
Abstract
Brain serotonergic signaling is coupled to arachidonic acid (AA)-releasing calcium-dependent phospholipase A2. Increased brain serotonin concentrations and disturbed serotonergic neurotransmission have been reported in the Flinders Sensitive Line (FSL) rat model of depression, suggesting that brain AA metabolism may be elevated. To test this hypothesis, 14C-AA was intravenously infused to steady-state levels into control and FSL rats derived from the same Sprague–Dawley background strain, and labeled and unlabeled brain phospholipid and plasma fatty acid concentrations were measured to determine the rate of brain AA incorporation and turnover. Brain AA incorporation and turnover did not differ significantly between controls and FSL rats. Compared to controls, plasma unesterified docosahexaenoic acid was increased, and brain phosphatidylinositol AA and total lipid linoleic acid and n-3 and n-6 docosapentaenoic acid were significantly decreased in FSL rats. Several plasma esterified fatty acids differed significantly from controls. In summary, brain AA metabolism did not change in FSL rats despite reported increased levels of serotonin concentrations, suggesting possible post-synaptic dampening of serotonergic neurotransmission involving AA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reduced serotonergic neurotransmission is one of many neurochemical disturbances in major depression [1]. This is evidenced by studies showing reduced serotonin (5-HT) receptor binding sites in post-mortem hippocampus from anti-depressant-free depressed suicide victims, compared to non-depressed controls [2], as well as reductions in regional binding of serotonin to 5-HT 1A and 2A receptor subtypes [3, 4]. Serotonin binding to the 5-HT transporter (5-HTT) is increased in patients with major depression compared to healthy controls, reflecting reduced synaptic serotonin clearance [5]. Selective serotonin reuptake inhibitors (SSRIs), such as fluoxetine, are clinically effective in the treatment of depression, because they increase synaptic serotonin levels by targeting 5-HTT, thus blocking 5-HT reuptake from the synaptic cleft [6, 7].
Serotonergic transmission is coupled to group IV calcium-dependent phospholipase A2 (cPLA2) [8], which selectively hydrolyzes omega-6 arachidonic acid (AA, 20:4n-6) from the sterospecifically numbered-2 (sn-2) position of membrane phospholipids [9]. Using quantitative autoradiography, radiolabeled 14C-AA incorporation into rodent brain was reported to increase following 5-HT2A/2C receptor activation by the partial agonist, 2,5-Dimethoxy-4-iodoamphetamine hydrochloride (DOI) [10]. Chronic fluoxetine, which increases synaptic serotonin levels by targeting 5-HTT, increased brain AA incorporation and turnover, in association with increased cPLA2 activity [11–13]. The increase in AA incorporation following activation of serotonergic neurotransmission reflects post-synaptic coupling between AA-releasing cPLA2 and 5-HT neuroreceptors [8, 14, 15].
The Flinders Sensitive Line (FSL) rat is a well-characterized genetic animal model of human depression obtained by selective breeding of Sprague Dawley (SD) rats for increased sensitivity to the anticholinesterase agent diisopropyl fluorophosphate (DFP) [16, 17]. This animal model shows face, construct and predictive validity, in that depressive-like symptoms and disturbances in serotonergic neurotransmission are reversed by SSRIs [18]. Behavioral characteristics of FSL rats include anhedonia, reduced locomotion and activity, sleep disorders and cognitive impairment, which are improved by chronic treatment with antidepressants such as desipramine, fluoxetine, imipramine or sertraline [17].
Serotonergic function is impacted in FSL rats. Compared to SD controls, FSL brains show reduced 5-HT synthesis [19], three to eightfold increased concentration of serotonin and its 5-hydroxyindoleacetic acid metabolite in the nucleus accumbens, prefrontal cortex, hippocampus and hypothalamus [20], reduced 5-HTT density [7], decreased 5HT1A receptors and increased 5HT1B receptors [21, 22]. Compared to a control strain less sensitive to DFP, the Flinders Resistant Line (FRL), FSL rats were reported to have reduced mRNA expression of 5-HT2A in perirhinal cortex, piriform cortex and medial anterodorsal amygdala nucleus, and increased 5-HT2A mRNA in hippocampal CA2 and CA3 regions [23]. The FSL rats also show reduced cholinergic [17, 24], noradrenergic [25] and dopaminergic [26] neurotransmission and increased glutamatergic transmission [27] compared to SD controls.
Brain AA composition is altered in FSL rats, suggesting a link between depression and brain lipid metabolism. Green et al. [28, 29] reported increased brain AA fractional concentrations (i.e. percent of total fatty acids) and AA-containing phosphatidylcholine in hypothalamus, nucleus accumbens, prefrontal cortex or striatum of FSL rats compared to controls. Changes in brain AA concentrations could be linked to disturbed AA-metabolizing cPLA2 and reduced serotonergic transmission in FSL rats.
