
Abstract
Recently, molecular studies have determined that the SLC17/type I phosphate transporters, a family of proteins initially characterized as phosphate carriers, mediate the transport of organic anions. While their role in phosphate transport remains uncertain, it is now clear that the transport of organic anions facilitated by this family of proteins is involved in diverse processes ranging from the vesicular storage of the neurotransmitter glutamate to the degradation and metabolism of glycoproteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview
The SLC17 (type I phosphate/vesicular glutamate transporter) family of transporters (see Table 1) is a group of proteins that mediate the transmembrane transport of organic anions. The members of this family first characterized, the type I phosphate transporters, were initially identified as Na+-dependent phosphate transporters. More recent work has, however, determined that all recognized members of this family, including the type I phosphate transporters, are involved in the transmembrane transport of organic anions. In addition to the type I phosphate transporters, identified mammalian members of this family now include vesicular glutamate transporters (VGLUT1–3), and a lysosomal sialic acid transporter (sialin). The proteins are multipass transmembrane proteins with 6–12 predicted transmembrane domains that reside primarily on synaptic vesicles (the VLGUTs), lysosomes (sialin) and the plasma membrane (type I phosphate transporters).
Phylogeny
The SLC17 are part of a large family that in addition to the mammalian proteins covered in this review (Fig. 1) includes bacterial, fungal and plant proteins. Although the SLC17 proteins have been implicated in functions similar to those of several other families of proteins, they have no significant homology to the other vesicular neurotransmitter transporter families (SLC18 which includes the VMAT and VAChT and SLC32 which includes VGAT/VIAAT), other lysosomal transport proteins (LYAAT1—a member of the SLC36 family, and cystinosin a lysosomal cystine transporter), the type II (SLC34) or type III (SLC20) phosphate transporters, or the organic anion transporters (SLC22). BLAST searches [2] of available NCBI databases with members of the SLC17 family indicate that related bacterial proteins are likely to be transporters for the anionic sugar glucarate, but no functional studies have been done on these proteins. Multiple putative proteins are also identified from plant and fungal sequence databases, but functions for these proteins have neither been directly determined, nor predicted.

Three distinct subfamilies are present in the SLC17A family of proteins. Computer-based phylogenetic analysis of the identified members of the SLC17A family proteins illustrates a high degree of homology among the VGLUT sequences (74–82% identity.) The homology of the VGLUTs to sialin (40% identity between VGLUT1 and sialin) and the type I phosphate transporters (30% identity between VGLUT1 and NPT1) is much lower. Sialin has similar homology to the VGLUTs and type I phosphate transporters (38% identity between NPT1 and sialin.) Among the type I phosphate transporters the identity is 38–60%
The type I phosphate transporters (SLC17A1–4)
The type I phosphate transporters were first identified in a functional screen for Na+-dependent phosphate transporters [47]. The screen led to the identification of a single sequence from rabbit kidney that when injected into Xenopus oocytes increased phosphate uptake by four- to sixfold with a K m~1 mM for inorganic phosphate (Pi). The cDNA predicts a multitransmembrane domain protein of 465 amino acids and was designated NaPi-1 as it was the first sequence shown to mediate Na+-dependent phosphate transport. The human orthologue NPT1 has similar transport characteristics but a slightly higher apparent affinity for Pi (0.3 mM) [29].
The measured affinity for phosphate transport by NaPi-1 is much lower than for transport by native tissues, suggesting that NaPi-1 is not the primary route for phosphate uptake. Supporting this possibility, two other structurally unrelated families of phosphate transporters (type II and type III) have been identified and shown to mediate phosphate uptake with higher affinity [30].
