
Abstract
Alzheimer’s disease (AD) is the most common type of neurodegenerative dementia that affects the elderly population. Nerve growth factor (NGF) contributes to the survival, regeneration and death of neurons during aging and in neurodegenerative diseases. Recently, research has shown that NGF is related to the pathology, mechanisms and symptoms of AD. Therefore, there is a need to summarize the new advancements in NGF research and its potential therapeutic implications in AD. In this review, we will focus on NGF distribution, production, and function; the interaction of Aβ and NGF; and the effect of different therapy methods on AD. In summary, we hope to describe the experimental and clinical data demonstrating the important roles of NGF for AD treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Overview
Clinically, AD presents as short term memory loss and is considered to be a progressive behavioral disorder. The neuropathological characteristics of AD are senile plaque deposition (mainly consisting of aggregated β amyloid), synaptic dysfunction, intracellular neurofibrillary tangles composed of hyperphosphorylated tau proteins, diffuse loss of neurons and dystrophic neurites [1, 2]. This ailment poses a severe threat to aging people across the world. Although great efforts have been made to slow the incidence of AD, little progress has been achieved. Over the last decade, the neurotrophin family has been shown to play important roles in neuronal development, plasticity and neurodegenerative disease therapy, such as AD. The neurotrophin family includes NGF and other recently identified members, namely, brain derived neurotrophic factor (BDNF), NT-3 (neurotrophin 3), NT-4/5 and the recently discovered NT-6 (neurotrophin 6) [3]. NGF is a secreted growth factor and was discovered in the 1950s [4, 5]. NGF is derived from the precursor Pro NGF, which is the major molecule found in the human brain [6]. ProNGF is cleaved by the matrix metalloproteinase-7 (MMP-7) and plasmin. Plasmin cleavage of ProNGF produces the mature form NGF, whereas MMP-7 cleavage results in a 17-kD intermediate [6–8]. Plasminogen is converted to plasmin via the activation of tissue plasminogen activator (tPA), a mechanism that is regulated by the protein neuroserpin [9]. The disassembly and consequent inactivation of the mature NGF is mediated via the activation of the precursor of matrix metalloproteinase 9 (MMP-9) by plasmin, and its activity is regulated by the tissue inhibitor of matrix metalloproteinase 1 (TIMP-1) [9]. Like other neurotrophins, NGF is composed of two domains, the prodomain and the NGF domain. NGF binds to the tyrosine kinase A receptor and the p75 pan neurotrophin receptor (p75NTR) [10]. ProNGF binds to its, receptor p75NTR, and sortilin (approximately 95 kDa), which results in the apoptosis of neurons [11]. In addition, the chronic depletion of NGF has been proposed to promote AD-type neurodegenerative pathologies, which include neuronal loss, cholinergic deficits, tau hyperphosphorylation and large amounts of Aβ plaques [12]. Accordingly, in this review, we attempt to describe the effect of NGF on AD and discuss current and future opportunities for NGF-based drug therapy.
NGF: Distribution, Production and Function
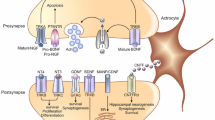
Nerve growth factor (NGF) is ~13 kD peptide. It was the first well-characterized neurotrophic factor in the neurotrophic family and act to promote nerve growth factors, which led to the formation of the neurotrophic factor hypothesis [13]. NGF has very important roles in the survival, growth and maintenance of various neurons in the mammalian nervous system [14]. For instance, NGF has an effect on the development of sympathetic and neural crest-derived sensory neurons in the peripheral nervous system and cholinergic neurons in the CNS. It has been demonstrated that NGF expression varies during the development of different rat CNS regions, but is significantly expressed in the adult hippocampus [15]. In addition to neurons, glial cells, such as microglial cells, olfactory ensheathing cells, astrocytes and oligodendrocytes, have all been shown to express NGF [16–21]. In the peripheral structures, NGF is stored in blood platelets, muscle cells, Schwann cells, and macrophages [22–25]. Additionally, NGF mediates its biological activity through binding at the plasma membrane to two distinct NGF high-affinity TrkA receptors (tyrosine kinase receptors) and the low-affinity p75NTR (a member of the tumor necrosis factor receptor superfamily) [26]. Trk-mediated NGF signaling activates intracellular signaling cascades involving pathways that are dependent on extracellular signal-regulated kinase (ERK) and phosphatidylinositol 3-kinase (PI3K), which act to prevent nuclear and mitochondrial cell death programs, resulting in neuronal survival and differentiation. [27]. Recently, Yu et al. [28] found that neurotrophic factors regulate the efficiency of the Trk-mediated NGF signaling pathway through ligand-independent interactions of Trk and GM1 (Fig. 1). It is well known that the fundamental neuromorphological changes of AD are the loss of cholinergic neurons and the widespread degeneration of cholinergic functions in the brain, which contribute to cognitive decline and dementia [29]. Ruberti et al. [30] demonstrated that knocking out NGF in adult mice by expressing transgenic anti-NGF antibodies resulted in basal forebrain and hippocampal cholinergic neuron reductions in adult mice (55 and 62 % reductions, respectively). In addition, adult mice showed deficits in spatial learning behavioral tasks. Increasing evidence supports that basal forebrain cholinergic metabolic dysfunction of NGF is involved in AD and that defective NGF processing leads to an increased accumulation of Aβ [10]. In sum, NGF may be a potential candidate in the pathogenesis of AD and a therapy that may alleviate Aβ deposition and improve clinical cognitive and behavioral performance.

