
Abstract
Glutamate accumulation into synaptic vesicles is a pivotal step in glutamate transmission. This process is achieved by a vesicular glutamate transporter (VGLUT) coupled to v-type proton ATPase. Normal synaptic transmission, in particular during intensive neuronal firing, would demand rapid transmitter re-filling of emptied synaptic vesicles. We have previously shown that isolated synaptic vesicles are capable of synthesizing glutamate from α-ketoglutarate (not from glutamine) by vesicle-bound aspartate aminotransferase for immediate uptake, in addition to ATP required for uptake by vesicle-bound glycolytic enzymes. This suggests that local synthesis of these substances, essential for glutamate transmission, could occur at the synaptic vesicle. Here we provide evidence that synaptosomes (pinched-off nerve terminals) also accumulate α-ketoglutarate-derived glutamate into synaptic vesicles within, at the expense of ATP generated through glycolysis. Glutamine-derived glutamate is also accumulated into synaptic vesicles in synaptosomes. The underlying mechanism is discussed. It is suggested that local synthesis of both glutamate and ATP at the presynaptic synaptic vesicle would represent an efficient mechanism for swift glutamate loading into synaptic vesicles, supporting maintenance of normal synaptic transmission.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the central nervous system, the majority of neural communication involves glutamate transmission. In glutamate transmission, glutamate loading into synaptic vesicles is an initial pivotal step, the gate for glutamate to enter the committed neurotransmitter pathway, away from the metabolic pathway [1–6]. This process enables synaptic vesicles to sufficiently concentrate glutamate prior to its release to the synaptic cleft. Glutamate thus concentrated, when released, serves as a signal to fire and regulate postsynaptic and presynaptic neurons via activation of its various receptors (AMPA-, kainate-, NMDA-preferring types, and the metabolic type).
Glutamate accumulation into synaptic vesicles is achieved by an energy-dependent, glutamate-specific vesicular uptake system, consisting of v-type proton-pump ATPase and a vesicular glutamate transporter (VGLUT) [1, 3, 6–18]. Vesicular proton-pump ATPase generates an electrochemical proton gradient, which is harnessed by VGLUT. Three isoforms (VGLUT1-3, SLC17A7, SLC17A6 and SLC17A8, respectively) are known [14, 15, 19–30], VGLUT1 being the major. They have different brain regional, cellular, and subcellular distributions. They are expressed not only in the brain and spinal cord, but also in “non-nervous tissues.” Gaged by their distributions, they perform different physiological functions [6]. However, they exhibit no significant difference in biochemical uptake function [18, 19, 23, 25, 27, 31]. The functional difference between VGLUT1 and VGLUT2 is thought to lie in membrane trafficking and transmitter release probability [19, 23, 28, 29]. VGLUT1 appears to have a role in regulating release probability [19, 23, 28, 29, 32, 33] and in synaptic plasticity [23, 34, 35]. The low release probability associated with VGLUT1 was shown due to its binding to endophilin A1 [32]. More recent studies however indicate that vesicular glutamate concentration itself affects release probability [33].
Synaptic vesicles are endowed with glycolytic ATP-generating enzymes, glyceraldehyde-3-phosphate dehydrogenase/3-phosphoglycerate kinase complex and pyruvate kinase, both capable of fueling VGLUT [36, 37]. Evidence suggests that vesicular glutamate loading in the presynaptic nerve ending is largely achieved at the expense of glycolytically produced ATP [36]. These observations suggest that local synthesis of ATP at the synaptic site plays an important role in accumulation of glutamate into synaptic vesicles. This could provide an efficient mechanism for vesicular glutamate loading into the nerve terminal, and hence in glutamate transmission.
In an effort to extend the local synthesis concept to glutamate synthesis, we have explored the possibility that synaptic vesicles could be capable of producing the VGLUT substrate. In eukaryotes, glutamate can be formed from α-ketoglutarate by glutamate dehydrogenase (GDH) or by aspartate aminotransferases (AAT), as well as from glutamine by glutaminase. Takeda et al. [38] have shown that isolated synaptic vesicles have the capacity to produce glutamate from α-ketoglutarate (α-KGA) by AAT, for immediate uptake by VGLUT. In this study, we provide evidence supporting the notion that α-KGA-derived glutamate, produced by AAT, is accumulated into synaptic vesicles within the nerve ending. The significance of these observations is discussed.
Materials and Methods
Animals
Sprague Dawley rats (180–220 g) were obtained from Charles River. All animal procedures were approved by the University of Michigan Committee for Use and Care of Animals (UCUCA) in accordance with National Institutes of Health Guide for the Use and Care of Laboratory Animals. Rats were sacrificed by decapitation, and cerebrum extracted.
Chemicals
[14C]α-KGA, [3H]glutamine, and [3H]glutamate were purchased from PerkinElmer, and bafilomycin from Fisher Scientific. All other chemicals were from Sigma-Aldrich.
Assay for Synaptosomal Uptake of Radioactive Ligands and Vesicular Radioactive Content
Crude synaptosomes (P2 fraction: 214–263 µg of protein) prepared from calf frontal cortex (obtained from a local slaughterhouse) were suspended in 200 µl oxygenated (95 % O2–5 % CO2) Krebs–Ringer buffer containing 171.5 mM NaCl, 1.84 mM K2HPO4, 2.45 mM MgSO4, 1.59 mM CaCl2, 6.13 mM glucose and 12.25 mM HEPES-NaOH (pH 7.4), and preincubated at 37 °C for 10 min in the absence or presence of 5 µM bafilomycin. After addition of 53 µM [14C]α-KGA (56.8 mCi/mmol), [3H]glutamine (94.7 or 190 mCi/mmol), or [3H]glutamate (190 mCi/mmol), synaptosomes were incubated for 30 min. Aliquots (40 µl) were removed and filtered on Whatman GF/C filters to determine the total amount of [14C]α-KGA, [3H]glutamine, or [3H]glutamate taken up by the synaptosomes, and the rest immediately frozen on dry ice. For determination of vesicular radioactive contents, the frozen synaptosomes (160 µl) were thawed, followed by addition of 1.5 ml of ice-cold hypotonic solution containing 6 mM Tris–maleate (pH 8.1) and 2 mM aspartate. The mixture was allowed to stand for 20 min on ice, and centrifuged at 4 °C for 10 min (Beckman microfuge, 17,600g max). The supernatant (1.2 ml) was filtered on Whatman GF/C filters, and radioactivity retained on filter determined in a Beckman LS 6500 scintillation spectrophotometer.