The uncertain role of type I transporters in phosphate uptake contrasts further with the other activities that they exhibit. Specifically, these proteins have the capacity to transport organic anions [12]. The transport of probenecid and penicillin, but not Pi, correlate directly with NaPi-1 expression. The apparent affinity of NaPi-1 for organic anions (0.22 mM for benzylpenicillin) is also higher than for Pi (1 mM for NaPi-1). Studies of NaPi-1 and human NPT1 further demonstrate that the transport of organic anions is electrogenic and that it is not dramatically influenced by pH [12, 43, 50]. However, the ionic coupling has not been determined and the nature of the endogenous substrate remains controversial. In addition, NaPi-1 mediates an ionic conductance with the apparent selectivity I−>Br−>Cl−, and this activity also correlates well with NaPi-1 expression [11, 12]. The ionic conductance is inhibited by substrates for the transport activity. In the case of benzylpenicillin, the EC50 is 0.02 mM, tenfold lower than the K m for transport [12]. Transport may thus negatively regulate the ionic conductance associated with NaPi-1.
The tissue distribution of type I phosphate transporters is relatively restricted. By northern analysis, the rabbit NaPi-1 mRNA is expressed in the kidney and to a lesser extent the liver [47]. In situ hybridization and immunohistochemistry indicate expression is limited to the brush border membrane of the proximal tubule in the kidney and the sinusoidal membrane of hepatocytes [10, 16, 50].
Three other proteins closely related to NPT1 have been identified through genomic analysis, and designated NPT3 (SLC17A2), NPT4 (SLC17A3) and Na+/PO4 2− cotransporter homologue (SLC17A4) [36, 38]. NPT2 is actually a type II transporter (SLC34A1) and structurally unrelated to NaPi-1. Interestingly, the four related genes (SLC17A1–4) all localize to 6p21.3-p23 near the hereditary hemochromatosis gene HFE, but no human disease has been linked genetically to the NPTs [36]. By Northern blot, NPT3 is expressed at high levels in the heart and muscle, with lower levels in brain, placenta, lung, liver and kidney, and NPT4 is restricted to the liver and kidney, but RTPCR also indicates expression in the small intestine and testis. RTPCR studies also indicate expression of the SCL17A4 gene in the liver, pancreas, small intestine and colon. Hepatocyte nuclear factor 1 alpha (HNF1α) has been shown to upregulate NPT1 and NPT4 (the other nearby genes (NPT3 and Na+/PO4 2− cotransporter homologue) were not analyzed) [15]. NPT1, 3 and 4 also undergo alternative splicing, but the physiological significance has not been determined. As of yet, there has been no functional characterization of SLC17A2–4.
Sialin (SLC17A5)
Lysosomes export sialic acid derived from the degradation of glycosylated membrane proteins through a process dependent on the H+ electrochemical gradient across the lysosomal membrane. The identification of free sialic acid as the material accumulated in the enlarged lysosomes of individuals with sialic acid storage disorders [Salla disease and infantile sialic acid storage disease (ISSD)] indeed led to the characterization of this transport system [27].
A lysosomal sialic acid transport activity was first described in lysosomes isolated from native tissue [27]. The activity saturates with a K m~0.24 mM, and depends on pH, with higher levels of transport associated with lower extralumenal pH [21, 28]. The mechanism has thus been proposed to involve the symport of acidic sugar with H+. Although transport has been measured by the uptake of radiolabelled sialic acid into lysosomes, the biologically relevant process is considered to be efflux, with the H+ electrochemical gradient (ΔμH+) generated by the lysosomal H+-ATPase as the driving force. Transport appears to depend on the chemical gradient (ΔpH) but not the electrical gradient (ΔΨ) across the lysosomal membrane, suggesting an electroneutral process, and this fits with the experimentally determined 1:1 (amino acid:H+) stoichiometry. In addition to sialic acid, this transport system recognizes monocarboxylic acids, including lactate, glucuronic acid and gluconate, but does not mediate transport of neutral sugars such as glucose and mannose.