In view of the coupling between serotonergic neurotransmission and AA-releasing cPLA2 [8], and disturbed serotonergic neurotransmission associated with regional three to eightfold increases in brain serotonin concentrations in FSL rats [20], we hypothesized that brain AA turnover, a marker of AA signaling, would increase in the FSL model of depression. Changes in brain AA metabolism were expected also because of the reported changes in brain AA fractional concentrations in FSL rats [28, 29].
We used the in vivo kinetic method developed by Rapoport and colleagues [30, 31] to measure brain AA incorporation and turnover in FSL and non-littermate SD control rats. Radiolabeled 14C-AA was infused intravenously for 5 min in unanaesthetized FSL and SD control rats, and brain and plasma unlabeled fatty acid and acyl-CoA concentrations, as well as labeled AA and arachidonoyl-CoA (AA-CoA) concentrations were measured to calculate turnover. We found no significant differences in brain AA incorporation or turnover between FSL and SD rats.
Materials and Methods
Animals
Experiments were conducted in accordance with the National Institutes of Health guidelines for animal care (Publication no. 86-23) and followed a protocol approved by the Animal Care and Use Committee of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Adult FSL rats were obtained from Duke University (Durham, NC, USA). Male SD rats (Harlan Laboratories, Indianapolis, IN, USA) were used as controls. The FSL colony was established by selective breeding of SD rats, based on their sensitivity to the cholinergic agent DFP [17, 32]. FSL rats were obtained from a breeding colony established at Duke University by Dr. Amir H. Rezvani.
Initially, seven control and seven FSL male rats were obtained at 8 weeks of age. Two of the FSL rats turned out to be females because they gave birth when they arrived at the NIH vivarium. They and their pups were therefore euthanized. The remaining animals were sexed and confirmed to be males.
SD and FSL rats were housed (3–4 per cage) in an animal facility with regulated temperature, humidity and light cycle (12 h dark cycle) until they reached 16–17 weeks of age. They had free access to water and food. Rats were fed the 5001 diet (LabDiet, Richmond, IN, USA) which contained (as % total fatty acid): 33.5 % saturated fatty acids, 32.1 % monounsaturated fatty acids, 29.6 % linoleic acid (LA, 18:2n-6), 2.2 % α-linolenic acid (α-LNA, 18:3n-3), 1.5 % eicosapentaenoic acid (EPA, 20:5n-3) and 1.1 % docosahexaenoic acid (DHA, 22:6n-3), which was the diet used in the Duke University facility. The fatty acid composition of the diet was determined by gas-chromatography following extraction of total lipids from food pellets using chloroform:methanol (2:1, v:v) and transesterification with 1 % H2SO4 in methanol as previously reported [33].
Surgical Procedure and Tracer Infusion
Rats were anesthetized with isofluorane (10 % O2) and polyethylene catheters (PE50, Clay Adams, Becton–Dickinson, Sparks, MD, USA) filled with heparinized saline were inserted surgically into the right femoral vein and artery [31]. Animals recovered from anesthesia for approximately 4 h in a temperature-controlled recovery chamber maintained at 25 °C, while body temperature was maintained at 37 °C with a rectal probe and a feedback heating element (TACT-2DF Temperature controller; Physitemp Instruments, Clifton, NJ, USA).
Before infusion, heart rate and blood pressure were monitored using a CyberSense monitor (BPM01 CyQ 302, Nicholasville, KY, USA). Rats were infused intravenously for 5 min with 170 µCi/kg [1-14C]AA (50 mCi/mmol, >98 % pure; Moravek Biochemicals, Brea, CA, USA) dissolved in HEPES buffer (pH 7.4) containing 50 mg/kg fatty acid-free bovine serum albumin (BSA, Sigma) [34]. The tracer was infused at a rate of 0.223(1 + e−0.032t) ml/min (t = s) with a computer-controlled variable rate infusion pump (No. 22; Harvard Apparatus, South Natick, MA, USA), to achieve a steady-state plasma specific activity within 1 min [35]. The 5-min infusion paradigm was used because a previous study showed that more than 90 % of radiolabeled AA that enters the brain is esterified within 1 min into brain phospholipids [35]. Blood samples were collected at different time points (0, 15, 30, 45, 90, 180, 240, and 300 s) during infusion. Five minutes after infusion, the rats were anesthetized using sodium pentobarbital (50 mg/kg, i.v.) and subjected to head-focused microwave irradiation (5.5 kW, 4.8 s, Cober Electronics, Norwalk, CT, USA) to stop brain lipid metabolism [36, 37]. The brains were quickly excised, separated into two hemispheres, frozen on dry ice and stored at −80 °C until further analysis.