Analysis of Radioactive Compounds Accumulated into the Synaptic Vesicle Fraction from Synaptosomes Which Have Taken Up [14C]α-KGA or [3H]Glutamine
The calf cerebral cortex P2 fraction was incubated with [14C]α-KGA or [3H]glutamine in 200 ml of Krebs-Ringer solution, followed by addition of 1.6 ml of ice-cold hypotonic solution containing 6 mM Tris–maleate (pH 8.1) and 2 mM aspartate, as described above. The crude synaptic vesicle fraction (the material retained on the filter) was collected, as described above, and subjected to 80 % ethanol extraction and analysis for radioactive compounds by HPLC, as previously described [37].
Purification of Synaptosomes
In some experiments (Fig. 2), crude synaptosomes (P2 fraction) in 0.32 M sucrose, 0.25 mM DTT, and 1 mM EDTA were subjected to Percoll-gradient centrifugation, as described by Dunkley et al. [39]. Two aliquots (3 ml each) of the P2 fraction derived from 10 g of fresh calf frontal cortex were layered on top of the gradient consisting of 3, 10, 15, and 23 % Percoll (4 ml each per Sorvall SS-34 rotor tube) and centrifuged at 20,000 rpm (47,800g max) for 5 min. Fractions 1–4 represent the gradient interfaces from top to bottom, respectively. Fraction 5 represents the pellet. Each fraction (collected from two tubes) was suspended in 20 ml of Krebs-Ringer buffer containing 140 mM NaCl, 1.5 mM K2HPO4, 2 mM MgSO4, 1.3 mM CaCl2, 5 mM glucose and 10 mM HEPES–NaOH (pH 7.4) and centrifuged at 14,000 rpm (23,400g max) for 10 min, and resulting pellets suspended in Krebs-Ringer buffer.
Results
Radioactive Material Derived from [14C]α-Ketoglutarate is Accumulated into Synaptic Vesicles in Synaptosomes as is that Derived from [14H]Glutamine
We have previously shown that isolated synaptic vesicles are associated with AAT that catalyzes the synthesis of glutamate from α-KGA for its immediate uptake [38]. In an effort to demonstrate that this could occur in the intra-nerve ending milieu, synaptosomes (pinched-off nerve terminals) were incubated with [14C]α-KGA in an oxygenated Krebs-Ringer solution in the absence or presence of the proton-pump ATPase inhibitor bafilomycin, and the amount of radioactive material incorporated into the synaptic vesicle fraction determined. Bafilomycin (1 µM) has previously been shown to abolish glutamate uptake into isolated synaptic vesicles [10]. Figure 1 shows that although bafilomycin (5 µM) had negligible effect on total uptake of [14C]α-KGA into synaptosomes (A), it led to markedly reduced accumulation of radioactive material into synaptic vesicles in synaptosomes (B). The majority of radioactive material accumulated in synaptic vesicles was determined to be glutamate (Fig. 2a). This is compatible with the observation by Shank and Campbell (40) that α-ketoglutarate taken up into synaptosomes is mainly converted to glutamate. In addition, radioactive material produced from α-KGA by, and accumulated into, isolated synaptic vesicles, is essentially all glutamate, and α-KGA itself (i.e., in the absence of aspartate) is not taken up into synaptic vesicles [38]. Moreover, glutamate has been shown to represent the major neurotransmitter taken up into highly purified synaptic vesicles in an energy-dependent manner [40], consistent with glutamate being the principal neurotransmitter in brain [41].

α-KGA-derived glutamate uptake into synaptic vesicles occurs in synaptosomes, as does glutamate uptake derived from glutamine and exogenous glutamate. Synaptosomes (214–263 µg of protein) were preincubated in the absence (black bars) or presence (white bars) of bafilomycin, followed by incubation with [14C]α-KGA, [3H]glutamine, or [3H]glutamate, and vesicular content of radioactive material (glutamate) derived from each (b), as well as for synaptosomal uptake of each (a), was determined, as described in “Materials and Methods” section. For calculation of the ratio of SV-Glu content/synaptosomal uptake (c), vesicular glutamate content values obtained in the presence of bafilomycin were corrected for the inhibitory effect of bafilomcin on synaptosomal uptake and subtracted from those obtained in the absence of bafilomycin (b), and the bafilomycin-sensitive vesicular content value was divided by total synaptosomal uptake value in the absence of bafilomycin (a). Data on experiments with α-KGA and glutamine are the mean ± SEM from seven independent experiments and, for exogenous glutamate, the mean ± SEM from four independent experiments

HPLC chromatogram of [14C]α-KGA and [3H]Gln-derived products accumulated into synaptic vesicles in synaptosomes. Crude synaptosomes (P2 fraction) were incubated with [14C]α-KGA (a) or [3H]Gln (b), and the synaptic vesicle fraction was collected and subjected to analysis for radioactive compounds by HPLC, as described in “Materials and Methods” section
Bafilomycin-sensitive incorporation of radioactive material was considered to reflect the vesicular content of largely glutamate. Taking into account the small effect of bafilomycin on synaptosomal uptake, the vesicular accumulation of glutamate was expressed as the ratio (Fig. 1c) of bafilomycin-sensitive vesicular content (Fig. 1b) to synaptosomal uptake (Fig. 1a). Thus, Fig. 1 indicates that [14C]glutamate derived from [14C]α-KGA in synaptosomes is accumulated into synaptic vesicles.
Since glutamine is generally thought to be the principal precursor of the neurotransmitter glutamate, we have also measured vesicular content of radioactive material derived from [3H]glutamine in synaptosomes to compare with that derived from [14C]α-KGA. Figure 1 also shows that vesicular accumulation of radioactive material derived from [3H]glutamine is bafilomycin-sensitive. The majority of the radioactive substance accumulated is glutamate (Fig. 2b). The metabolic product of glutamine taken up into synaptosomes has also been shown to be largely glutamate [42]. Of interest, Fig. 1 indicates that α-KGA is more effective than glutamine as a precursor of glutamate in synaptosomes from calf frontal cortex. Surprisingly, when compared with vesicular [3H]glutamate accumulation in synaptosomes exogenously loaded with [3H]glutamate, vesicular accumulation of α-KGA-derived putative glutamate appears at least as efficient. This suggests that conversion of α-KGA to glutamate is rapid in synaptosomes, consistent with the high rates of AAT and the malate/aspartate shuttle in synaptosomes [43].
In order to confirm that vesicular accumulation of α-KGA- and glutamine-derived glutamate indeed occurs in synaptosomes, the crude synaptosome preparation was purified by Percoll-gradient centrifugation [39] and various fractions analyzed for vesicular content of glutamate derived from α-KGA and glutamine. Figure 3 shows that vesicular accumulation of glutamate derived from both α-KGA and glutamine is most abundant in the fraction richest in synaptosomes (fraction 4).