The export of sialic acid out of lysosomes is defective in patients with both Salla disease and ISSD [5]. Genetic linkage studies of patients with Salla disease led to the identification of a gene associated with the disease on chromosome 6q14-q15 [45]. As anticipated, the mRNA is expressed ubiquitously but no further anatomical studies have been performed. In addition, the protein, designated sialin, has strong sequence similarity to type I Na+-dependent phosphate transporters, but biochemical evidence for sialic acid transport by the protein has not yet been provided. Nonetheless, a total of 16 disease-causing mutations have now been identified in the sialin genes, and different mutations produce different phenotypes [3]. The most common mutation, R39C, causes Salla disease with findings that include developmental delay, ataxia and marked cognitive impairment with intelligence quotients of 20–30, but a normal life expectancy. The ISSD mutations are associated with dysmorphic features, more severe neurologic symptoms and death by 2 years of age. Interestingly, the Salla disease mutant and one ISSD missense mutant localize predominantly to the Golgi apparatus rather than to lysosomes. Mistargeting of the protein may thus contribute to the disease phenotype [4].
The vesicular glutamate transporters (SLC17A6–8)
The first two vesicular glutamate transporters to be identified (VGLUT1 and VGLUT2) were initially characterized as phosphate transporters. VGLUT1 was identified in a screen for cDNAs upregulated in cerebellar granule cells in response to subtoxic levels of the glutamate receptor agonist NMDA [31]. The predicted multitransmembrane domain protein was designated BNPI for brain specific Na+-dependent phosphate transporter. VGLUT2 was isolated in a screen for cDNAs upregulated during differentiation of rat pancreatic AR42J cells to a neuroendocrine phenotype in response to a growth factor, and was designated DNPI for differentiation associated Na+-dependent phosphate transporter [1]. This protein has 82% identity to VGLUT1. In both cases, the structural similarity to NaPi-1 (~32% identity) initially led to a focus on phosphate transport. Heterologous expression of these proteins in Xenopus oocytes indeed increases Na+-dependent phosphate uptake very similar to NaPi-1. However, both localize exclusively to glutamatergic neurons of the brain and specifically to synaptic vesicles, not the plasma membrane [8, 17, 44].
Genetic studies in Caenorhabditis elegans implicated the BNPI orthologue eat-4 in glutamate release [26]. Eat-4 mutants are defective in a number of behaviors involving glutamate, and EAT-4 localizes presynaptically, indicating a role in glutamate release. Together with sequence similarity to proteins that transport organic anions, and its localization to the synaptic vesicles of glutamatergic neurons, this suggested that BNPI might have a role entirely different from Na+-dependent phosphate transport at the plasma membrane—transport of the organic anion glutamate into synaptic vesicles. Indeed, both BNPI and DNPI have been shown to mediate the transport of glutamate into secretory vesicles with kinetic and pharmacological characteristics indistinguishable from those of glutamate transport by native synaptic vesicles and were hence renamed VGLUT1 and 2 [6, 9, 17, 24, 40, 41, 44]. A third protein with 78% identity to VGLUT1 and 74% identity to VGLUT2 has more recently been shown to mediate vesicular glutamate transport and been designated VGLUT3 [18, 19, 37, 42].
The three VGLUT isoforms exhibit saturable glutamate transport with a K m~1 mM that is driven primarily by the electrical component (Δψ) of the proton electrochemical gradient across the vesicle membrane: valinomycin, a K+ ionophore that dissipates Δψ reduces transport to a greater extent than nigericin, a H+-K+-exchanging ionophore that dissipates the pH gradient [6, 9, 17, 18, 19, 24, 37, 40, 41, 42, 44]. Residual transport in the absence of Δψ (but presence of ΔpH) suggests, nonetheless, that transport is coupled to proton exchange. However, the stoichiometry of coupling both in native synaptic vesicles and in heterologous systems with the cloned transporters remains undetermined and it has been suggested that coupling may differ for the different isoforms [6, 19, 42]. Transport by native membranes and in heterologous expression systems is stimulated by low concentrations and inhibited by high concentrations of chloride. In contrast to plasma membrane excitatory amino acid transporters that recognize aspartate as well as glutamate, the VGLUTs do not recognize aspartate. They also prefer l-glutamate over d-glutamate. Pharmacological studies indicate that the two structurally related dyes Evans blue and Chicago skye blue, and the fluorescein derivative Rose Bengal inhibit glutamate uptake by synaptic vesicles [32, 35]. Other inhibitors include several glutamate analogues and substituted quinoline-2,4-dicarboxylic acids [13, 48]. In addition, the relatively non-selective anion channel blocker 4,4'-diisothiocyanatostilbene-2,2'-disulfonic acid (DIDS) inhibits synaptic vesicle glutamate uptake [20, 35]. Interestingly, an endogenous calpain-derived fragment of α-fodrin also inhibits vesicular glutamate transport [33]. The extremely high affinity (IC50=26 nM) suggests that this protein may have a biological role, but IPF also inhibits GABA transport into synaptic vesicles, suggesting that it may not interact directly with the VGLUTs. DIDS, Evans blue and closely related compounds also inhibit the VGLUTs expressed in heterologous systems [6, 9, 17, 18, 19, 24, 37, 44], but characterization of the other inhibitors have not been reported.