Signaling pathtway of NGF, involving GM1 receptor, besides the “traditional” receptors p75NTR and TrkA. NGF complexes with TrkA to activate cell survival mechanisms in the aged human healthy brain,which are dependent on ERK and PI3K signaling pathtway. Additionally, Aβ binding to p75NTR could result into neuron death
Association of NGF with Aβ in AD
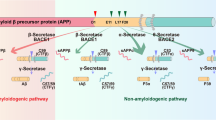
The major components of AD senile plaques is a 4-kDa peptide of 39–43 amino acids named amyloid (Aβ), which originates from a larger precursor protein named amyloid precursor protein (APP). Recently studies have demonstrated that NGF deprivation results in AD-like pathologies, such as Aβ accumulation/deposition, tau hyperphosphorylation, and synaptic dysfunction in mice, and that NGF treatment can ameliorate the changes of Aβ pathologies and inhibit memory impairment in AD animal models [31]. Aβ is derived from APP after cleavage by β-and γ-secretases [12]. Aβ is known to bind to p75NTR and may act as a neurotrophic factor that mimics the activity of NGF, depending on its concentration [32, 33]. Wang et al. [34] showed that the expression of p75NTR in the brain of APPswe transgenic mice was increased, illustrating that Aβ is a crucial factor in the regulation of p75NTR over-expression in AD and could increase Aβ production and attenuate its aggregation (Fig. 1). Additionally, APP serves to mediate the internalization of NGF receptors via direct interactions and therefore regulates the internalization of NGF and NGF-induced signaling transduction for neuronal survival and differentiation [35]. During early AD pathological events, which begin with intracellular Aβ accumulation and end with plaque deposition, studies have reported that Aβ-induced inflammation leads to nerve growth factor deregulation in transgenic models of Alzheimer’s disease-like amyloid pathology [36, 37]. In vitro, the NGF-induced differentiation of PC12 cells into neuron-like cells caused a marked increase in both gangliosides and cholesterol and thereby greatly potentiated the accumulation and cytotoxicity of Aβ (1–42). NGF-differentiated cells exposed to Aβ (1–42) have been shown to display degenerated neurites; however, a cholesterol synthesis inhibitor prevented the formation of Aβ (1–42) in PC12 cells [38]. More interestingly, NGF signaling controls the amyloidogenic route and Aβ production in hippocampal neurons, and deprivation of NGF leads to an increase in the production of Aβ aggregates and progressive release of Aβ into culture medium, which could cause cell death [39].