Vesicular accumulation of [14C]α-KGA- and [3H]glutamine-derived glutamate in various fractions obtained upon Percoll-gradient centrifugation of the crude synaptosome (P2) fraction. An aliquot of each fraction, which had been preincubated in the absence or presence of bafilomycin, was assayed for synaptosomal uptake of [14C]α-KGA and [3H]glutamine and for vesicular content of [14C]α-KGA-derived glutamate (black bars) and [3H]glutamine-derived glutamate (white bars), as described in “Materials and Methods” section, except that the incubation medium contained 53 µM [14C]α-KGA plus 53 µM [3H]glutamine (double-labeling). Fractions 1–4 denote the gradient interfaces from top to bottom, respectively. Fraction 5 denotes the pellet. Vesicular glutamate content (a) represents bafilomycin-sensitive vesicular glutamate content and expressed as the ratio (b) of vesicular glutamate content/synaptosomal uptake. PDG Percoll-density gradient
Vesicular α-KGA- and glutamine-derived glutamate accumulation in synaptosomes occurs in a time-dependent and bafilomycin-sensitive manner (Fig. 4). Bafilomycin greatly decreased vesicular accumulation of α-KGA- and glutamine-derived glutamate at all incubation times. Bafilomycin-sensitive vesicular accumulation of glutamate derived from α-KGA was 2.5- to threefold faster than that of glutamate derived from glutamine. This is likely due to the multi steps involved in generating glutamine-derived α-KGA available for extra-mitochondrial AAT: metabolism of glutamine, first to glutamate by mitochondrial glutaminase, then to α-KGA by mitochondrial AAT and GDH, followed by translocation of α-KGA to the cytoplasm. In addition, not all the α-KGA produced from glutamine may be transported out of mitochondria; part could be metabolized via the TCA cycle.

Time course of accumulation of [14C]α-KGA- and [3H]glutamine-derived glutamate in synaptic vesicles within synaptosomes. Synaptosomes (256 µg of protein) were preincubated in the absence (open circles) or presence (filled circles) of 5 µM bafilomycin, and assayed for uptake of [14C]α-KGA (a) and [3H]glutamine (d) and for vesicular content of [14C]α-KGA- and [3H]glutamine-derived glutamate (b, e), as described in Fig. 3, with the following modifications. The incubation mixture contained 140 mM NaCl, 1.5 mM K2HPO4, 2 mM MgSO4, 1.3 mM CaCl2, 5 mM glucose 10 mM HEPES–NaOH (pH 7.4), and 53 µM [14C]α-KGA plus 53 µM [3H]glutamine (double labeling). The ratio of vesicular content/synaptosomal uptake (c, f) was calculated as described in Fig. 1
Vesicular Loading of α-KGA-Derived Glutamate in Synaptosomes is Mediated by AAT and Fueled by Glycolytic ATP
In an effort to show that α-KGA-derived glutamate accumulation into synaptic vesicles in synaptosomes is mediated by AAT, we have examined the effect of hydroxylamine, a membrane-permeable AAT inhibitor, on that process. As shown in Fig. 5, hydroxylamine resulted in decreased vesicular content of α-KGA-derived “glutamate” in a dose-dependent manner, confirming this supposition.

Accumulation of α-KGA-derived glutamate in synaptic vesicles within synaptosomes is abolished by the AAT inhibitor hydroxylamine. Experimental conditions and procedures were essentially the same as described in Fig. 3, except that preincubation was carried out in the presence of various concentrations of hydroxylamine. The data represent the average with variation of two independent experiments
Evidence suggests that glutamate uptake into vesicles in synaptosomes is harnessed largely by glycolysis-generated ATP, a minor source of cellular energy [36]. In order to see whether this also applies to vesicular uptake of α-KGA-derived glutamate, we have examined the effect of the GAPDH inhibitor iodoacetate on this process and compared it with that of the mitochondrial ATP synthase inhibitor oligomycin. As shown in Fig. 6, vesicular accumulation of α-KGA-derived glutamate is substantially reduced in the presence of iodoacetate but little affected by oligomycin. On the other hand, overall ATP levels in synaptosomes are decreased in a similar fashion under these conditions [36]. These observations suggest that glycolytically produced ATP plays a major role in fueling vesicular loading of α-KGA-derived glutamate as well.

The glycolytic ATP synthase inhibitor iodoacetate, but not the mitochondrial ATP synthase inhibitor oligomycin, blocks vesicular accumulation of α-KGA-derived glutamate in synaptosomes. Experimental conditions and procedures were essentially the same as described in Fig. 3, except that preincubation was carried out in the presence of various concentrations of iodoacetate (a) or oligomycin (b). The data represent the mean ± SD from three independent experiments
Vesicular Loading of α-KGA-Derived Glutamate is Increased by Depolarization, as is that of Glutamine-Derived Glutamate
Memory formation involves high frequency neuronal firing, which leads to neuronal membrane depolarization. In order to examine whether vesicular loading of α-KGA- and/or glutamine-derived glutamate is modulated by neuronal activity, synaptosomes were depolarized and vesicular content of α-KGA- and glutamine-derived glutamate determined. In this experiment, we used synaptosomes prepared from the fresh cerebrum of matured rats. Depolarization inhibited synaptosomal uptake of both α-KGA and glutamine (Fig. 7a), as expected. Nonetheless, the measured amount of α-KGA-derived glutamate accumulated in synaptic vesicle was hardly changed (Fig. 7b). When the latter was corrected for the reduced uptake into synaptosomes, vesicular content (per amount of α-KGA taken up into synaptosomes) of both α-KGA- and glutamine-derived glutamate was actually increased (Fig. 7c). This indicates that while depolarization suppresses uptake into synaptosomes of α-KGA and glutamine, it leads to enhanced vesicular loading of glutamate derived from α-KGA and glutamine, perhaps in order to maintain vesicular glutamate content homeostasis.

Depolarization facilitates vesicular accumulation of α-KGA- and glutamine-derived glutamate. Depolarization enhances vesicular α-KGA- or glutamine-derived glutamate content in synaptosomes. Synaptosomes (P2 fraction: 374–387 µg) from fresh rat cerebrum were incubated at 37 °C for 30 min with 53 μM [14C]α-KGA plus [3H]glutamine (double labeling), each under depolarizing (with 56 mM K+ replacing 56 mM NaCl) or the non-depolarizing conditions described in Fig. 3, and bafilomycin-sensitive vesicular glutamate content (b) as well as synaptosomal uptake (a) determined. The ratio of vesicular content/synaptosomal uptake (c) was calculated as described in Fig. 1. The data represent the average with variation from two independent experiments
It is noted that in this experiment, which employed synaptosomes prepared from fresh rat cerebrum, that the vesicular content of glutamate derived from α-KGA is lower than that derived from glutamine. The reason is not known. This could be due to the difference in brain region, species, or developmental stages.