Lipid Extraction and Separation
Total lipids from plasma samples collected at 5 min infusion, and from one brain hemisphere, were extracted using the Folch procedure [38]. After addition of unesterified heptadecanoic acid (17:0) as an internal standard, lipid extracts from plasma were separated into neutral lipid classes by thin layer chromatography (TLC) on silica plates (Silica gel 60A TLC plates, Whatman, Clifton, NJ, USA) using heptane:diethylether:acetic acid (60:40:3, v:v) [39]. Phospholipid classes from brain lipid extracts were separated using chloroform:methanol:H2O:acetic acid (60:50:4:1, v:v) [40], and choline glycerophospholipids (ChoGpls), phosphatidylserine (PtdSer), ethanolamine glycerophospholipids (EtnGpls) and phosphatidylinositol (PtdIns) were identified by running standards in parallel with the samples. The lipid bands were visualized under ultraviolet light after spraying the plates with 0.03 % (w/v) 6-p-toluidine-2-naphthalene sulfonic acid (Acros, Fairlawn, NJ, USA) in 50 mM Tris buffer (pH 7.4). Each band was scraped, and the silica gel was used directly to quantify radioactivity by scintillation counting or to prepare fatty acid methyl esters (FAMEs) as described below.
Quantification of Plasma and Brain Radioactivity
Radioactivity in plasma total lipid extracts collected over the course of the 5 min infusion and brain phospholipids and acyl-CoAs was determined using a liquid scintillation analyzer (2200CA, TRI-CARB®; Packard Instruments, Meriden, CT, USA).
FAME Preparation and Analysis by Gas Chromatography
Scrapes collected from TLC were heated in 1 % H2SO4 in methanol at 70 °C for 3 h to produce FAMEs, which were separated on a SP-2330 fused silica capillary column (30 m × 0.25 mm inner diameter, 0.25 µm film thickness) (Supleco, Bellefonte, PA, USA), using gas chromatography (GC) equipped with a flame ionization detector (6890 N, Agilent Technologies, Palo Alto, CA, USA). The run temperature started at 80 °C, was ramped up at 10 °C/min to 150 °C, and 6 °C/min to 200 °C, held at 200 °C for 10 min and increased to 240 °C for a total run time of 38 min. Fatty acid concentrations (nmol/g brain or nmol/ml plasma) were calculated by comparison of the GC peak areas to the 17:0 internal standard area.
Quantification of Acyl-CoA
Acyl-CoAs were extracted from the remaining microwaved half-brains by the method of Deutsch et al. [41]. 10 nmol 17:0-CoA was added as a surrogate standard to a weighed half-brain before homogenization by sonication in 2 mL of 25 mM potassium phosphate. Proteins were precipitated with saturated ammonium sulphate and gentle shaking. Four mL acetonitrile were added and the sample was shaken for 10 min before centrifugation. The upper phase was collected and diluted with 10 mL of 25 mM potassium phosphate and passed three times though an activated oligonucleotide purification cartridge (ABI Masterpiece™, OPC®, Applied Biosystems, Foster City, CA, USA). The cartridge was washed with 10 mL of 25 mM potassium phosphate. Acyl-CoAs were then eluted using 500 µL elution buffer containing 75 % isopropanol/25 % 1 mM glacial acetic acid (v:v). Samples were dried under N2 and reconstituted in 100 µL elution buffer for high performance liquid chromatography (HPLC) analysis. Extracted acyl-CoA were separated on a reverse phase HPLC column (Symmetry C-18, 5 µm particle size, 250 × 4.6 mm, Waters Millipore, Milford, MA, USA), using HPLC (Beckman, Fullerton, CA, USA) coupled with a UV/VIS detector (Model 168, System Gold, Beckman). HPLC conditions were set to a 1 ml/min gradient system composed of (A) 75 mM potassium phosphate and (B) acetonitrile. The gradient was set initially at 44 % B for 1 min, then 48 % over 25 min, and 70 % for 5 min. The gradient was maintained at 70 % for 9 min, decreased to 44 % over 4 min and was held at 44 % for 4 min. Peak identification was performed with UV set at 280 and 260 nm. Acyl-CoA species were identified by running authentic standards, and quantitated by integration of the HPLC peaks relative to the 17:0-CoA standard (UV detection at 260 nm). The arachidonoyl-CoA (AA-CoA) peak was collected to measure its radioactivity by scintillation counting.