Effect of Different Therapeutic Modalities on the NGF Metabolic Levels in AD
Advancing Cell Therapy for Alzheimer’s Disease
Another disadvantage to the use of NGF as a treatment of AD pathology is its inability to penetrate the blood–brain barrier (BBB); thus, NGF brain bioavailability is challenging. Moreover, intra-ventricular administration of NGF leads to adverse effects, including aberrant sympathetic and sensory neurite sprouting [40]. However, when NGF is administered intranasally to APP/PS1 double-transgenic mice, the results show that NGF enhances the APP non-amyloidogenic cleavage pathway and reduces the Aβ generation in transgenic mouse brains [41]. Neural stem cells (NSCs) are defined as CNS progenitor cells that have the capacity for self-renewal and a multipotent potential to become neurons or glial cells. Because of their ability to self-renew and differentiate into neuronal and glial phenotypes, NSCs are a promising candidate for cell-based therapy for CNS injury [42]. A genetically modified cell line, HB1.F3, transfected with cholinergic acetyltransferase (ChAT), HB1.F3.ChAT cells, have shown favorable safety and efficacy profiles in AD models [43]. The upregulation of ChAT would enhance the potency of NGF in the brain. The HB1 parent cell could differentiate into neurons and glial cells. Marei et al. [40] showed that human olfactory bulb NSCs expressing hNGF could restore cognitive deficits in an AD rat model; these results are relevant to neuronal and glial cell replacement and the trophic influence exerted by the secreted NGF. Because the differentiation of neural stem cells (NSCs) into cholinergic neurons in vivo would be an important advancement for AD therapy, Gu et al. [29] demonstrated that the transplantation of NSC-derived cholinergic neuron-like cells improves cognitive function in APP/PS1 transgenic mice and that these cholinergic neuron-like cells were induced by NGF. Additionally, Aβ inhibits proliferation, promotes apoptosis of human cortical stem cells in culture and impairs neurogenesis, which are altered in the brains of AD patients. However, recent data also suggest that Aβ does not impair the neurogenic rate in NSC progeny but increases the total number of neurons in vitro [44]. These findings show that Aβ may be involved in neurogenesis regulation and that the therapy mechanism of NSC grafts for the treatment of AD need further exploration.
Acupuncture and Herbal Supplements
Acupuncture is a potential strategy for the treatment of AD with few side effects and is easy to administer. Although acupuncture is a promising intervention for the treatment of AD [45, 46], the effect is still controversial [47]. Recent data reveals that electroacupuncture treatment protects cholinergic neurons in the hippocampus of an AD rat model by increasing the NGF levels [48]. In addition, cholinergic neuron degeneration is found in the basal forebrain in postmortem AD brains [49, 50]. The Bushen–Yizhi formula (BSYZ) is a traditional Chinese medicine compound recipe that has been shown to ameliorate behavior deficits in a rat model of dementia induced by d-galactose and ibotenic acid (IBO) by up-regulating the expression of ChAT and NGF in the hippocampus and cortex [51]. Because Chinese herbal formulas and acupuncture usually target multiple targets, pathways, and systems, elucidation of the detailed mechanisms needs further study.
Natural products from traditional medicine are also referred to as herbal extracts. In recent decades, a number of agents have been separated from herbs that are used in traditional medicine, and their efficacies against AD have been assayed. For instance, huperzine A (HupA), a novel alkaloid isolated from the Chinese herb Huperzia serrata, is a potent, highly specific and reversible inhibitor of acetylcholinesterase (AChE). HupA has been found to improve cognitive deficits and upregulate NGF expression in AD models [52]. Several experimental studies have suggested that 6-shogaol, a bioactive component of ginger, may play an important role in ameliorating scopolamine-induced memory impairment in animal models of dementia by elevating NGF levels, PSD-95 and synaptophysin expression in hippocampal tissues [53]. Soy isoflavones have estrogen-like effects and play a role in the association between NGF gene polymorphisms and executive dysfunction in Japanese patients with early stage AD and amnestic mild cognitive impairment. Postmenopausal women who participated in estrogen replacement treatment had a significantly lower risk of developing AD than women who did not [54]. Another randomized, double-blind, cross-over and placebo controlled trial suggested that soy isoflavones were safe and had positive effects on cognitive function, especially verbal memory, in postmenopausal women. Hence, the authors speculated that the underlying mechanisms of the favorable effects of soy isoflavones on cognitive function may be related to enhanced NGF levels [54].
Cholinesterase Inhibitors (ChEI) and Anti-depressants
In AD, many neurons disappear in the superficial cortex, and the loss of cholinergic transmission in the temporal lobe and other cortical brain regions is responsible for the degree of memory impairment [55]. ChEI induces a modest improvement in memory and/or delays the rate of memory decline [56, 57]. To date, four ChEI, tacrine, donepezil, galanthamine and rivastigmine, have been approved by the US Food and Drug Administration for the treatment of AD, and several new ChEI are being studied [52]. Simona Capsoni et al. [58] reported that the intraperitoneal and oral administration of ganstigmine and donepezil can reverse the cholinergic and behavioral deficits in AD11 mice, which express anti-nerve growth factor (anti-NGF) antibodies and are a comprehensive model for AD, but not amyloid and phospho-tau accumulation, revealing different mechanisms leading to neurodegeneration in AD11 mice.
The simultaneous incidence of AD and depression is approximately 30–50 % [59]. Depressed adults have an increased risk of developing AD. Anti-depressant treatments have been hot topics for the administration of AD [60]. NGF specifically has important effects on hippocampal neurons that are involved in the pathogenesis and clinical features of AD. Therefore, the administration of antidepressants could improve the symptoms of AD patients, and this improvement may be associated with the elevation of NGF [59].
