physiol 588(8):1227–1240
Kiselyov K, Colletti GA, Terwilliger A, Ketchum K, CWP L, Quinn J et al (2011) TRPML: transporters of metals in lysosomes essential for cell survival? Cell Calcium 50:288–294
Cao Q, Zhong XZ, Zou Y, Murrell-Lagnado R, Zhu MX, Dong XP (2015) Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Biol 209(6):879–894
Haller T, Dietl P, Deetjen P, Völkl H (1996) The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+release. Cell Calcium 19(2):157–165
McGuinness L, Bardo SJ, Emptage NJ (2007) The lysosome or lysosome-related organelle may serve as a Ca2+store in the boutons of hippocampal pyramidal cells. Neuropharmacology 52(1):126–135
Repnik U, Česen MH, Turk B (2013) The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol [Internet] 5(1):a008755. Jan 1 [cited 2017 Oct 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23284043
Ferguson SM (2018) Neuronal lysosomes. Neurosci Lett [Internet]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S030439401830260X
Goo MS, Sancho L, Slepak N, Boassa D, Deerinck TJ, Ellisman MH et al (2017) Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J Cell Biol 216(8):2499–2513
Galione A, Morgan AJ, Arredouani A, Davis LC, Rietdorf K, Ruas M et al (2010) NAADP as an intracellular messenger regulating lysosomal calcium-release channels. Biochem Soc Trans [Internet] 38(6):1424–1431. Available from: http://biochemsoctrans.org/lookup/doi/10.1042/BST0381424
Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S et al (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3:731
Macgregor A, Yamasaki M, Rakovic S, Sanders L, Parkesh R, Churchill GC et al (2007) NAADP controls cross-talk between distinct Ca2+ Stores in the Heart. J Biol Chem [Internet] 282(20):15302–15311. May 18 [cited 2018 July 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17387177
Galione A, Parrington J, Funnell T (2011) Physiological roles of NAADP-mediated Ca2+ signaling. Sci China Life Sci [Internet] 54(8):725–732. Available from: http://springerlink.bibliotecabuap.elogim.com/10.1007/s11427-011-4207-5
Putney JW (1986) A model for receptor-regulated calcium entry. Cell Calcium 7(1):1–12
Putney JW (2009) Capacitative calcium entry: from concept to molecules. Immunol Rev 231:10–22
Hui L, Geiger NH, Bloor-Young D, Churchill GC, Geiger JD, Chen X (2015) Release of calcium from endolysosomes increases calcium influx through N-type calcium channels: evidence for acidic store-operated calcium entry in neurons. Cell Calcium [Internet] 58:617–627. [cited 2017 May 31]. Available from: http://ac.els-cdn.com/S0143416015001529/1-s2.0-S0143416015001529-main.pdf?_tid=d155fae0-460a-11e7-a743-00000aab0f02&acdnat=1496239973_5d092efe55666657b3cd6c5dc4881cf0
Arredouani A, Ruas M, Collins SC, Parkesh R, Clough F, Pillinger T et al (2015) Nicotinic acid adenine dinucleotide phosphate (NAADP) and endolysosomal two-pore channels modulate membrane excitability and stimulus-secretion coupling in mouse pancreatic β cells. J Biol Chem [Internet] 290(35):21376–21392. Aug 28 [cited 2018 July 14]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26152717
Moccia F, Lim D, Nusco GA, Ercolano E, Santella L (2003) NAADP activates a Ca2+ current that is dependent on F-actin cytoskeleton. FASEB J 17(13):1907–1909
Moccia F, Billington RA, Santella L (2006) Pharmacological characterization of NAADP-induced Ca2+ signals in starfish oocytes. Biochem Biophys Res Commun 348(2):329–336
Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A et al (2009) Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol 5(4):220–226
Churchill GC, O’Neill JS, Masgrau R, Patel S, Thomas JM, Genazzani AA et al (2003) Sperm deliver a new second messenger: NAADP. Curr Biol 13(2):125–128
Yogalingam G, Bonten EJ, van de Vlekkert D, Hu H, Moshiach S, Connell SA et al (2008) Neuraminidase 1 is a negative regulator of Lysosomal exocytosis. Dev Cell 15(1):74–86
Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W et al (2016) A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol 18(4):404–417
Herrera-Cruz MS, Simmen T (2017) Over six decades of discovery and characterization of the architecture at mitochondria-associated membranes (MAMs). Adv Exp Med Biol 997:13–31
Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF et al (2017) Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci [Internet] 114(24):E4859–E4867. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1701078114
Schon EA, Area-Gomez E (2013) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci [Internet] 55:26–36. July [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22922446
Joshi AU, Kornfeld OS, Mochly-Rosen D (2016) The entangled ER-mitochondrial axis as a potential therapeutic strategy in neurodegeneration: a tangled duo unchained. Cell Calcium 60:218–234
Soto-Heredero G, Baixauli F, Mittelbrunn M (2017) Interorganelle communication between mitochondria and the Endolysosomal system. Front Cell Dev Biol [Internet] 5:95. Available from: http://journal.frontiersin.org/article/10.3389/fcell.2017.00095/full
Demers-Lamarche J, Guillebaud G, Tlili M, Todkar K, Bélanger N, Grondin M et al (2016) Loss of mitochondrial function impairs lysosomes∗. J Biol Chem [Internet] 291(19):10263–10276. May 6 [cited 2017 Dec 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26987902
Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S (2013) Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci [Internet] 126(Pt 1):60–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23108667%5Cnhttp://jcs.biologists.org/content/ joces/126/1/60.full.pdf
Penny CJ, Kilpatrick BS, Eden ER, Patel S (2015) Coupling acidic organelles with the ER through Ca2+ microdomains at membrane contact sites. Cell Calcium 58:387–396
Hariri H, Ugrankar R, Liu Y, Henne WM (2016) Inter-organelle ER-endolysosomal contact sites in metabolism and disease across evolution. Communicative and Integrative Biology 9(3):e1156278
Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H (2016) The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife [Internet] 5:e15887. May 23 [cited 2017 May 11]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27213518
Zbidi H, Jardin I, Woodard GE, Lopez JJ, Berna-Erro A, Salido GM et al (2011) STIM1 and STIM2 are located in the acidic Ca2+ stores and associates with Orai1 upon depletion of the acidic stores in human platelets. J Biol Chem 286(14):12257–12270
Kilpatrick BS, Yates E, Grimm C, Schapira AH, Patel S (2016) Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J Cell Sci [Internet]. 129(20):3859–3867. [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27577094
Ronco V, Potenza DM, Denti F, Vullo S, Gagliano G, Tognolina M et al (2015) A novel Ca2+−mediated cross-talk between endoplasmic reticulum and acidic organelles: implications for NAADP-dependent Ca2+signalling. Cell Calcium [Internet] 57(2):89–100. Feb 1 [cited 2018 July 14]. Available from: https://www.sciencedirect.com/science/article/pii/S0143416015000020
Tate BA, Mathews PM (2006) Targeting the role of the endosome in the pathophysiology of Alzheimer’s disease: a strategy for treatment. Sci Aging Knowl Environ [Internet] 2006(10):re2–re2. June 28 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16807486
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH et al (2008) Autophagy induction and Autophagosome clearance in neurons: relationship to Autophagic pathology in Alzheimer’s disease. J Neurosci [Internet] 28(27):6926–6937. July 2 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18596167
Gelman BB, Soukup VM, Holzer CE 3rd, Fabian RH, Schuenke KW, Keherly MJ et al (2005) Potential role for white matter lysosome expansion in HIV-associated dementia. J Acquir Immune Defic Syndr 39(4):422–425
Spector SA, Zhou D (2008) Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy [Internet] 4(5):704–706. July [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18424919
Cysique LA, Hewitt T, Croitoru-Lamoury J, Taddei K, Martins RN, Chew CS et al (2015) APOE ε4 moderates abnormal CSF-abeta-42 levels, while neurocognitive impairment is associated with abnormal CSF tau levels in HIV+ individuals – a cross-sectional observational study. BMC Neurol [Internet] 15(1):51. Dec 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25880550
Ellis RJ, Rosario D, Clifford DB, McArthur JC, Simpson D, Alexander T et al (2010) Continued high prevalence and adverse clinical impact of human immunodeficiency virus–associated sensory neuropathy in the era of combination antiretroviral therapy. Arch Neurol [Internet] 67(5):552. May 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20457954
Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F et al (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: charter study. Neurol Int 75(23):2087–2096. Dec 7 [cited 2017 Sept 13]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21135382
Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A et al (1991) Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol [Internet] 65(2):961–967. Feb [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1898974
Weeks BS, Lieberman DM, Johnson B, Roque E, Green M, Loewenstein P et al (1995) Neurotoxicity of the human immunodeficiency virus type 1 tat transactivator to PC12 cells requires the tat amino acid 49-58 basic domain. J Neurosci Res [Internet] 42(1):34–40. Sept 1 [cited 2018 July 27]. Available from: http://doi.wiley.com/10.1002/jnr.490420105
Haughey NJ, Holden CP, Nath A, Geiger JD (1999) Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem [Internet] 73(4):1363–1374. Oct [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10501179
Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD (2000) Synergistic neurotoxicity by human immunodeficiency virus proteins tat and gp120: protection by memantine. Ann Neurol [Internet] 47(2):186–194. Feb [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10665489
Pérez A, Probert AW, Wang KK, Sharmeen L (2001) Evaluation of HIV-1 tat induced neurotoxicity in rat cortical cell culture. J Neurovirol [Internet] 7(1):1–10. Feb 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11519477
King JE, Eugenin EA, Buckner CM, Berman JW (2006) HIV tat and neurotoxicity. Microbes Infect [Internet] 8(5):1347–1357. Apr [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16697675
Buscemi L, Ramonet D, Geiger JD (2007) Human immunodeficiency virus type-1 protein tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis [Internet] 26(3):661–670. June [cited 2018 June 1]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17451964