Calculations
The Rapoport in vivo model used to quantify brain fatty acid kinetic parameters is described in detail elsewhere [30, 42]. Unidirectional incorporation coefficients, k * i (ml s−1 g−1) of AA, representing incorporation from plasma into brain lipid i, were calculated as follows:
C * br,i (T) (nCi g−1) represents radioactivity of brain lipid i at the end of the infusion T = 5 min, t is time after starting infusion, and C * pl (nCi ml−1) is the plasma concentration of labeled unesterified AA during infusion. Integrals of plasma radioactivity were determined by trapezoidal integration. Since brain AA synthesis from its precursor LA (18:2n-6) is minimal [43], the rate of incorporation J in,i (nmol s−1 g−1) of plasma unesterified AA into brain lipid i, represents the rate of metabolic loss of AA by the brain, and is calculated as follows:
C pl (nmol ml−1) is the concentration of unlabeled unesterified AA in plasma. The “dilution factor” λ, defined as the steady-state ratio during [1-14C]AA infusion of the specific activity of the brain arachidonoyl-CoA (AA-CoA) pool to the specific activity of plasma unesterified AA, was determined as follows:
Net rates of incorporation of unlabeled unesterified AA from brain AA-CoA into brain lipid i, \( J_{FA,i } \) (nmol s−1 g−1) equals:
The fractional turnover of AA within phospholipid i, due to deacylation and reacylation, \( F_{FA,i } \) (% h−1) is defined as:
Data Handling and Statistical Analysis
One FSL rat died during the 14C-AA infusion of unknown causes. Two control rats were not successfully infused to steady-state, as evidenced by the lack of radioactivity in the plasma of one rat, and another rat which showed delayed appearance of the tracer in its plasma (after 30 s) due to the presence of blood clots in the venous catheter line. These animals were therefore excluded from the study.
Results were expressed as mean ± SD, for n = 5 controls and 4 FSL rats. Data were compared using Student t test (GraphPad Prism software, San Diego, CA, USA) and statistical significance was considered at P < 0.05.
Results
Physiological Parameters
Body weight was significantly higher in FSL rats than control SD rats (Table 1). Mean arterial blood pressure, heart rate and body temperature did not differ significantly between the two groups (Table 1).
Plasma Kinetics
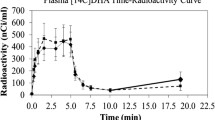
Plasma radioactivity, measured over the 5 min 14C AA infusion period, reached steady state for both groups after 45 s (Fig. 1). The total area under the curve (AUC) for plasma 14C-AA concentration did not significantly differ between FSL and control rats (147,467 ± 27,017 for 4 FSL rats and 177,188 ± 18,840 for 5 controls, P = 0.092).

Plasma radioactivity during the 5 min of [1-14C] AA intravenous infusion
Plasma Fatty Acid Concentrations
As shown in Table 2, plasma unesterified fatty acid concentrations did not significantly differ between control and FSL rats, except for DHA concentration which was significantly higher in FSL rats compared to controls (+36 %, P < 0.05).
Supplementary Table 1 shows esterified plasma fatty acid concentrations. Total phospholipid and cholesteryl ester fatty acid concentrations were higher by 74 and 39 %, respectively, in FSL rats than controls (P < 0.05). Within plasma phospholipids, stearic acid (18:0), palmitoleic acid (18:1n-7), AA, n-6 DPA and DHA were significantly increased by 34–98 % in FSL compared to control rats. Cholesteryl ester α-LNA was decreased by 45 % and AA was increased by 57 %.
Brain Fatty Acid Concentrations
Brain total lipid LA, n-3 DPA and n-6 DPA concentrations were significantly decreased by 11–21 % in FSL rats. PtdIns AA concentration was significantly reduced by 5.2 % in FSL rats compared to controls, as was total n-6 PUFA concentration (P < 0.05). There were no significant differences in fatty acid concentrations within brain ChoGpls, EtnGpls, PtdSer and unesterified fatty acids, between control and FSL rats (Table 3).
Brain Acyl-CoA Concentrations
There were no significant differences in 14:0, 16:0, 18:0, 18:1 n-9, 18:2 n-6, AA or DHA acyl-CoA concentrations between FSL rats and controls (Table 4). There was no change in specific activity of 14C-AA-CoA between the groups. The dilution factor λ, which measures the ratio of specific activity of brain 14C-AA-CoA to that of plasma unesterified 14C-AA, did not significantly differ.
Brain AA Kinetics
The brain AA incorporation coefficient k* (Eq. 1) did not differ significantly between FSL and control rats, in total phospholipids and individual phospholipid subclasses (Table 5). The incorporation rate of AA from plasma J in,I was calculated from Eq. 2, and did not differ significantly between the groups.
There was no significant change in the rate of incorporation J FA of brain unesterified AA from the AA-CoA pool into phospholipids (Eq. 5) or in brain AA turnover F FA (Table 6).
Discussion
This study showed no significant differences in brain AA incorporation or turnover in the FSL rat model of depression compared to SD controls. Plasma unesterified DHA, cholesteryl ester AA, and phospholipid stearic acid, palmitoleic acid, AA, n-6 DPA and DHA concentrations were significantly increased in FSL compared to control rats, whereas plasma cholesteryl ester α-LNA and brain PtdIns AA and total lipid LA, n-3 DPA and n-6 DPA concentrations were decreased in FSL rats. These concentration differences likely reflect peripheral and central changes in PUFA metabolism.