NGF Gene Polymorphism
Several previous studies have reported that variations in NGF single-nucleotide polymorphisms (SNPs) are significantly correlated to the progression of multiple sclerosis symptoms in a gender-dependent manner [61, 62]. One common SNP is rs6330, located in the NGF locus in coding exon 3, in which the C/T (104C → T) allele changes a nonsynonymous amino acid (Ala35Val) at position 35 and may be related to early stage AD and amnestic mild cognitive deficits in Japanese patients [62, 63]. Another study has revealed that the minor allele of rs6330 causes genetic variations in Pro NGF and may influence the occurrence of sporadic, late-onset AD [64]. However, other studies have suggested that β-NGF is not the gene responsible for late-onset familial Alzheimer’s disease (FAD) in the families analyzed [65].
Finally, the role of NGF gene polymorphisms on patients with AD is controversial, which suggests that the association between them is very complicated. In the future, we should consider more factors in clinical trials, such as age, gender, living conditions and so on, to address the questions of gene susceptibility related to the onset of AD.
Diet, Environment and Exercise
The pathogenic mechanism of AD is not clear, but environmental factors, such as advancing age, family history, presence of chronic diseases such as cardiovascular disease (CVD) and diabetes, poor diet and lifestyle, are believed to contribute to the development of AD [66]. Epidemiological studies have revealed an association between nutritional deficiency and AD risk. Recently, it was reported that vitamin D3-enriched diets in AD transgenic mice could decrease Aβ peptide levels and increase NGF levels [67]. These observations suggest that a vitamin D3-enriched diet may benefit AD patients. Caloric restriction has been demonstrated to activate multiple neuroprotective benefits and reduce Aβ pathology in various AD mouse models [68, 69]. Furthermore, 2-deoxy-d-glucose treatment has been shown to reduce the β-amyloid burden and significantly increase the expression of neurotrophic growth factors, such as BDNF and NGF [70].
Additionally, physical exercise has been shown to contribute to the recovery of patients with AD. The transition from a sedentary life-style to a simple active life-style has been shown to inhibit neuronal cell death in both animal models and patients of AD [71], indicating that physical activity or an active life might protect against AD. Treadmill exercise has also been shown to represses neuronal cell death and up-regulate the expression of NGF in an aged transgenic mouse model of AD [71]. Similarly, AD11 mice develop age-dependent neurodegeneration, which includes all features of human AD. Environmental enrichment (EE) has been shown to restrain the progression of neurodegeneration in a mouse model of AD, which may be related to elevated NGF levels [72].
Mimetics of NGF and TrkA
Much evidence has suggested that the NGF-TrkA signaling pathway is involved in the pathogenesis and treatment of AD. Agents that can strengthen the pathway signaling may be therapeutic for AD. Recently, data has shown that D 7,8-dihydroxyflavone (7,8-DHF) is a potent TrkB agonist and has been validated to have therapeutic efficacy for AD [73]. Although agonists of the TrkA neurotrophin receptor and mimetics of NGF have been identified [74–76], there are no reports on the effect of these agents on AD.
NGF-Based Gene Therapy for AD
Administration of NGF directly into the CNS is a promising route to intervene in the disruption of NGF maturation or degradation in clinical trials. Work from Tuszynski et al. [77] showed that 22 months following the implantation of autologous fibroblasts genetically modified to express human NGF into the forebrain of six subjects, an improvement in the rate of cognitive decline was observed. This trial suggests that NGF might reduce the cholinergic neuron loss in AD. Another study on gene therapy for AD reported on the delivery of NGF via a NGF-producing, genetically engineered encapsulated human cell line. At 12 months, the implants were successfully removed and persistent NGF secretion was assayed in half of the patients [78]. Additionally, adeno-associated virus (AAV) is the most widely used vector for gene therapy in clinical trials focused on AD. One study reported on the use of AAV2 as the vector to deliver the NGF gene into the bilateral nucleus basalis of Meynert of AD patients. After 2 years, positron emission tomographic imaging and neuropsychological testing showed no evidence of accelerated decline [79]. Although limited progress has been achieved in NGF-based gene therapy for AD, so far, the safety concerns of this therapy may be serious.
Concluding Remarks
In this review, we highlight the important roles of NGF in the development and treatment of AD and summarize the different therapy methods (Table 1). Hence, there are many of reasons to consider NGF as an adjunct treatment in AD patients. However, in addition to cholinergic systems, the etiology of AD involves various neurotransmitter systems, such as the glutamatergic, serotonergic, and peptidergic systems. Therefore, attention should be given to the adverse effects of NGF-related agents on other systems.