Like the type 1 phosphate transporters, VLGUT1 has been shown to mediate a chloride conductance [9]. Unlike the conductance associated with NaPi-1, which was detected by electrophysiological measurements, the VGLUT1 chloride conductance was inferred from the rate of vesicle acidification in the presence of chloride. Since the activity of the vesicular proton ATPase is inhibited by the development of an electrical gradient, the influx of a counter-anion facilitates acidification. The expression VGLUT1 increased the rate of ATP dependent acidification in the presence of chloride, suggesting that the protein exhibits an anion conductance similar to NaPi1. Analogous to the NaPi-1 associated conductance, the VGLUT1 conductance is inhibited by substrate, in this case glutamate. A similar conductance has not yet been demonstrated for the other isoforms. The physiological significance of the chloride conductance in VGLUT1 remains unclear, but it would tend to diminish ΔΨ, the driving force for glutamate transport and might thus serve to regulate synaptic release of transmitter. The conductance may also contribute to the previously observed biphasic dependence of vesicular glutamate uptake on chloride: 2–10 mM is optimal, with substantially less transport at 0 or 140 mM [20]. However, the chloride dependence of vesicular glutamate transport does not reflect changes in the driving force—chloride modulates uptake independent of changes in ΔpH and Δψ [49] supporting an allosteric mechanism for this form of regulation.
The distributions of the different VGLUT isoforms are striking. VGLUT1 and VGLUT2 exhibit an almost completely complementary distribution by essentially all neurons that have been well-characterized as glutamatergic [17, 24, 25, 44]. By in situ hybridization, all cerebral cortical layers label strongly for VGLUT1, whereas only layer IV of frontal and parietal cortex and layers IV and VI of temporal cortex label for VGLUT2. In the hippocampus, dentate gyrus granule cells contain only VGLUT1 mRNA, whereas pyramidal neurons from CA1 through CA3 also express abundant VGLUT1, and low levels of VGLUT2. In the amygdala, the medial and central nuclei contain abundant mRNA for VGLUT2, whereas the lateral and basolateral nuclei express VGLUT1. The thalamus expresses much more VGLUT2 than VGLUT1, but certain thalamic nuclei express VGLUT1.VGLUT2 is the predominant isoform expressed by brainstem and deep cerebellar nuclei, whereas VGLUT1 is expressed in the cerebellar cortex.