Despite the three to eightfold reported increase in regional brain serotonin concentrations in FSL rats [20], brain AA incorporation (k*), incorporation rate (Jin) and turnover (FFA) did not significantly differ relative to controls, suggesting post-synaptic down-regulation of serotonergic transmission involving AA. This could be related to altered 5-HT receptor subtype (1A and 1B) density as reported in FSL rats [21], or changes in 5-HT2A/2C receptor density, internalization [44] or affinity states [45], which remains to be confirmed in this model. 5-HT2A mRNA was reported to decrease or increase in various brain regions of FSL rats compared to FRL controls, although protein expression was not measured [23].
The absence of change in AA turnover may also be due to other neurotransmission systems coupled to AA. Dopamine release was reported to be reduced in nucleus accumbens of FSL rats following 5HT2C receptor activation [26], suggesting inverse coupling of 5-HT receptors to dopaminergic neurotransmission, which is also coupled to AA-releasing cPLA2 [46, 47]. A similar lack of change in AA metabolism was reported in dopamine-transporter knockout mice, in which brain AA incorporation did not differ from wildtype controls despite a tenfold increase in extracellular dopamine concentrations relative to controls [48].
The reported elevations in brain serotonin concentrations in FSL rats were based on whole brain homogenates, and therefore do not necessarily reflect extracellular levels [20]. Dremencov et al. [26] reported no difference in extracellular serotonin concentration in nucleus accumbens between FSL and SD rats, although extracellular serotonin levels were not measured in other brain regions. Regional extracellular brain serotonin distribution and turnover in relation to post-synaptic AA signaling could be tested in future studies with micro-dialysis [26] and quantitative autoradiography [10–12].
Total lipid AA fractional concentrations in hypothalamus, nucleus accumbens, prefrontal cortex and striatum were reported to be elevated in FSL compared to control rats [29], as was phosphatidylcholine containing AA in striatum and hypothalamus [28]. We found reduced AA concentration (nmol/g wet weight) within PtdIns, but not in other phospholipid fractions. AA fractional concentrations, calculated by dividing brain total lipid AA by total fatty acid concentrations, also did not significantly change in our study (10.7 ± 1.4 FSL vs. 10.7 ± 0.1 SD). Differences in study outcomes could be related to the fact that we analyzed whole brain instead of regional AA distribution, or to differences in methodology, since Green et al. used electrospray ionization tandem mass spectra (ESI/MS/MS), whereas we used TLC/GC to determine fatty acid profiles. Regional changes in brain AA incorporation could be measured with quantitative autoradiography in future studies to assess whether brain AA metabolism changes in specific brain regions [48].
Body weight was significantly higher in FSL rats than controls, which is in contrast with previous studies that reported lower body weight in FSL rats than SD controls [17]. FSL and SD rats were fed the same diets at our animal facility for 2 months before starting the experiment. However, FSL rats were obtained from the Duke University colony, whereas SD rats were purchased from a vendor because control FSL littermates were not available. Because of these differences in rat source, the animals were exposed to a different diet from conception until they arrived at our facility. The SD rats were born and weaned to the 2018S Teklad diet, which has 6.2 g of fat containing 31 % LA and 3 % α-LNA, and no EPA or DHA [49]. The FSL rats were maintained on the 5001 diet (LabDiet, Richmond, IN, USA) since birth, which has 6.4 g of fat, composed of 29.6 % LA, 2.2 % α-LNA, 1.5 % EPA and 1.1 % DHA. Differences in fatty acid intake prior to the arrival of the animals to our facility may have contributed to the difference in body weight.
The significant increase in plasma unesterified DHA and esterified stearic acid, palmitoleic acid, AA, n-6 DPA and DHA, and decrease in plasma cholesteryl ester α –LNA and brain PtdIns AA and total lipid LA, n-3 DPA and n-6 DPA concentrations in FSL rats, could be a type I error associated with multiple comparisons and a low sample size. They may also be related to the differences in dietary fatty acid intake prior to the arrival of the rats at our facility, which are likely to have affected adipose tissue fatty acid composition and release, and liver synthesis-secretion rates.
Limitations of this work include the low sample size and the lack of measurement of various AA cascade enzymes (cPLA2, COX-2, among many others) and AA-derived metabolites such as PGE2. We did not quantify enzyme protein level or activity in brains collected from this study because the animals were subjected to high-energy microwave fixation, which denatures proteins and stops brain lipid metabolism [36, 37]. AA-derived mediators, which are typically measured in microwaved brain samples, were not quantified in this study because both half-brains were used for either AA or AA-CoA measurements. It would be worthwhile to measure AA cascade enzymes and metabolites in better-powered studies, to better assess whether specific pathways within the AA cascade are altered in the FSL model of depression.