Furthermore, NGF does not significantly penetrate the BBB, which makes its clinical utility dependent on invasive neurosurgical procedures [80]. Therefore, how to modify the molecular structure of NGF to allow it to cross the BBB easily and to safely exert its therapeutic effects in AD patients is an important question. Moreover, NGF is essential for the survival of both the peripheral ganglion cells and the central cholinergic neurons of the basal forebrain. The BBB could help regulate the passage of NGF from the periphery to the CNS and from the CNS to the periphery [81]. Thus, it is important to study the dynamic roles of the BBB following NGF therapy to determine if it may affect communication between the periphery and the CNS.
In conclusion, the advancement of AD includes multiple and concomitant processes. These processes can be partially reversed by the administration of NGF. Therefore, a combined treatment should be applied to all of the aspects of AD neurodegeneration.
References
Wang YJ, Valadares D, Sun Y, Wang X, Zhong JH, Liu XH, Majd S, Chen L, Gao CY, Chen S, Lim Y, Pollard A, Salegio EA, Gai WP, Yang M, Zhou XF (2010) Effects of ProNGF on neuronal viability, neurite growth and amyloid-β metabolism. Neurotox Res 17:257–267
Ayton S, Lei P, Bush AI (2015) Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 12:109–120
Lad SP, Neet KE, Mufson EJ (2003) Nerve growth factor: structure, function and therapeutic implications for Alzheimer’s disease. Curr Drug Targets CNS Neurol Disord 2:315–334
Cohen S, Levi-Montalcini R, Hamburger V (1954) A nerve growth-stimulating factor isolated from sarcom as 37 and 180. Proc Natl Acad Sci USA 40:1014–1018
Levi-Montalcini R, Hamburger V (1951) Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool 116:321–361
Fahnestock M, Yu G, Coughlin MD (2004) ProNGF: a neurotrophic or an apoptotic molecule? Prog Brain Res 146:101–110
Hempstead BL (2009) Commentary: regulating ProNGF action: multiple targets for therapeutic intervention. Neurotox Res 16:255–260
Lee R, Kermani P, Teng KK, Hempstead BL (2001) Regulation of cell survival by secreted proneurotrophins. Science 294:1945–1948
Cuello AC, Bruno MA, Allard S, Leon W, Iulita MF (2010) Cholinergic involvement in Alzheimer’s disease. A link with NGF maturation and degradation. J Mol Neurosci 40:230–235
Iulita MF, Cuello AC (2014) Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol Sci 35:338–348
Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM (2004) Sortilin is essential for ProNGF-induced neuronal cell death. Nature 427:843–848
Capsoni S, Giannotta S, Cattaneo A (2002) β-Amyloid plaques in a model for sporadic Alzheimer’s disease based on transgenic anti-nerve growth factor antibodies. Mol Cell Neurosci 21:15–28
Yuen EC, Howe CL, Li Y, Holtzman DM, Mobley WC (1996) Nerve growth factor and the neurotrophic factor hypothesis. Brain Dev 18:362–368
Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK (2013) GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther 138:155–175
Cirulli F, Alleva E, Antonelli A, Aloe L (2000) NGF expression in the developing rat brain: effects of maternal separation. Dev Brain Res 123:129–134
Woodhall E, West AK, Chuah MI (2001) Cultured olfactory ensheathing cells express nerve growth factor, brain-derived neurotrophic factor, glia cell line-derived neurotrophic factor and their receptors. Mol Brain Res 88:203–213
Lu B, Yokoyama M, Dreyfus CF, Black I (1991) NGF gene expression in actively growing brain glia. J Neurosci 11:318–326
Heese K, Fiebich BL, Bauer J, Otten U (1997) Nerve growth factor (NGF) expression in rat microglia is induced by adenosine A 2a-receptors. Neurosci Lett 231:83–86
Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H (1992) Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci 12:4793–4799
Du Y, Dreyfus CF (2002) Oligodendrocytes as providers of growth factors. J Neurosci Res 68:647–654
Byravan S, Foster LM, Phan T, Verity AN, Campagnoni AT (1994) Murine oligodendroglial cells express nerve growth factor. Proc Natl Acad Sci USA 91:8812–8816