Discussion
Synaptic Vesicles Have the Capacity to Synthesize the VGLUT Substrate Glutamate
We have provided evidence that synaptic vesicles are capable of generating the neurotransmitter glutamate from α-KGA with aspartate as the amino donor, providing the VGLUT substrate for immediate uptake into vesicles, and resulting in an increased vesicular concentration of glutamate [38]. The vesicular concentration step is a process essential for glutamate to function as a neurotransmitter signal. We propose that this glutamate synthesis is efficiently achieved in large part by vesicle-bound AAT, not by glutaminase. This vesicle-bound aminotransferase is highly specific to L-aspartate, and its affinity for aspartate (Km = 0.9 mM) is similar to that of mitochondrial AAT, but substantially higher than that of cytosolic AAT (Km = 6.7 mM) [44]. Not only in kinetic properties but also in immunoreactivity, vesicle-bound AAT is distinct from cytosolic AAT and indistinguishable from mitochondrial AAT. Our finding is consistent with the mass spectrometric detection of mitochondrial AAT peptide fragments in highly purified synaptic vesicles [45, 46].
There are only two genes known to encode for AAT, GOT1 and GOT2, the former for cytosolic and the latter for mitochondrial AAT. Thus, vesicle-bound AAT is likely to be encoded by GOT2, belonging to the mitochondrial type of AAT. Mitochondrial AAT is bound to the matrix side of the inner membrane of mitochondria [47–52]. In contrast, vesicular AAT is likely to be bound to the vesicle exterior, rather than its lumen side, since it is easily dissociated with salt (data not shown). It would function as a generator of the VGLUT substrate to be taken up into synaptic vesicles, whereas mitochondrial AAT would function as a generator of α-KGA to be transported out of mitochondria and/or to be metabolized via the TCA cycle.
Mitochondrial AAT is synthesized in a precursor form containing a 29-amino acid-presequence peptide, whereas cytosolic AAT lacks this peptide. The N-terminal 30-amino acid segment of the processed precursor is critical for recognition by the peptidase which removes the presequence peptide, forming the mature form of mitochondrial AAT [53]. Transport of the mitochondrial AAT precursor to mitochondria involves binding to hsp 70; the binding requires both the presequence element and the N-terminal region of mature form of AAT [54]. These factors are also required for import of the precursor protein into mitochondria [53]. The mechanism of transport and binding of the mitochondrial type of AAT to synaptic vesicles remains to be elucidated.
Physiological Significance
In order to maintain normal synaptic transmission, after synaptic vesicles release neurotransmitters, emptied vesicles require rapid transmitter refilling. The evidence that synaptic vesicles bear AAT producing glutamate readily available for VGLUT suggests an efficient mechanism for swiftly accumulating glutamate into vesicles. That is proposed to be through its local synthesis at the synaptic vesicle site in the nerve ending, as opposed to the synthesis in the cytosol or mitochondria distant from synaptic vesicles. This is supported by the observation that vesicle-bound AAT has kinetic advantage over the cytosolic AAT described above. Having an aspartate Km value substantially lower than its physiological concentrations [55, 56], vesicle-bound AAT would function at a reaction rate close to Vmax in situ, whereas cytosolic AAT, with much lower aspartate affinity, would function at a rate far below Vmax. Cytosolic AAT is likely to be functional at locations where aspartate concentration is high, such as those close to mitochondria, where it would play a role in the malate/aspartate shuttle. Cytosolic glutamate produced by this isoform would be transported into mitochondria in exchange for the efflux of aspartate from mitochondria, mediated by the aspartate/glutamate carrier [57–60].
Synaptosomes are capable of taking up α-KGA (Figs. 1a, 3a, 4a), as initially shown by Shank and Campbell [42, 61, 62]. We have provided evidence here that α-KGA taken up into synaptosomes results in vesicular storage of putative glutamate (Figs. 3c, 4b, 5c). This is compatible with the observation that exogenously supplied α-KGA produces releasable glutamate in a neuronal culture [63]. Shank and Campbell [42] have also shown that exogenous α-KGA is converted largely to glutamate in synaptosomes, suggesting a role for extraneuronally derived α-KGA as a precursor of the transmitter glutamate. One source of α-KGA in vivo could be astrocytes. Astrocytes in culture have been shown to release α-KGA [64, 65]. This release was increased when the culture was incubated in the presence of bicarbonate, suggesting that this α-KGA is largely produced through pyruvate carboxylation prominent in astrocytes, followed by the TCA cycle [64, 65]. These observations together raise the possibility that α-KGA supplied by astrocytes in vivo could serve in part as a precursor for synthesis of the vesicular pool of glutamate. Shank et al. [66] suggested that α-KGA derived from the oxaloacetate de novo synthesized by astrocyte-specific pyruvate carboxylase could serve as a precursor of the neurotransmitter pool of glutamate. Moreover, glutamate taken up into astrocytes has been shown to be converted substantially to α-KGA, as well as to glutamine [67–71]. Thus, the glutamate-glutamine cycle is not stoichiometric [41].
Another source of α-KGA would be nerve terminal mitochondria, where it is produced via the TCA cycle fueled by glucose and glutamine, the latter being supplied by astrocytes as well as from cerebrospinal fluid [47, 72–74]. Palaiologos et al. [75] have provided evidence consistent with the notion that α-KGA transported out of mitochondria, mediated by the α-KGA-malate exchanger [76–78], is utilized to synthesize releasable glutamate.
Evidence presented here suggests that α-KGA, either exogenously supplied or endogenously produced from glutamine by glutaminase coupled with mitochondrial AAT and/or GDH, is converted to glutamate by extra-mitochondrial AAT in nerve terminals, and that glutamate, particularly the one re-synthesized by vesicle-bound AAT, is readily loaded into synaptic vesicles. This implies that vesicle-bound AAT has an important role in glutamate synaptic transmission. This notion is supported by a preliminary observation that injection of the AAT-selective inhibitor pyrazine-2,3-dicarboxylate [38] into the calyx of Held, a giant nerve terminal in the auditory brainstem, attenuated glutamate synaptic transmission (L. Y. Wang, personal communication). Cytosolic AAT could also contribute in part to production of glutamate to be taken up by synaptic vesicles. However, this mechanism would be less efficient because of its much lower affinity for aspartate, as described earlier. Nonetheless, the relative contribution of vesicle-bound and cytosolic AAT to the synthesis of the neurotransmitter pool of glutamate remains to be determined, particularly in view of the immunocytochemical localization of cytosolic AAT in nerve terminals [79, 80].