Another limitation of this study is that we did not validate our kinetic method by pharmacologically stimulating brain AA metabolism. However, values for brain AA concentration, incorporation and turnover in control rats are consistent with a previous study which used the same rat strain, thus confirming the validity and reproducibility of our in vivo kinetic method [35].
Using quantitative autoradiography, Qu et al. [50] reported that acute administration of DOI to 5-HTT wildtype and knockout mice produced an AA signal in wildtype but not knockout mice, suggesting a downregulation of 5-HT2A/2C receptor mediated signaling via AA. In this regard, it would be worthwhile to test whether challenging FSL rats with DOI would elicit an AA response. In view of the reported reduction in 5-HTT density in FSL rats [7], it is likely that AA signaling would be dampened following DOI administration, similar to what had been reported in 5-HTT knockout mice [50].
In summary, brain AA incorporation and turnover did not significantly differ in the FSL model of depression compared to controls. The absence of differences may be related to post-synaptic dampening of serotonergic transmission in response to elevated brain serotonin levels [20], similar to what was reported in a dopamine reuptake transporter knockout model of depression [48]. Future studies should examine the involvement of AA signaling in FSL rats after receiving a challenge such as DOI. Whether brain AA metabolism is altered in clinical depression could be explored with positron-emitting tomography imaging using 11C-AA, as had been done for Alzheimer’s disease patients [51].
Abbreviations
- 5-HT:
-
Serotonin
- 5-HTT:
-
5-HT transporter
- α-LNA:
-
Alpha-linolenic acid
- AA:
-
Arachidonic acid
- AA-CoA:
-
Arachidonoyl-CoA
- AUC:
-
Area under the curve
- ChoGpl:
-
Choline glycerophospholipid
- cPLA2 :
-
Cytosolic phospholipase A2
- CSF:
-
Cerebrospinal fluid
- DFP:
-
Diisopropyl fluorophosphate
- DHA:
-
Docosahexaenoic acid
- DOI:
-
2,5-Dimethoxy-4-iodoamphetamine hydrochloride
- GC :
-
Gas Chromatography
- EtnGpl:
-
Ethanolamine glycerophospholipid
- DPA:
-
Docosapentaenoic acid
- EPA:
-
Eicosapentaenoic acid
- FAME:
-
Fatty Acid Methyl Ester
- FRL:
-
Flinders Resistant Line
- FSL:
-
Flinders Sensitive Line
- LA:
-
Linoleic acid
- PtdIns:
-
Phosphatidylinositol
- PdtSer:
-
Phosphatidylserine
- SFA:
-
Saturated fatty acids
- MUFA:
-
Monounsaturated fatty acids
- PUFA:
-
Polyunsaturated fatty acids
- SD:
-
Sprague–Dawley
- SSRI:
-
Selective serotonin reuptake inhibitors
- TLC:
-
Thin Layer Chromatography
References
Owens MJ, Nemeroff CB (1994) Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem 40:288–295
Cheetham SC, Crompton MR, Katona CL, Horton RW (1988) Brain 5-HT2 receptor binding sites in depressed suicide victims. Brain Res 443:272–280
Mintun MA, Sheline YI, Moerlein SM, Vlassenko AG, Huang Y, Snyder AZ (2004) Decreased hippocampal 5-HT2A receptor binding in major depressive disorder: in vivo measurement with [18F]altanserin positron emission tomography. Biol Psychiatry 55:217–224
Sargent PA, Kjaer KH, Bench CJ, Rabiner EA, Messa C, Meyer J, Gunn RN, Grasby PM, Cowen PJ (2000) Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry 57:174–180
Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N, Goulding V, Kennedy J, Wilson AA (2004) Brain serotonin transporter binding potential measured with carbon 11-labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry 61:1271–1279
Meyer JH, Wilson AA, Sagrati S, Miler L, Rusjan P, Bloomfield PM, Clark M, Sacher J, Voineskos AN, Houle S (2009) Brain monoamine oxidase A binding in major depressive disorder: relationship to selective serotonin reuptake inhibitor treatment, recovery, and recurrence. Arch Gen Psychiatry 66:1304–1312
Kovacevic T, Skelin I, Diksic M (2010) Chronic fluoxetine treatment has a larger effect on the density of a serotonin transporter in the Flinders Sensitive Line (FSL) rat model of depression than in normal rats. Synapse 64:231–240
Garcia MC, Kim HY (1997) Mobilization of arachidonate and docosahexaenoate by stimulation of the 5-HT2A receptor in rat C6 glioma cells. Brain Res 768:43–48
Strokin M, Sergeeva M, Reiser G (2003) Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+. Br J Pharmacol 139:1014–1022
Qu Y, Chang L, Klaff J, Balbo A, Rapoport SI (2003) Imaging brain phospholipase A2 activation in awake rats in response to the 5-HT2A/2C agonist (±)2,5-dimethoxy-4-iodophenyl-2-aminopropane (DOI). Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 28:244–252