Freund V, Pons F, Joly V, Mathieu E, Martinet N, Frossard N (2002) Upregulation of nerve growth factor expression by human airway smooth muscle cells in inflammatory conditions. Eur Respir J 20:458–463
Heumann R, Lindholm D, Bandtlow C, Meyer M, Radeke MJ, Misko TP, Shooter E, Thoenen H (1987) Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proc Natl Acad Sci USA 84:8735–8739
Taniuchi M, Clark HB, Schweitzer JB, Johnson E (1988) Expression of nerve growth factor receptors by Schwann cells of axotomized peripheral nerves: ultrastructural location, suppression by axonal contact, and binding properties. J Neurosci 8:664–681
Kawamoto K, Aoki J, Tanaka A, Itakura A, Hosono H, Arai H, Kiso Y, Matsuda H (2002) Nerve growth factor activates mast cells through the collaborative interaction with lysophosphatidylserine expressed on the membrane surface of activated platelets. J Immunol 168:6412–6419
Aloe L, Rocco ML (2015) NGF and therapeutic prospective: what have we learned from the NGF transgenic models? Ann Ist Super Sanita 51:5–10
Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11:272–280
Fukuda Y, Fukui T, Hikichi C, Ishikawa T, Murate K, Adachi T, Imai H, Fukuhara K, Ueda A, Kaplan AP (2015) Neurotropin promotes NGF signaling through interaction of GM1 ganglioside with Trk neurotrophin receptor in PC12 cells. Brain Res 1596:13–21
Gu G, Zhang W, Li M, Ni J, Wang P (2015) Transplantation of NSC-derived cholinergic neuron-like cells improves cognitive function in APP/PS1 transgenic mice. Neuroscience 291:81–92
Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J, Gonfloni S, Rossi G, Berardi N, Cattaneo A (2000) Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J Neurosci 20:2589–2601
Zhang YW, Chen Y, Liu Y, Zhao Y, Liao FF, Xu H (2013) APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS One 8:e80571
Arevalo MÁ, Roldan PM, Chacón PJ, Rodríguez-Tebar A (2009) Amyloid β serves as an NGF-like neurotrophic factor or acts as a NGF antagonist depending on its concentration. J Neurochem 111:1425–1433
Xu C-J, Wang J-L, Jin W-L (2015) The neural stem cell microenvironment: focusing on axon guidance molecules and myelin-associated factors. J Mol Neurosci 56:887–897. doi:10.1007/s12031-015-0538-1
Wang Y-J, Wang X, Lu J-J, Li Q-X, Gao C-Y, Liu X-H, Sun Y, Yang M, Lim Y, Evin G (2011) p75NTR regulates Aβ deposition by increasing Aβ production but inhibiting Aβ aggregation with its extracellular domain. J Neurosci 31:2292–2304
Zhang Y-W, Chen Y, Liu Y, Zhao Y, Liao F-F, Xu H (2013) APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS One 8:e80571. doi:10.1371/journal.pone.0080571
Cuello A, Ferretti M, Iulita M (2012) Preplaque (‘preclinical’) Aβ-induced inflammation and nerve growth factor deregulation in transgenic models of Alzheimer’s disease-like amyloid pathology. Neurodegener Dis 10:104–107. doi:10.1159/000333339
Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC (2009) Amyloid β-induced nerve growth factor dysmetabolism in Alzheimer disease. J Neuropathol Exp Neurol 68:857–869
Wakabayashi M, Matsuzaki K (2007) Formation of amyloids by Aβ-(1–42) on NGF-differentiated PC12 cells: roles of gangliosides and cholesterol. J Mol Biol 371:924–933
Matrone C, Ciotti MT, Mercanti D, Marolda R, Calissano P (2008) NGF and BDNF signaling control amyloidogenic route and Aβ production in hippocampal neurons. Proc Natl Acad Sci USA 105:13139–13144
Marei HE, Farag A, Althani A, Afifi N, Abd-Elmaksoud A, Lashen S, Rezk S, Pallini R, Casalbore P, Cenciarelli C (2015) Human olfactory bulb neural stem cells expressing hNGF restore cognitive deficit in Alzheimer’s disease rat model. J Cell Physiol 230:116–130
Yang C, Liu Y, Ni X, Li N, Zhang B, Fang X (2014) Enhancement of the nonamyloidogenic pathway by exogenous NGF in an Alzheimer transgenic mouse model. Neuropeptides 48:233–238