We have previously shown that synaptic vesicles are endowed with the glycolytic ATP-generating enzymes glyeraldehyde-3-phosphate dehydrogenase/3-phospoglycerate kinase and pyruvate kinase [36, 37], and that ATP produced through glycolysis is preferentially harnessed to fuel vesicular loading of glutamate into synaptosomes [36]. Evidence is presented here that α-KGA-derived glutamate uptake into synaptic vesicles in synaptosomes is also fueled by glcolytically generated ATP. This is consistent with the notion of an efficient mechanism for rapid vesicular glutamate refilling [36, 81]; glycolytic ATP is produced prior to ATP synthesized in mitochondria.
The cellular model of memory long-term potentiation, observed under high frequency neuronal stimulation, involves modification of both pre- and post-synaptic elements [82, 83]. Evidence indicates that neuronal activity presynaptically regulates neurotransmitter quantal size under physiological conditions [84]. Wilson et al. [85] have demonstrated that chronic increase in neuronal activity regulates VGLUT expression and the amount of glutamate released per vesicle. We have observed that the process of vesicular accumulation of α-KGA-derived and glutamine-derived glutamate in synaptosomes is enhanced by depolarization, which however suppresses synaptosomal uptake. This is relevant to activity-dependent alteration in the presynaptic nerve terminal. Depolarization is known to induce reversal of Na+-dependent plasma membrane transport [86]. The enhancing effect of depolarization on glutamate accumulation in synaptic vesicles could reflect a presynaptic acute homeostatic response in order to sustain glutamate transmission. These results are consistent with our previous observation that pre-exposure of synaptosomes to depolarization conditions leads to augmented vesicular glutamate content [87]. The mechanism underlying this acute depolarization-induced alteration in the synaptic vesicular content of α-KGA/glutamine-derived glutamate remains to be elucidated.
Vesicular Accumulation of Glutamine-Derived Glutamate vs. α-KGA-Derived Glutamate
Bradford et al. [72] have previously shown that glutamine is taken up into synaptosomes, where it is transformed to releasable glutamate by glutaminase. Our studies suggest that glutamine taken up into synaptosomes is incorporated into synaptic vesicles largely as glutamate. This occurs despite the fact that synaptic vesicles are essentially free of glutaminase [38, 45, 46]. Five pathways are possible for this occurrence: (a) Glutamate produced by mitochondrial glutaminase [88] is transferred to the cytoplasm and taken up by synaptic vesicles. (b) Glutaminase-generated glutamate is first transaminated by AAT in mitochondria, producing α-KGA and aspartate, both of which are then transported out of mitochondria and utilized by extra-mitochondrial AAT, reforming glutamate and oxaloacetate. Glutamate thus re-synthesized is taken up into synaptic vesicles. (c) Glutamine-derived glutamate is converted to α-KGA in the mitochondria matrix through oxidative deamination mediated by GDH. α-KGA thus formed is then transported out of mitochondria and transformed by extra-mitochondrial AAT to glutamate, which is then taken up into synaptic vesicles. (d) Combination of (b) and (c). (e) Combination of (a), (b) and/or (c).
Pathway a is unlikely since mitochondrial glutaminase is localized to the mitochondrial inner membrane [89] and its matrix side [48, 49, 52]; this implies that the glutamine to glutamate conversion occurs in the matrix. Palaiologos et al. [75] have provided evidence, using cultured cerebellar granule neurons, suggesting that AAT activity and α-KGA efflux from mitochondria are required for re-synthesis of releasable glutamate from glutamine. Yudkoff et al. [43] have demonstrated that reformation of glutamate from glutamine-derived α-KGA, as well as the malate/aspartate shuttle, is rapid in synaptosomes. Synaptosomal mitochondria have high AAT activity [47, 74, 90], which would produce substantial amounts of α-KGA and aspartate from glutamine-derived glutamate for export to the cytoplasm, supplying the substrates for extra-mitochondrial AAT. These lines of evidence support pathway b. On the other hand, Tildon et al. [91] and McKenna et al. [92] have provided evidence that in synaptosomes GDH plays a major role in converting glutamine-derived glutamate to α-KGA for the TCA cycle. GDH activity is significantly higher in synaptic mitochondria than in non-synaptic mitochondria, in particular the heavier synaptic mitochondrial fraction [47, 74, 90, 93]. These observations support pathway c. Moreover, over-expression of GDH has recently been shown to cause increased synaptic release of glutamate [94]. Overall pathway d would be the more likely possibility. Pathway b generates not only α-KGA but also aspartate, which is not directly produced by pathway c. α-KGA generated by pathway c could be transported out of mitochondria and/or enter the TCA cycle, in parallel leading to replenishment of the oxaloacetate which had exited from the TCA cycle as a result of its conversion to aspartate. The relative contribution of these pathways remains to be investigated.
Bradford et al. [72] and Hamberger et al. [73] have provided biochemical evidence that glutamine serves as the major precursor for synthesis of the neurotransmitter glutamate. This, together with the localization of glutamine synthetase to astrocytes [95] and other evidence [55, 96, 97], has led to the concept that the glutamate-glutamine cycle plays a central role in synthesis of the neurotransmitter pool of glutamate [65, 98, 99]. According to this model, glutamate released from the nerve terminal is taken up by adjacent astrocytes, where it is converted to glutamine by glutamine synthetase, which is highly enriched in astrocytes. Glutamine thus produced is released from astrocytes and taken up by adjacent nerve terminals, where it is hydrolyzed to glutamate by mitochondrial glutaminase. Glutamate thus produced provides the neurotransmitter pool of glutamate and is released. Although this model is well recognized, and the glutamine-glutamate conversion is essential for maintaining activity-dependent release of glutamate and the function of active synapses, the role of the glutamate-glutamine cycle in glutamate synaptic transmission, in particular spontaneous glutamate release and baseline glutamatergic synaptic transmission is not entirely clear [84, 100]. Kam and Nicoll [100] and Masson et al. [101] have provided evidence that spontaneous glutamate transmission occurs even in the absence of glia cells or of an exogenous supply of glutamine, as well as in the absence of mitochondrial glutaminase. This raises the possibility that glutamate synthesized from part of the α-KGA derived from glucose via the neuronal TCA cycle could be responsible for spontaneous glutamate transmission. This α-KGA is constantly produced in nerve terminals, independent of astrocyte metabolism or synaptically released glutamate, a major source of glutamine synthesis in astrocytes. Alternative carbon sources such as other amino acids, fatty acids, and ketone bodies, however, cannot be ruled out [41, 71].