Unlike VGLUT1 and VGLUT2, VGLUT3 is expressed in neurons not classically considered glutamatergic. Immunohistochemical and in situ studies indicate that inhibitory cells in layer II of the parietal cortex express VGLUT3, as do scattered GAD (glutamic acid decarboxylase) containing interneurons in stratum radiatum of CA1-CA3 of the hippocampus [18, 19, 37] and immuno-electron microscopy studies indicate that VGLUT3 localizes to asymmetric, and symmetric synapses [18, 19]. In addition, dopaminergic cells in the substantia nigra pars compacta and ventral tegmental area contain VGLUT3 mRNA, as do serotonergic cells in the dorsal raphe, and neurons in the pontine raphe nuclei and olivary nuclei [18, 19, 37]. Double label immunofluorescence studies suggest colocalization of VGLUT3 with VMAT2 [37]. Cholinergic interneurons in the dorsal striatum also express VGLUT3 [18, 37] and VGLUT3 has been colocalized with VAChT by immunofluorescence [37] and immuno-electron microscopy [19]. In the cerebellum, the granule cell layer labels most strongly for VGLUT3, but the molecular layer also contains scattered hybridizing cells [18]. Interestingly, immuno-electron microscopy also demonstrates that VGLUT3 occurs in astrocytes, on both processes surrounding synapses and endfeet abutting capillaries [18].
Outside the central nervous system, VGLUT2 has been identified in the intrinsic and extrinsic primary afferent neurons of the gut, α and β cells in the pancreatic islets, and pinealocytes [22, 23], whereas VGLUT1 has only been identified in β cells [23]. The expression of VGLUT1 and VGLUT2 on secretory vesicles [23] and the regulation of transport activity and VGLUT2 mRNA by glucose levels in cultured pancreatic cells [7] is consistent with a role for glutamate in intercellular signaling in the pancreas and glucose regulation [39]. VGLUT3 is expressed in the liver and to a lesser extent the kidney by RTPCR [18, 19, 37, 42], but expression of the protein has not been confirmed. The role of vesicular glutamate transport in these tissues remains to be determined. However, the expression of plasma membrane glutamate (EAAC1 (SLC1 family)) and glutamine (SN1, SA1/SAT2, SA2/SAT1 (SLC38 family)) transporters in liver and kidney suggests the presence of intercellular glutamine-glutamate cycles in tissues other than the brain that rely on the vesicular storage and hence possibly the exocytotic release of glutamate [14, 34, 46].
Although all VGLUT isoforms localize to synaptic vesicles, each appears to have a different distribution among other cell membranes. Subcellular fractionation of rat brain membranes demonstrates that VGLUT1 segregates most closely with synaptic vesicle markers and to a lesser extent the plasma membrane [8]. VGLUT2 cofractionates with crude membranes lighter than synaptosomes in addition to synaptic vesicle and plasma membrane markers [17]. Immuno-electron microscopy has further demonstrated that VGLUT3 has a very different subcellular localization. In addition to synaptic vesicles, VGLUT3 occurs in vesicular structures of astrocytes and neuronal dendrites, whereas VGLUT1 and 2 appear only at nerve terminals [18]. The novel tissue distribution and subcellular localization of these different isoforms suggests novel modes of signaling by glutamate. While coexpression of different isoforms has not been demonstrated, the differences in subcellular localization also suggest that although biochemical functions of the isoforms are similar, coexpression would suggest multiple physiological roles for vesicular glutamate storage and release, rather than a redundancy of function.
SLC17 transporters as pharmaceutical targets.
No drugs targeting SLC17 family members are currently used to treat human diseases. However, given the varied functions and substrate specificity of the SCL17 transporters (Fig. 2), the potential for pharmacological targeting is great and the implications diverse. The vesicular glutamate transporters, for example might be targets for the treatment of neurodegenerative diseases such as amyotrophic lateral sclerosis, Alzheimer's disease and Huntington's disease in which chronic excitotoxicity has been implicated. Reducing synaptic glutamate storage through inhibition of the VGLUTs would in principle lower the synaptic concentration of glutamate, reduce binding to postsynaptic glutamate receptors and thus mitigate excitotoxicity. Several compounds have been shown to block synaptic vesicle glutamate uptake, but efficacy may be limited by delivery to the central nervous system and synaptic vesicles, as only Rose Bengal appears to be membrane permeant [32]. Since impairment of sialin function is associated with a lysosomal storage disorder, inhibitors of sialin are unlikely to be of benefit as pharmaceutical agents. However, data suggesting that sialin may be mislocalized in Salla disease and in one mutation associated with ISSD, indicates that if the mutated proteins are still functional, drugs that increase their trafficking to the lysosome may be of interest. With a still poorly defined physiological role, pharmacological targeting of the type I phosphate transporters and related proteins is of unclear significance. The recognition of penicillin by NaPi1 does, however, suggest that pharmacological modulation of these proteins may have implications for the metabolism and secretion of drugs with other actions.