Xu CJ, Xu L, Huang LD, Li Y, Yu PP, Hang Q, Xu XM, Lu PH (2011) Combined NgR vaccination and neural stem cell transplantation promote functional recovery after spinal cord injury in adult rats. Neuropathol Appl Neurobiol 37:135–155
Borlongan CV (2012) Recent preclinical evidence advancing cell therapy for Alzheimer’s disease. Exp Neurol 237:142–146
Heese K, Low JW, Inoue N (2006) Nerve growth factor, neural stem cells and Alzheimer’s disease. Neurosignals 15:1–12
Yu J, Zhang X, Liu C, Meng Y, Han J (2006) Effect of acupuncture treatment on vascular dementia. Neurol Res 28:97–103
Chen J-H, Liang J, Wang G-B, Han J-S, Cui C-L (2005) Repeated 2 Hz peripheral electrical stimulations suppress morphine-induced CPP and improve spatial memory ability in rats. Exp Neurol 194:550–556
Lee M, Shin BC, Ernst E (2009) Acupuncture for Alzheimer’s disease: a systematic review. Int J Clin Pract 63:874–879
Guo H-D, Tian J-X, Zhu J, Li L, Sun K, Shao S-J, Cui G-H (2015) Electroacupuncture suppressed neuronal apoptosis and improved cognitive impairment in the AD model rats possibly via downregulation of notch signaling pathway. Evid Based Complement Alternat Med 2015:393569. doi:10.1155/2015/393569
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215:1237–1239
Lehéricy S, Hirsch ÉC, Cervera-Piérot P, Hersh LB, Bakchine S, Piette F, Duyckaerts C, Hauw JJ, Javoy-Agid F, Agid Y (1993) Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer’s disease. J Comp Neurol 330:15–31
Hou X-Q, Zhang L, Yang C, Rong C-P, He W-Q, Zhang C-X, Li S, Su R-Y, Chang X, Qin J-H (2015) Alleviating effects of Bushen–Yizhi formula on ibotenic acid-induced cholinergic impairments in rat. Rejuvenation Res 18:111–127
Wang R, Yan H (2006) Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol Sin 27:1–26
Moon M, Kim HG, Choi JG, Oh H, Lee PK, Ha SK, Kim SY, Park Y, Huh Y, Oh MS (2014) 6-Shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia. Biochem Biophys Res Commun 449:8–13
Gao J, Inagaki Y, Li X, Kokudo N, Tang W (2013) Research progress on natural products from traditional Chinese medicine in treatment of Alzheimer’s disease. Drug Discov Ther 7:46–57
Geula C, Mesulam M-M (1995) Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis Assoc Disord 9:23–28
Weinstock M, Kirschbaum-Slager N, Lazarovici P, Bejar C, Youdim MB, Shoham S (2001) Neuroprotective effects of novel cholinesterase inhibitors derived from rasagiline as potential anti-Alzheimer drugs. Ann N Y Acad Sci 939:148–161
Liston DR, Nielsen JA, Villalobos A, Chapin D, Jones SB, Hubbard ST, Shalaby IA, Ramirez A, Nason D, White WF (2004) Pharmacology of selective acetylcholinesterase inhibitors: implications for use in Alzheimer’s disease. Eur J Pharmacol 486:9–17
Capsoni S, Giannotta S, Stebel M, Garcia AA, De Rosa R, Villetti G, Imbimbo BP, Pietra C, Cattaneo A (2004) Ganstigmine and donepezil improve neurodegeneration in AD11 antinerve growth factor transgenic mice. Am J Alzheimers Dis Other Dement 19:153–160
Aboukhatwa M, Dosanjh L, Luo Y (2010) Antidepressants are a rational complementary therapy for the treatment of Alzheimer’s disease. Mol Neurodegener 5:10. doi:10.1186/1750-1326-5-10
Russo-Neustadt A, Beard RC, Cotman CW (1999) Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology 21:679–682
Lang UE, Hellweg R, Kalus P, Bajbouj M, Lenzen KP, Sander T, Kunz D, Gallinat J (2005) Association of a functional BDNF polymorphism and anxiety-related personality traits. Psychopharmacology 180:95–99
Akkad DA, Kruse N, Arning L, Gold R, Epplen JT (2008) Genomic NGFB variation and multiple sclerosis in a case control study. BMC Med Genet 9:107
Nagata T, Shinagawa S, Nukariya K, Nakayama R, Nakayama K, Yamada H (2011) Association between nerve growth factor gene polymorphism and executive dysfunction in Japanese patients with early-stage Alzheimer’s disease and amnestic mild cognitive impairment. Dement Geriatr Cogn Disord 32:379–386
Di Maria E, Giorgio E, Uliana V, Bonvicini C, Faravelli F, Cammarata S, Novello MC, Galimberti D, Scarpini E, Zanetti O (2012) Possible influence of a non-synonymous polymorphism located in the NGF precursor on susceptibility to late-onset Alzheimer’s disease and mild cognitive impairment. J Alzheimers Dis 29:699