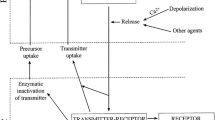
Palaiologos et al. [75] have provided evidence suggesting that the conversion of glutamine-derived glutamate to α-KGA by mitochondrial AAT, as well as the efflux of α-KGA and aspartate from mitochondria, is essential for re-synthesis of releasable glutamate in the extra-mitochondrial compartment. This notion is consistent with our observations presented here. We propose that α-KGA is the principal immediate precursor for synthesis of the neurotransmitter pool of glutamate, regardless of whether α-KGA is derived from glutamine (through the actions of glutaminase and AAT/GDH) or from glucose via the TCA cycle, or of whether it is directly supplied from astrocytes (Fig. 8). Vesicle-bound AAT-coupled glutamate biosynthesis from α-KGA, together with local synthesis of ATP by vesicle-bound GAPDH/3-phosphoglycerate kinase, could represent an effective mechanism for rapid refilling of synaptic vesicles with glutamate to sustain normal synaptic transmission.

Model for synthesis and loading of neurotransmitter glutamate. Proposed is an efficient mechanism for synthesis of the VGLUT substrate glutamate for immediate uptake into synaptic vesicles. The VGLUT substrate is locally produced by vesicle-bound AAT from α-KGA and aspartate and readily transported into synaptic vesicles, fueled by vesicle-bound glycolytic ATP-generating enzymes. α-KGA derives from nerve terminal mitochondria, where it is generated (1) thorough the TCA cycle, (2) by AAT and (3) by GDH from glutamate, which is produced by glutaminase from astrocyte-derived glutamine. α-KGA is also supplied by adjacent astrocytes. Aspartate derives from mitochondria, where it is produced by AAT from oxaloacetate with glutamate as the amino donor. AAT aspartate aminotransferase, α-KGA α-ketoglutarate, Asp aspartate, 1,3-BPG 1,3-bisphosphoglycerate, GAP glyceraldehyde-3-phophate, GAPDH glyceraldehyde-3-phophate dehydrogenase, GDH glutamate dehydrogenase, Glu glutamate, Gln glutamine, IA iodoacetate, OAA oxaloacetate, P i inorganic phosphate, 2,3-PDC 2,3-pirazinedicarboxylate, 3-PG 3-phophoglycerate, 3-PGK 3-phosphoglycerate kinase, SV synaptic vesicle, VGLUT vesicular glutamate transporter, v-H + -ATPase v-type proton-pump ATPase
Abbreviations
- AAT:
-
Aspartate aminotransferase
- ACPD:
-
1-Aminocyclopentane-1,3-dicarboxylic acid
- α-KGA:
-
α-Ketoglutarate
- FCCP:
-
Carbonyl cyanide p-(trifluoromethoxy)-phenylhydrazone
- GAPDH:
-
Glyceraldehyde-3-phophate dehydrogenase
- GABA:
-
γ-Aminobutyric acid
- GDH:
-
Glutamate dehydrogenase
- Gln:
-
Glutamine
- GOT:
-
Glutamate oxaloacetate aminotransferase
- HPLC:
-
High pressure liquid chromatography
- hsp:
-
Heat shock protein
- SV:
-
Synaptic vesicle
- TCA:
-
Tricarboxylic acid
- VGLUT:
-
Vesicular glutamate transporter
References
Ueda T (1986) Glutamate transport in the synaptic vesicle. In: Roberts PJ, Storm-Mathisen J, Bradford HF (eds) Excitatory amino acids. Macmillan, London, pp 173–195
Maycox PR, Hell JW, Jahn R (1990) Amino acid neurotransmission: spotlight on synaptic vesicles. Trends Neurosci 13:83–87
Ozkan ED, Ueda T (1998) Glutamate transport and storage in synaptic vesicles. Jpn J Pharmacol 77:1–10
Otis TS (2001) Vesicular glutamate transporters in cognito. Neuron 29:11–14
Reimer RJ, Fremeau RT Jr, Bellocchio EE, Edwards RH (2001) The essence of excitation. Curr Opin Cell Biol 13:417–421
Ueda T (2016) Vesicular glutamate uptake. In: Schousboe A, Sonnewald U (eds) Advanced neurobiology. Springer, New York (in press)
Naito S, Ueda T (1985) Characterization of glutamate uptake into synaptic vesicles. J Neurochem 44:99–109
Maycox PR, Deckwerth T, Hell JW, Jahn R (1988) Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem 263:15423–15428
Fykse EM, Christensen H, Fonnum F (1989) Comparison of the properties of γ-aminobutyric acid and L-glutamate uptake into synaptic vesicles isolated from rat brain. J Neurochem 52:946–951
Tabb JS, Ueda T (1991) Phylogenetic studies on the synaptic vesicle glutamate transport system. J Neurosci 11:1822–1828
Tabb JS, Kish PE, Van Dyke R, Ueda T (1992) Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient, and intravesicular pH. J Biol Chem 267:15412–15418
Wolosker H, de Souza DO, de Meis L (1996) Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem 271:11726–11731
Lewis SM, Ueda T (1998) Solubilization and reconstitution of synaptic vesicle glutamate transport system. Methods Enzymol 296:125–144
Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407:189–194
Bellocchio EE, Reimer RJ, FremeauTJ, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289:957–960
Juge N, Yoshida Y, Yatsushiro S, Omote H, Moriyama Y (2006) Vesicular glutamate transporter contains two independent transport machineries. J Biol Chem 281:39499–39506
Schenck S, Wojcik SM, Brose N, Takamori S (2009) A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosci.12: 156–162
Preobraschenski J, Zander JF, Suzuki T, Ahnert-Hilger G, Jahn R (2014) Vesicular glutamate transporters use flexible anion and cation binding sites for efficient accumulation of neurotransmitter. Neuron 84:1287–1301
Bai L, Xu H, Collins JF, Ghishan FK (2001) Molecular and functional analysis of a novel neuronal vesicular glutamate transporter. J Biol Chem 276:36764–36769
Fremeau RT Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31:247–260
Takamori S, Rhee JS, Rosenmund C, Jahn R (2001) Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2). J Neurosci 21:RC182
Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S (2001) The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci 21:RC181
Varoqui H, Schafer MK-H, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na+/Pi transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22:142–155
Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S (2002) A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci 22:5442–5451
Fremeau RT Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci USA 99:14488–14493
Takamori S, Malherbe P, Broger C, Jahn R (2002) Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep 3:798–803
Schafer MK-H, Varoqui H, Defamie N, Weihe E, Erickson JD (2002) Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem 277:50734–50748
Fremeau RT Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304:1815–1819