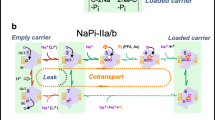
A The human type I phosphate transporter NPT1 is associated with three different transport activities. NPT1 was first described as a Na+-dependent phosphate (PO4 2−) transporter. Although this activity does not correlate with expression level of NPT1, organic anion (OA −) transport and an inorganic anion (Cl−) conductance do correlate with its expression. Saturating concentrations of organic anion substrates inhibit the chloride conductance, whereas high concentrations of Cl− inhibit organic anion transport, suggesting that NPT1 may not simultaneously mediate both activities. The localization of NPT1 to renal proximal tubular brush border membranes suggests that it may secrete organic anions for urinary excretion. B Sialin mediates the efflux of sialic from the lysosome. The degradation of oligosaccharides and glycoproteins leads to the generation of free sialic acid (SA −) in lysosomes. Transport of sialic acid out of the lysosome is coupled to proton cotransport and mediated by sialin. Efflux of sialic acid thus depends on the proton electrochemical gradient generated by the lysosomal H+-ATPase. The free sialic acid released into the cytoplasm is then linked to CMP, transported into the Golgi via a CMP-SA−-CMP exchange transporter and incorporated into newly synthesized glycoproteins. C The VGLUTs concentrate glutamate into neurosecretory vesicles for regulated release. Although glutamate (glu −) is present in all cells, its release through exocytosis requires active transport into secretory vesicles. This activity is mediated by the VGLUTs. Like sialin, VGLUT activity is coupled to the proton electrochemical gradient generated by a vaculolar type H+-ATPase. It is presumed that the VGLUTs function as glutamate-H+ antiporters. VGLUT is depicted here on synaptic vesicles, but the expression of the VGLUT3 on astrocytes as well as in liver and kidney suggests that exocytotic release of glutamate is not limited to neurons or to the nervous system. Release mediated by the VGLUTs may thus participate in metabolism as well as signaling
References
Aihara Y, Mashima H, Onda H, Hisano S, Kasuya H, Hori T, Yamada S, Tomura H, Yamada Y, Inoue I, Kojima I, Takeda J (2000) Molecular cloning of a novel brain-type Na(+)-dependent inorganic phosphate cotransporter. J Neurochem 74:2622–5
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Aula N, Salomaki P, Timonen R, Verheijen F, Mancini G, Mansson JE, Aula P, Peltonen L (2000) The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am J Hum Genet 67:832–840
Aula N, Jalanko A, Aula P, Peltonen L (2002) Unraveling the molecular pathogenesis of free sialic acid storage disorders: altered targeting of mutant sialin. Mol Genet Metab 77:99
Aula P, Gahl WA (2001) Disorders of free sialic acid storage. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 5109–5120
Bai L, Xu H, Collins JF, Ghishan FK (2001) Molecular and functional analysis of a novel neuronal vesicular glutamate transporter. J Biol Chem 276:36764–36769
Bai L, Zhang X, Ghishan FK (2003) Characterization of vesicular glutamate transporter in pancretic alpha and beta cells and its regulation by glucose. Am J Physiol 284:G808–G814
Bellocchio EE, Hu H, Pohorille A, Chan J, Pickel VM, Edwards RH (1998) The localization of the brain-specific inorganic phosphate transporter suggests a specific presynaptic role in glutamatergic transmission. J Neurosci 18:8648–8659
Bellocchio EE, Reimer RJ, Fremeau RT, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289:957–960
Biber J, Custer M, Werner A, Kaissling B, Murer H (1993) Localization of Na/Pi cotransporter, in rabbit kidney proximal tubules. II. Localization by immunohistochemistry. Pflugers Arch 424:210–215