Alberts MJ, Pericak-Vance MA, Royal V, Bebout J, Gaskell P, Thomas J, Hung WY, Clark C, Earl N, Roses AD (1991) Genetic linkage analysis of nerve growth factor (β) in familial Alzheimer’s disease. Ann Neurol 30:216–219
Qosa H, Mohamed LA, Batarseh YS, Alqahtani S, Ibrahim B, LeVine H, Keller JN, Kaddoumi A (2015) Extra-virgin olive oil attenuates amyloid-β and tau pathologies in the brains of TgSwDI mice. J Nutr Biochem 26:1479–1490. doi:10.1016/j.jnutbio.2015.07.022
Yu J, Gattoni-Celli M, Zhu H, Bhat NR, Sambamurti K, Gattoni-Celli S, Kindy MS (2011) Vitamin D3-enriched diet correlates with a decrease of amyloid plaques in the brain of AβPP transgenic mice. J Alzheimers Dis 25:295–307. doi:10.3233/JAD-2011-101986
Lee J, Duan W, Long JM, Ingram DK, Mattson MP (2000) Dietary restriction increases the number of newly generated neural cells, and induces BDNF expression, in the dentate gyrus of rats. J Mol Neurosci 15:99–108
Duan W, Lee J, Guo Z, Mattson MP (2001) Dietary restriction stimulates BDNF production in the brain and thereby protects neurons against excitotoxic injury. J Mol Neurosci 16:1–12
Yao J, Chen S, Mao Z, Cadenas E, Brinton RD (2011) 2-Deoxy-d-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS One 6:e21788
Um H-S, Kang E-B, Koo J-H, Kim H-T, Kim E-J, Yang C-H, An G-Y, Cho I-H, Cho J-Y (2011) Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci Res 69:161–173
Berardi N, Braschi C, Capsoni S, Cattaneo A, Maffei L (2007) Environmental enrichment delays the onset of memory deficits and reduces neuropathological hallmarks in a mouse model of Alzheimer-like neurodegeneration. J Alzheimers Dis 11:359–370
Zhang Z, Liu X, Schroeder JP, Chan C-B, Song M, Yu SP, Weinshenker D, Ye K (2014) 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 39:638–650
Maliartchouk S, Feng Y, Ivanisevic L, Debeir T, Cuello AC, Burgess K, Saragovi HU (2000) A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol Pharmacol 57:385–391
Scarpi D, Cirelli D, Matrone C, Castronovo G, Rosini P, Occhiato E, Romano F, Bartali L, Clemente A, Bottegoni G (2012) Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis 3:e339
Massa SM, Xie Y, Longo FM (2003) Alzheimer’s therapeutics. J Mol Neurosci 20:323–326
Tuszynski MH, Thal L, Pay M, Salmon DP, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11:551–555
Wahlberg LU, Lind G, Almqvist PM, Kusk P, Tornøe J, Juliusson B, Söderman M, Selldén E, Seiger Å, Eriksdotter-Jönhagen M (2012) Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: a technology platform for restorative neurosurgery: clinical article. J Neurosurg 117:340–347
Rafii MS, Baumann TL, Bakay RA, Ostrove JM, Siffert J, Fleisher AS, Herzog CD, Barba D, Pay M, Salmon DP (2014) A phase 1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement 10:571–581
Friden PM, Walus LR, Watson P, Kozarich J, Backman C, Bergman H, Hoffer B, Bloom F, Granholm A (1993) Blood–brain barrier penetration and in vivo activity of an NGF conjugate. Science 259:373–377
Kastin AJ, Pan W, Maness LM, Banks WA (1999) Peptides crossing the blood–brain barrier: some unusual observations. Brain Res 848:96–100
Acknowledgments
This project is supported by the Zhejiang Provincial Natural Science Foundation of China (LY13H090007) and the National Natural Science Foundation of China (No. 31171033).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
We declare that we did not receive financial support or maintain relationships that may have posed a conflict of interest.
Additional information
Chao-jin Xu and Jun-Ling Wang have contributed equally to this paper.
Rights and permissions
About this article

Cite this article
Xu, CJ., Wang, JL. & Jin, WL. The Emerging Therapeutic Role of NGF in Alzheimer’s Disease. Neurochem Res 41, 1211–1218 (2016). https://doi.org/10.1007/s11064-016-1829-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-1829-9