Fremeau RT Jr, Voglmaier S, Seal RP, Edwards RH (2004) VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci 27:98–103
Reimer RJ, Edwards RH (2004) Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch 447:629–635
Kaneko T, Fujiyama F (2002) Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci Res 42:243–250
Weston MC, Nehring RB, Wojcik SM, Rosenmund C (2011) Interplay betweenVGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron 69:1147–1159
Herman MA, Ackermann F, Trimbuch T, Rosenmund C (2014) Vesicular glutamate transporter expression level affects synaptic vesicle release probability at hippocampal synapses in culture. J Neurosci 34:11781–11791
Tordera R, Totterdell S, Wojcik S, Brose N, Elizalde N, Lasheras B, Rio J (2007) Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (VGLUT1). Eur J Neurosci 25:281–290
Balschun D, Moechars D, Callaerts-Vegh Z, Vermaercke B, Van Acker N, Andries L, D’Hooge R (2010) Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb Cortex 20:684–693
Ikemoto A, Bole DG, Ueda T (2003) Glycolysis and glutamate accumulation into synaptic vesicles: role of glyceraldehyde phosphate dehydrogenase and 3-phosphoglycerate kinase. J Biol Chem 278:5929–5940
Ishida A, Noda Y, Ueda T (2009) Synaptic vesicle-bound pyruvate kinase can support vesicular glutamate uptake. Neurochem Res 34:807–818
Takeda K, Ishida A, Takahash K, Ueda T (2012) Synaptic vesicles are capable of synthesizing the VGLUT substrate glutamate from α-ketoglutarate for vesicular loading. J Neurochem 121:184–196
Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JAP (1986) A rapid method for isolation of synaptosomes on percoll gradients. Brain Res 372:115–129
Naito S, Ueda T (1983) Adenosine triphosphate-dependent uptake of glutamate into protein I-associated synaptic vesicles. Biol Chem 258:696–699
McKenna MC (2007) The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 85:3347–3358
Shank RP, Campbell GL (1982) Glutamine and alpha-ketoglutarate uptake and metabolism by nerve terminals enriched material from mouse cerebellum. Neurochem Res 7:601–616
Yudkoff M, Nelson D, Daikhin Y, Erecinska M (1994) Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J Biol Chem 269:27414–27420
Magee SC, Phillips AT (1971) Molecular properties of the multiple aspartate aminotransferases purified from rat brain. Biochemistry 31:3397–3405
Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, Jahn R (2006) Molecular anatomy of a trafficking organelle. Cell 127:831–846
Burré J, Beckhaus T, Schägger H, Corvey C, Hofmann S, Karas M, Zimmermann H, Volknandt W (2006) Analysis of the synaptic vesicle proteome using three gel-based protein separation techniques. Proteomics 6:6250–6262
McKenna MC, Stevenson JH, Huan X, Hopkins IB (2000) Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem Int 37:229–241
Shapiro RA, Haser WG, Curthoys NP (1985) The orientation of phosphate-dependent glutaminase on the inner membrane of rat renal mitochondria. Arch Biochem Biophys 243:1–7
Aledo JC, de Pedro E, Gomez-Fabre PM, de Castro IN, Marquez J (1997) Submitochondrial localization and membrane topography of Ehrlich ascitic tumour cell glutaminase. Biochim Biophys Acta 1323:173–184
Albrecht J, Dolinska M, Hilgier W, Lipkowski AW, Nowacki J (2000) Modulation of glutamine uptake and phosphate-activated glutaminase activity in rat brain mitochondria by amino acids and their synthetic analogues. Neurochem Int 36:341–347
Zieminska E, Hilgier W, Waagepetersen HS, Hertz L, Sonnewald U, Schousboe A, Albrecht J (2004) Analysis of glutamine accumulation in rat brain mitochondria in the presence of a glutamine uptake inhibitor, histidine, reveals glutamine pools with a distinct access to deamidation. Neurochem Res 29:2121–2123
Bak LK, Zieminnska E, Waagepetersen HS, Schousboe A, Albrecht J (2008) Metabolism of [U-13C] glutamine and [U-13C]glutamate in isolated rat brain mitochondria suggests functional phosphate-activated glutaminase activity in matrix. Neurochem Res 33:273–278
Lain B, Yañez A, Iriarte A, Martinez-Carrion M (1998) Aminotransferase variants as probes for the role of the N-terminal region of a mature protein in mitochondrial precursor import and processing. J Biol Chem 273:4406–4415
Lain B, Iriarte A, Mattingly JR Jr, Moreno JI, Martinez-Carrion M (1995) Structural features of the precursor to mitochondrial aspartate minotransferase responsible for binding to hsp70. J Biol Chem 270:24732–24739
Benjamin AM, Quastel JH (1972) Locations of amino acids in brain slices from the rat. Tetrodotoxin-sensitive release of amino acids. Biochem J 128:631–646
Taguchi T, Miyake K, Tanonaka K, Okada M, Takagi N, Fujimori K, Takeo S (1993) Sustained changes in acetylcholine and amino acid contents of brain regions following microsphere embolism in rats. Jpn J Pharmacol 62:269–278
LaNoue KF, Schoolwerth AC (1979) Metabolite transport in mitochondria. Annu Rev Biochem 48:871–922
Cheeseman AJ, Clark JB (1988) Influence of the malate-aspartate shuttle on oxidative metabolism in synaptosomes. J Neurochem 50:1559–1565
Napolioni V, Persico AM, Porcelli V, Palmieri L (2011) The mitochondrial aspartate/glutamate carrier AGC1 and calcium homeostasis: physiological links and abnormalities in autism. Mol Neurobiol 44:83–92
Rueda CB, Llorente-Folch I, Amigo I, Contreras L, González-Sánchez P, Martínez-Valero P, Juaristi I, Pardo B, Del Arco A, Satrústegui J (2014) Ca2+ regulation of mitochondrial function in neurons. Biochim Biophys Acta 1837:1617–1624
Shank RP, Campbell GL (1981) Avid Na+-dependent, high-affinity uptake of alpha-ketoglutarate by nerve terminal enriched material from mouse cerebellum. Life Sci 28:843–850
Shank RP, Campbell GL (1984) Alpha-ketoglutarate and malate uptake and metabolism by synaptosomes: further evidence for an astrocyte-to-neuron metabolic shuttle. J Neurochem 42:1153–1161
Peng L, Schousboe A, Hertz L (1991) Utilization of alpha-ketoglutarate as a precursor for transmitter glutamate in cultured cerebellar granule cells. Neurochem Res 16:29–34
Westergaard N, Sonnewald U, Schousboe A (1994) Release of α-ketoglutarate, malate and succinate from cultured astrocytes: possible role in amino acid neurotransmitter homeostasis. Neurosci Lett 176:105–109