Bröer S, Schuster A, Wagner CA, Bröer A, Forster I, Biber J, Murer H, Werner A, Lang F, Busch AE (1998) Chloride conductance and Pi transport are separate functions induced by the expression of NaPi-1 in Xenopus oocytes. J Membr Biol 164:71–77
Busch AE, Schuster A, Waldegger S, Wagner CA, Zempel G, Broer S, Biber J, Murer H, Lang F (1996) Expression of a renal type I sodium/phosphate transporter (NaPi-1) induces a conductance in Xenopus oocytes permeable for organic and inorganic anions. Proc Natl Acad Sci USA 93:5347–5351
Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, Thompson CM (2002) Synthesis and in vitro pharmacology of substituted quinoline-2,4-dicarboxylic acids as inhibitors of vesicular glutamate transport. J Med Chem 45:2260–2276
Chaudhry FA, Reimer RJ, Krizaj D, Barber D, Storm-Mathisen J, Copenhagen DR, Edwards RH (1999) Molecular analysis of system N suggests novel physiological roles in nitrogen metabolism and synaptic transmission. Cell 99:769–780
Cheret C, Doyen A, Yaniv M, Pontoglio M (2002) Hepatocyte nuclear factor 1 alpha controls renal expression of the Npt1-Npt4 anionic transporter locus. J Mol Biol 322:929–941
Custer M, Meier F, Schlatter E, Greger R, Garcia-Perez A, Biber J, Murer H (1993) Localization of NaPi-1, a Na-Pi cotransporter, in rabbit kidney proximal tubules. I. mRNA localization by reverse transcription/polymerase chain reaction. Pflugers Arch 424:203–209
Fremeau RT, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31:247–60
Fremeau RT, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci USA 99:14488–14493
Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S (2002) A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci 22:5442–5451
Hartinger J, Jahn R (1993) An anion binding site that regulates the glutamate transporter of synaptic vesicles. J Biol Chem 268:23122–23127
Havelaar AC, Mancini GM, Beerens CE, Souren RM, Verheijen FW (1998) Purification of the lysosomal sialic acid transporter. Functional characteristics of a monocarboxylate transporter. J Biol Chem 273:34568–34574
Hayashi M, Otsuka M, Morimoto R, Hirota S, Yatsushiro S, Takeda J, Yamamoto A, Moriyama Y (2001) Differentiation-associated Na+-dependent inorganic phosphate cotransporter (DNPI) is a vesicular glutamate transporter in endocrine glutamatergic systems. J Biol Chem 276:43400-43406
Hayashi M, Yamada H, Uehara S, Morimoto R, Muroyama A, Yatsushiro S, Takeda J, Yamamoto A, Moriyama Y (2003) Secretory granule-mediated co-secretion ofl-glutamate and glucagon triggers glutamatergic signal transmission in islets of langerhans. J Biol Chem (in press)
Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S (2001) The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci 21:RC181
Kaneko T, Fujiyama F (2002) Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci Res 42:243–50
Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L (1999) EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci 19:159–67
Mancini GM, Jonge HR de, Galjaard H, Verheijen FW (1989) Characterization of a proton-driven carrier for sialic acid in the lysosomal membrane. Evidence for a group-specific transport system for acidic monosaccharides. J Biol Chem 264:15247–15254
Mancini GM, Beerens CE, Galjaard H, Verheijen FW (1992) Functional reconstitution of the lysosomal sialic acid carrier into proteoliposomes. Proc Natl Acad Sci USA 89:6609–6613
Miyamoto K, Tatsumi S, Sonoda T, Yamamoto H, Minami H, Taketani Y, Takeda E (1995) Cloning and functional expression of a Na(+)-dependent phosphate co-transporter from human kidney: cDNA cloning and functional expression. Biochem J 305:81–85
Murer H, Hernando N, Forster I, Biber J (2000) Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev 80:1373–1409