Agrawal L, Louboutin J-P, Reyes BAS, Van Bockstaele EJ, Strayer DS (2012) HIV-1 tat neurotoxicity: a model of acute and chronic exposure, and neuroprotection by gene delivery of antioxidant enzymes. Neurobiol Dis [Internet] 45(2):657–670. Feb [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22036626
Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R et al (2013) Induction of IL-17 and nonclassical T-cell activation by HIV-tat protein. Proc Natl Acad Sci U S A [Internet] 110(33):13588–13593. Aug 13 [cited 2017 July 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23898208
DSK M, Jsnudsen BE, Geiger JD, Brownstone RM, Nath A (1995) Human lmmunodehclency V m s lype 1 tat activates non-N-Methyla-aspartate excitatory ammo acid receptors and causes neurotoxicity. Ann Neurol [Internet] 37(3):373–380. [cited 2017 Nov 20]. Available from: https://med-und.illiad.oclc.org/illiad/illiad.dll?Action=10&Form=75&Value=116096
Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N et al (1996) Identification of a human immunodeficiency virus type 1 tat epitope that is neuroexcitatory and neurotoxic. J Virol [Internet] 3(70):1475–1480. [cited 2018 June 13]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC189968/pdf/701475.pdf
Haughey NJ, Mattson MP (2002) Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins tat and gp120. J Acquir Immune Defic Syndr [Internet] 31(Suppl 2):S55–S61. Oct 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12394783
Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci [Internet] 28(48):12604–12613. Nov 26 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19036954
Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A et al (2010) Interactive comorbidity between opioid drug abuse and HIV-1 tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol [Internet] 177(3):1397–1410. Sept [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20651230
Hui L, Chen X, Haughey NJ, Geiger JD (2012) Role of Endolysosomes in HIV-1 tat-induced neurotoxicity. ASN Neuro [Internet] 4(4):AN20120017. Apr 3 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22591512
Rajendran L, Annaert W (2012) Membrane trafficking pathways in Alzheimer’s disease. Traffic [Internet] 13(6):759–770. June [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22269004
Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C et al (2013) Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun [Internet] 4(1):2250. Dec 2 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23907271
Jiang S, Li Y, Zhang X, Bu G, Xu H, Zhang Y (2014) Trafficking regulation of proteins in Alzheimer’s disease. Mol Neurodegener [Internet] 9:6. Jan 11 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24410826
Nixon RA (2005) Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging [Internet] 26(3):373–382. Mar [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15639316
Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T et al (2008) Efficient inhibition of the Alzheimer’s disease -Secretase by membrane targeting. Science (80- ) [Internet] 320(5875):520–523. Apr 25 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18436784
Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N (2008) Crystal structure of an active form of BACE1, an enzyme responsible for amyloid protein production. Mol Cell Biol [Internet] 28(11):3663–3671. June 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18378702
Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L et al (2011) ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci [Internet] 108(34):E559–E568. Aug 23 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21825135
Hamano T, Gendron TF, Causevic E, Yen S-H, Lin W-L, Isidoro C et al (2008) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci [Internet] 27(5):1119–1130. Mar [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18294209
Chesser AS, Pritchard SM, GVW J (2013) Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol [Internet] 4:122. Sept 3 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24027553
Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, GVW J (2014) Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun [Internet] 5(1):3496. Dec 25 [cited 2018 July 27]. Available from: http://www.nature.com/articles/ncomms4496
Bi X, Liao G (2007) Erratum: Autophagic-lysosomal dysfunction and neurodegeneration in Niemann-pick type C mice: lipid starvation or indigestion? (autophagy) [internet]. Autophagy 3:646–648. [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17921694
Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y et al (2014) Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med [Internet] 6(9):1142–1160. Sept 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25069841
Masgrau R, Churchill GC, Morgan AJ, Ashcroft SJH (2003) Galione a. NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic beta cells. Curr Biol [Internet] 13(3):247–251. Feb 4 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12573222