Schousboe A, Westergaard N, Waagepetersen H, Larsson OM, Bakken IJ, Sonnewald U (1997) Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 21:99–105
Shank RP, Bennett GS, Freytag SO, Campbell G (1985) Pyruvate carboxylase: astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res 329:364–367
Sonnewald U, Westergaard N, Petersen SB, Unsgard G, Schousboe A (1993) Metabolism of [U-13C] glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem 61:1179–1182
McKenna M, Sonnewald U, Huang X, Stevenson J, Zielke HR (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66:386–393
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145
Dienel GA, McKenna MC (2014) A dogma-breaking concept: glutamate oxidation in astrocytes is the source of lactate during aerobic glycolysis in resting subjects. J Neurochem 131:395–398
Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J Neurochem 131:399–406
Bradford HF, Ward HK, Thomas AJ (1978) Glutamine—a major substrate for nerve endings. J Neurochem 30:1453–1459
Hamberger AC, Chiang GH, Nylén ES, Scheff SW, Cotman CW (1979) Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res 168:513–530
Dennis SC, Lai JCK, Clark JB (1977) Comparative studies on glutamate metabolism in synaptic and non-synaptic rat brain mitochondria. Biochem J 164:727–736
Palaiologos G, Hertz L, Schousboe A (1988) Evidence that aspartate aminotransferase activity and ketodicarboxylate carrier function are essential for biosynthesis of transmitter glutamate. J Neurochem 51:317–320
Bisaccia F, Indiveri C, Palmieri F (1985) Purification of reconstitutively active α-ketoglutarate carrier from pig heart mitochondria. Biochim Biophys Acta 810:362–369
Bolli R, Narecz KA, Azzi A (1989) Monocarboxylate and α-ketoglutarate carriers from bovine heart mitochondria. J Biol Chem 264:18024–18030
Palaiologos G, Hertz L, Schousboe A (1989) Role of aspartate aminotransferase and mitochondrial dicarboxylate transport for release of endogenously and exogenously supplied neurotransmitter in glutamatergic neurons. Neurochem Res 14:359–366
Altschuler RA, Neises GR, Harmison GG, Wenthold RJ, Fex J (1981) Immunocytochemical localization of aspartate aminotransferase immunoreactivity in cochlear nucleus of the guinea pig. Proc Natl Acad Sci USA 78:6553–6657
Altschuler RA, Mosinger JL, Harmison GG, Parakkal MH, Wenthold RJ (1982) Aspartate aminotransferase-like immunoreactivity as a marker for aspartate/glutamate in guinea pig photoreceptors. Nature 298:657–659
Ueda T, Ikemoto A (2007) Cytoplasmic glycolytic enzymes. Synaptic vesicle-associated glycolytic ATP-generating enzymes: coupling to neurotransmitter accumulation. In: Gibson G, Dienel G (eds) Handbook of neurochemistry and molecular neurobiology, 3rd edn. Brain energetics, cellular and molecular integration. Springer, Heidelberg, pp 241–259
Malinow R, Tsien RW (1990) Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature 346:177–180
Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39
Edwards RH (2007) The neurotransmitter cycle and quantal size. Neuron 55:835–858
Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G (2005) Presynaptic regulation of quantal size by VGLUT1. J Neurosci 25:6221–6234
Nicholls D, Attwell D (1990) The release and uptake of excitatory amino acids. Trends Pharmacol Sci 11:462–468
Bole DG, Hirata K, Ueda T (2002) Prolonged depolarization of rat cerebral synaptosomes leads to an increase in vesicular glutamate content. Neurosci Lett 322:17–20
Bradford HF, Ward HK (1976) On glutaminase activity in mammalian synaptosomes. Brain Res 110:115–125
Laake JH, Takumi Y, Eidet J, Torgner IA, Roberg B, Kvamme E, Ottersen OP (1999) Postembedding immunogold labelling reveals subcellular localizationand pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience 88:1137–1151
Lai JC, Walsh JM, Dennis SC, Clark JB (1977) Synaptic and non-synaptic mitochondria from rat brain: isolation and characterization. J Neurochem 28:625–631
Tildon JT, Roeder LM, Stevenson JH (1985) Substrate oxidation by isolated rat brain mitochondria and synaptosomes. J Neurosci Res 14:207–215
McKenna MC, Tildon JT, Stevenson JH, Boatright R, Huang S (1993) Regulation of energy metabolism in synaptic terminals and cultured rat brain astrocytes: differences revealed using aminooxyacetate. Dev Neurosci 15:320–329
Dennis SC, Land JM, Clark JB (1976) Glutamate metabolism and transport in rat brain mitochondria. Biochem J 156:323–331
Michaelis EK, Wang X, Pal R, Bao X, Hascup KN, Wang Y, Wang WT, Hui D, Agbas A, Choi IY, Belousov A, Gerhardt GA (2011) Neuronal Glud1 (glutamate dehydrogenase 1) over-expressing mice: increased glutamate formation and synaptic release, loss of synaptic activity, and adaptive changes in genomic expression. Neurochem Int 59:473–481
Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195:1356–1358
van den Berg CJ, Garfinkel D (1971) A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J 123:211–218
Shank RP, Aprison MH (1981) Present status and significance of the glutamine cycle in neural tissues. Life Sci 28:837–842
Cotman CW, Foster AC, Lanthorn TH (1981) An overview of glutamate as a neurotransmitter. In: Di Chiara G, Gessa GL (eds) Glutamate as a neurotransmitter. Raven Press, New York, pp 1–27
Shank RP, Aprison MH (1988) Glutamate as a neurotransmitter. In: Kvamme E (ed) Glutamine and glutamate in mammals, vol II. CRC Press, Boca Raton, pp 3–20
Kam K, Nicoll R (2007) Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27:9192–9200
Masson J, Darmon M, Conjard A, Chuhma N, Ropert N, Thoby-Brisson M, Foutz AS, Parrot S, Miller GM, Jorisch R, Polan J, Hamon M, Hen R, Rayport S (2006) Mice lacking brain/kidney phosphate-activated glutaminase have impaired glutamatergic synaptic transmission, altered breathing, disorganized goal-directed behavior and die shortly after birth. J Neurosci 26:4660–4671
Acknowledgments
This work was supported by National Institutes of Health Grant MH 071384 (TU).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest regarding the work reported here.
Rights and permissions
About this article

Cite this article
Takeda, K., Ueda, T. Effective Mechanism for Synthesis of Neurotransmitter Glutamate and its Loading into Synaptic Vesicles. Neurochem Res 42, 64–76 (2017). https://doi.org/10.1007/s11064-016-2037-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2037-3