Qu Y, Chang L, Klaff J, Seemann R, Rapoport SI (2003) Imaging brain phospholipase A2-mediated signal transduction in response to acute fluoxetine administration in unanesthetized rats. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 28:1219–1226
Qu Y, Chang L, Klaff J, Seemann R, Greenstein D, Rapoport SI (2006) Chronic fluoxetine upregulates arachidonic acid incorporation into the brain of unanesthetized rats. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol 16:561–571
Lee HJ, Rao JS, Ertley RN, Chang L, Rapoport SI, Bazinet RP (2007) Chronic fluoxetine increases cytosolic phospholipase A(2) activity and arachidonic acid turnover in brain phospholipids of the unanesthetized rat. Psychopharmacology 190:103–115
Axelrod J (1990) Receptor-mediated activation of phospholipase A2 and arachidonic acid release in signal transduction. Biochem Soc Trans 18:503–507
Felder CC, Kanterman RY, Ma AL, Axelrod J (1990) Serotonin stimulates phospholipase A2 and the release of arachidonic acid in hippocampal neurons by a type 2 serotonin receptor that is independent of inositolphospholipid hydrolysis. Proc Natl Acad Sci USA 87:2187–2191
Overstreet DH, Friedman E, Mathe AA, Yadid G (2005) The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev 29:739–759
Overstreet DH (1993) The Flinders Sensitive Line rats: a genetic animal model of depression. Neurosci Biobehav Rev 17:51–68
Yadid G, Nakash R, Deri I, Tamar G, Kinor N, Gispan I, Zangen A (2000) Elucidation of the neurobiology of depression: insights from a novel genetic animal model. Prog Neurobiol 62:353–378
Hasegawa S, Nishi K, Watanabe A, Overstreet DH, Diksic M (2006) Brain 5-HT synthesis in the Flinders Sensitive Line rat model of depression: an autoradiographic study. Neurochem Int 48:358–366
Zangen A, Overstreet DH, Yadid G (1997) High serotonin and 5-hydroxyindoleacetic acid levels in limbic brain regions in a rat model of depression: normalization by chronic antidepressant treatment. J Neurochem 69:2477–2483
Nishi K, Kanemaru K, Diksic M (2009) A genetic rat model of depression, Flinders Sensitive Line, has a lower density of 5-HT(1A) receptors, but a higher density of 5-HT(1B) receptors, compared to control rats. Neurochem Int 54:299–307
Iritani S, Tohgi M, Arai T, Ikeda K (2006) Immunohistochemical study of the serotonergic neuronal system in an animal model of the mood disorder. Exp Neurol 201:60–65
Osterlund MK, Overstreet DH, Hurd YL (1999) The Flinders Sensitive Line rats, a genetic model of depression, show abnormal serotonin receptor mRNA expression in the brain that is reversed by 17beta-estradiol. Brain Res Mol Brain Res 74:158–166
Benca RM, Overstreet DE, Gilliland MA, Russell D, Bergmann BM, Obermeyer WH (1996) Increased basal REM sleep but no difference in dark induction or light suppression of REM sleep in flinders rats with cholinergic supersensitivity. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 15:45–51
Zangen A, Overstreet DH, Yadid G (1999) Increased catecholamine levels in specific brain regions of a rat model of depression: normalization by chronic antidepressant treatment. Brain Res 824:243–250
Dremencov E, Newman ME, Kinor N, Blatman-Jan G, Schindler CJ, Overstreet DH, Yadid G (2005) Hyperfunctionality of serotonin-2C receptor-mediated inhibition of accumbal dopamine release in an animal model of depression is reversed by antidepressant treatment. Neuropharmacology 48:34–42
Gomez-Galan M, De Bundel D, Van Eeckhaut A, Smolders I, Lindskog M (2013) Dysfunctional astrocytic regulation of glutamate transmission in a rat model of depression. Mol Psychiatry 18:582–594
Green P, Anyakoha N, Yadid G, Gispan-Herman I, Nicolaou A (2009) Arachidonic acid-containing phosphatidylcholine species are increased in selected brain regions of a depressive animal model: implications for pathophysiology. Prostaglandins Leukot Essent Fatty Acids 80:213–220
Green P, Gispan-Herman I, Yadid G (2005) Increased arachidonic acid concentration in the brain of Flinders Sensitive Line rats, an animal model of depression. J Lipid Res 46:1093–1096
Robinson PJ, Noronha J, DeGeorge JJ, Freed LM, Nariai T, Rapoport SI (1992) A quantitative method for measuring regional in vivo fatty-acid incorporation into and turnover within brain phospholipids: review and critical analysis. Brain Res Brain Res Rev 17:187–214