Mitchell KJ, Lai FA, Rutter GA (2003) Ryanodine receptor type I and nicotinic acid adenine dinucleotide phosphate receptors mediate Ca2+ release from insulin-containing vesicles in living pancreatic β-cells (MIN6). J Biol Chem [Internet] 278(13):11057–11064. Mar 28 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12538591
Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X et al (2009) Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res [Internet] 104(3):288–291. Feb 13 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19179659
Pandey V, Chuang C-C, Lewis AM, Aley PK, Brailoiu E, Dun NJ et al (2009) Recruitment of NAADP-sensitive acidic Ca2+ stores by glutamate. Biochem J [Internet] 422(3):503–512. Sept 15 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19548879
Dickinson GD, Churchill GC, Brailoiu E, Patel S (2010) Deviant nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated Ca2+ signaling upon lysosome proliferation. J Biol Chem [Internet] 285(18):13321–13325. Apr 30 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20231291
Rempel HC, Pulliam L (2005) HIV-1 tat inhibits neprilysin and elevates amyloid β. AIDS [Internet] 19(2):127–135. Jan 28 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15668537
Giunta B, Hou H, Zhu Y, Rrapo E, Tian J, Takashi M et al (2009) HIV-1 tat contributes to Alzheimer’s disease-like pathology in PSAPP mice. Int J Clin Exp Pathol [Internet] 5(2):433–443. [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19294002
Aksenov MY, Aksenova MV, Mactutus CF, Booze RM (2010) HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci Lett [Internet] 475(3):174–178. May 21 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20363291
Chen X, Hui L, Geiger NH, Haughey NJ, Geiger JD (2013) Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol Aging [Internet] 34(10):2370–2378. Oct [cited 2017 Aug 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23673310
Kim J, Yoon JH, Kim YS (2013) HIV-1 tat interacts with and regulates the localization and processing of amyloid precursor protein. Chauhan A, editor. PLoS One [Internet] 8(11):e77972. Nov 29 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24312169
Fields JA, Dumaop W, Crews L, Adame A, Spencer B, Metcalf J et al (2015) Mechanisms of HIV-1 tat neurotoxicity via CDK5 translocation and hyper-activation: role in HIV-associated neurocognitive disorders. Curr HIV Res [Internet] 13(1):43–54. [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25760044
Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE et al (2000) Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med [Internet] 6(12):1380–1387. Dec 1 [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11100124
Deshmane SL, Mukerjee R, Fan S, Sawaya BE (2011) High-performance capillary electrophoresis for determining HIV-1 tat protein in neurons. Kashanchi F, editor. PLoS One [Internet] 6(1):e16148. Jan 7 [cited 2018 July 27]. Available from: http://dx.plos.org/10.1371/journal.pone.0016148
Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B (2004) HIV-1 tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell [Internet] 15(5):2347–2360. May [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15020715
Mangieri LR, Mader BJ, Thomas CE, Taylor CA, Luker AM, Tse TE et al (2014) ATP6V0C knockdown in Neuroblastoma cells alters autophagy-lysosome pathway function and metabolism of proteins that accumulate in neurodegenerative disease. Srinivasula SM, editor. PLoS One [Internet] 9(4):e93257. Apr 2 [cited 2018 July 27]. Available from: http://dx.plos.org/10.1371/journal.pone.0093257
Bendiske J, Caba E, Brown QB, Bahr BA (2002) Intracellular deposition, microtubule destabilization, and transport failure: an “early” pathogenic Cascade leading to synaptic decline. J Neuropathol Exp Neurol [Internet] 61(7):640–650. July 1 [cited 2018 July 27]. Available from: https://academic.oup.com/jnen/article-lookup/doi/10.1093/jnen/61.7.640
Bendiske J, Bahr BA (2003) Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis--an approach for slowing Alzheimer disease? J Neuropathol Exp Neurol [Internet] 62(5):451–463. May [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12769185
Kanju PM, Parameshwaran K, Vaithianathan T, Sims CM, Huggins K, Bendiske J et al (2007) Lysosomal dysfunction produces distinct alterations in synaptic alpha-amino-3-hydroxy-5-methylisoxazolepropionic acid and N-methyl-D-aspartate receptor currents in hippocampus. J Neuropathol Exp Neurol [Internet] 66(9):779–788. Sept [cited 2018 July 27]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17805008
Acknowledgements
The authors gratefully acknowledge the funding provided by the NIH for our work; P30GM103329, R01MH100972, R01MH105329, R01MH119000, R01NS065957, and R01DA032444.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lakpa, K.L., Halcrow, P.W., Chen, X., Geiger, J.D. (2020). Readily Releasable Stores of Calcium in Neuronal Endolysosomes: Physiological and Pathophysiological Relevance. In: Islam, M. (eds) Calcium Signaling. Advances in Experimental Medicine and Biology, vol 1131. Springer, Cham. https://doi.org/10.1007/978-3-030-12457-1_27
Download citation
DOI: https://doi.org/10.1007/978-3-030-12457-1_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-12456-4
Online ISBN: 978-3-030-12457-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)