Ni B, Rosteck PR, Nadi NS, Paul SM (1994) Cloning and expression of a cDNA encoding a brain-specific Na+-dependent inorganic phosphate cotransporter. Proc Natl Acad Sci USA 91:5607–5611
Ogita K, Hirata K, Bole DG, Yoshida S, Tamura Y, Leckenby AM, Ueda T (2001) Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal. J Neurochem 77:34–42
Ozkan ED, Lee FS, Ueda T (1997) A protein factor that inhibits ATP-dependent glutamate and γ-aminobutyric acid accumulation into synaptic vesicles: purification and initial characterization. Proc Natl Acad Sci USA 94:4137–4142
Reimer RJ, Chaudhry FA, Gray AT, Edwards RH (2000) Amino acid transport System A resembles System N in sequence but differs in mechanism. Proc Natl Acad Sci USA 97:7715–7720
Roseth S, Fykse EM, Fonnum F (1995) Uptake of l-glutamate into rat brain synaptic vesicles: effect of inhibitors that bind specifically to the glutamate transporter. J Neurochem 65:96–103
Ruddy DA, Kronmal GS, Lee VK, Mintier GA, Quintana L, Domingo R Jr, Meyer NC, Irrinki A, McClelland EE, Fullan A, Mapa FA, Moore T, Thomas W, Loeb DB, Harmon C, Tsuchihashi Z, Wolff RK, Schatzman RC, Feder JN (1997) A 1.1-Mb transcript map of the hereditary hemochromatosis locus. Genome Res 7:441–456
Schafer MK, Varoqui H, Defamie N, Weihe E, Erickson JD (2002) Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem 277:50734–50748
Shibui A, Tsunoda T, Seki N, Suzuki Y, Sugane K, Sugano S (1999) Isolation and chromosomal mapping of a novel human gene showing homology to Na+/PO4 cotransporter. J Hum Genet 44:190–192
Skerry TM, Genever PG (2001) Glutamate signalling in non-neuronal tissues. Trends Pharmacol Sci 22:174–181
Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407:189–194
Takamori S, Rhee JS, Rosenmund C, Jahn R (2001) Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2). J Neurosci 21:RC182
Takamori S, Malherbe P, Broger C, Jahn R (2002) Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep 3:798–803
Uchino H, Tamai I, Yamashita K, Minemoto Y, Sai Y, Yabuuchi H, Miyamoto K, Takeda E, Tsuji A (2000) p-Aminohippuric acid transport at renal apical membrane mediated by human inorganic phosphate transporter NPT1. Biochem Biophys Res Commun 270:254–259
Varoqui H, Schafer MK, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na+/Pi transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22:142–55
Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, Spek PJ van der, Mancini GM (1999) A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet 23:462–465
Welbourne TC, Matthews JC (1999) Glutamate transport and renal function. Am J Physiol 277:F501–505
Werner A, Moore ML, Mantei N, Biber J, Semenza G, Murer H (1991) Cloning and expression of cDNA for a Na/Pi cotransport system of kidney cortex. Proc Natl Acad Sci USA 88:9608–9612
Winter HC, Ueda T (1993) Glutamate uptake system in the presynaptic vesicle: glutamic acid analogs as inhibitors and alternate substrates. Neurochem Res 18:79–85
Wolosker H, Souza DO de, Meis L de (1996) Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem 271:11726–11731
Yabuuchi H, Tamai I, Morita K, Kouda T, Miyamoto K, Takeda E, Tsuji A (1998) Hepatic sinusoidal membrane transport of anionic drugs mediated by anion transporter Npt1. J Pharmacol Exp Ther 286:1391–1396
Acknowledgements
The authors are supported in part by funding from the NINDS (R.H.E. and R.J.R.), NIDA (R.H.E.) and NIMH (R.H.E.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reimer, R.J., Edwards, R.H. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch - Eur J Physiol 447, 629–635 (2004). https://doi.org/10.1007/s00424-003-1087-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-003-1087-y