DeGeorge JJ, Noronha JG, Bell J, Robinson P, Rapoport SI (1989) Intravenous injection of [1-14C]arachidonate to examine regional brain lipid metabolism in unanesthetized rats. J Neurosci Res 24:413–423
Overstreet DH, Russell RW, Helps SC, Messenger M (1979) Selective breeding for sensitivity to the anticholinesterase DFP. Psychopharmacology 65:15–20
Igarashi M, Gao F, Kim HW, Ma K, Bell JM, Rapoport SI (2009) Dietary n-6 PUFA deprivation for 15 weeks reduces arachidonic acid concentrations while increasing n-3 PUFA concentrations in organs of post-weaning male rats. Biochim Biophys Acta 1791:132–139
Modi HR, Taha AY, Kim HW, Chang L, Rapoport SI, Cheon Y (2013) Chronic clozapine reduces rat brain arachidonic acid metabolism by reducing plasma arachidonic acid availability. J Neurochem 124:376–387
Washizaki K, Smith QR, Rapoport SI, Purdon AD (1994) Brain arachidonic acid incorporation and precursor pool specific activity during intravenous infusion of unesterified [3H]arachidonate in the anesthetized rat. J Neurochem 63:727–736
Deutsch J, Rapoport SI, Purdon AD (1997) Relation between free fatty acid and acyl-CoA concentrations in rat brain following decapitation. Neurochem Res 22:759–765
Bazinet RP, Lee HJ, Felder CC, Porter AC, Rapoport SI, Rosenberger TA (2005) Rapid high-energy microwave fixation is required to determine the anandamide (N-arachidonoylethanolamine) concentration of rat brain. Neurochem Res 30:597–601
Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509
Skipski VP, Good JJ, Barclay M, Reggio RB (1968) Quantitative analysis of simple lipid classes by thin-layer chromatography. Biochim Biophys Acta 152:10–19
Skipski VP, Barclay M, Reichman ES, Good JJ (1967) Separation of acidic phospholipids by one-dimensional thin-layer chromatography. Biochim Biophys Acta 137:80–89
Deutsch J, Grange E, Rapoport SI, Purdon AD (1994) Isolation and quantitation of long-chain acyl-coenzyme A esters in brain tissue by solid-phase extraction. Anal Biochem 220:321–323
Rapoport SI, Chang MC, Spector AA (2001) Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J Lipid Res 42:678–685
DeMar JC Jr, Lee HJ, Ma K, Chang L, Bell JM, Rapoport SI, Bazinet RP (2006) Brain elongation of linoleic acid is a negligible source of the arachidonate in brain phospholipids of adult rats. Biochim Biophys Acta 1761:1050–1059
Berry SA, Shah MC, Khan N, Roth BL (1996) Rapid agonist-induced internalization of the 5-hydroxytryptamine2A receptor occurs via the endosome pathway in vitro. Mol Pharmacol 50:306–313
Egan C, Grinde E, Dupre A, Roth BL, Hake M, Teitler M, Herrick-Davis K (2000) Agonist high and low affinity state ratios predict drug intrinsic activity and a revised ternary complex mechanism at serotonin 5-HT(2A) and 5-HT(2C) receptors. Synapse 35:144–150
Bhattacharjee AK, Chang L, White L, Bazinet RP, Rapoport SI (2008) Imaging apomorphine stimulation of brain arachidonic acid signaling via D2-like receptors in unanesthetized rats. Psychopharmacology 197:557–566
Vial D, Piomelli D (1995) Dopamine D2 receptors potentiate arachidonate release via activation of cytosolic, arachidonate-specific phospholipase A2. J Neurochem 64:2765–2772
Ramadan E, Chang L, Chen M, Ma K, Hall FS, Uhl GR, Rapoport SI, Basselin M (2012) Knocking out the dopamine reuptake transporter (DAT) does not change the baseline brain arachidonic acid signal in the mouse. Int J Neurosci 122:373–380
Qu Y, Villacreses N, Murphy DL, Rapoport SI (2005) 5-HT2A/2C receptor signaling via phospholipase A2 and arachidonic acid is attenuated in mice lacking the serotonin reuptake transporter. Psychopharmacology 180:12–20
Esposito G, Giovacchini G, Liow JS, Bhattacharjee AK, Greenstein D, Schapiro M, Hallett M, Herscovitch P, Eckelman WC, Carson RE, Rapoport SI (2008) Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J Nucl Med Off Publ Soc Nucl Med 49:1414–1421
Acknowledgments
We thank Dr. Epolia Ramadan and Dr. Mireille Basselin for writing the animal study protocol and Dr. Mei Chen for technical assistance.
Source of Funding
Research was supported by the Intramural Research Program of the National Institute on Aging.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Blanchard, H., Chang, L., Rezvani, A.H. et al. Brain Arachidonic Acid Incorporation and Turnover are not Altered in the Flinders Sensitive Line Rat Model of Human Depression. Neurochem Res 40, 2293–2303 (2015). https://doi.org/10.1007/s11064-015-1719-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1719-6