
Abstract
Numerous studies have shown robust neuroprotective effects of paeoniflorin (PF), a natural compound derived from the herbal medicine Paeony radix. In the present study, we determined associations of PF neuroprotection with its modulation of various apoptotic and anti-apoptotic pathways. PF (50–400 μM) pretreatment significantly improved viability of differentiated PC12 cells exposed to methyl-4-phenylpyridine ion (MPP+), a neurotoxin, and inhibited over-release of lactate dehydrogenase, a biomarker of neuronal cell death. PF also ameliorated MPP+-induced nuclear and mitochondrial apoptotic alteration and intracellular calcium overload. PF treatment reversed MPP+ suppression of activity of B cell lymphoma-extra large, which is a mitochondrial membrane molecule that protects cells from DNA damage-induced apoptosis, and strikingly inhibited the enhanced level of cleaved poly(ADP-ribose)polymerase, which is involved in the process of apoptosis. PF alone and coadministration with MPP+ enhanced phospho activation of extracellular signal-regulated kinases, Akt, and its downstream element glycogen synthase kinase-3, but the effects were completely abolished in the presence of their blockers PD98059 and LY294002. The presence of the blockers also diminished the potency of PF in improving viability of MPP+-exposed cells. These results indicate that neuroprotective effects of PF are related to its modulation of multiple anti-apoptotic and pro-apoptotic pathways, including blockade of intracellular calcium overload, prevention of mitochondrial membrane integrity, inhibition of pro-apoptotic molecules, and up-regulation of anti-apoptotic proteins associated with cell survival and proliferation. The study provides evidence supporting PF as a potential therapeutic agent used for the treatment of neurodegenerative diseases and neural injury.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The root of Paeonia lactiflora Pall (family Ranunculaceae) or peony root is a commonly used Chinese herbal medicine, which has been suggested to have broad therapeutic benefits for pain, muscle spasm, inflammation, menstrual dysfunction, and degenerative disorders (Wu 1985; Wu et al. 2010). It is thought that these therapeutic benefits are mainly derived from its major constituents which possess diverse pharmacological properties (Wu et al. 2010). Paeoniflorin (PF) is a principal ingredient found in peony root (Wu et al. 2010). Animal studies have shown that PF has remarkable effects in alleviating inflammatory pain (Zhang et al. 2009) and cerebral ischemia (Xiao et al. 2005). PF is also effective in improving cognitive impairment (Xiao et al. 2005), depression, and Parkinson’s-like behavior in rats (Liu et al. 2007). Meanwhile, numerous studies have shown robust protective effects of PF against various neurotoxicities (Cao et al. 2010). Nevertheless, the subtle mechanisms underlying neuroprotective effects of PF are not yet fully delineated.
It is believed that traumatic injury and neurotoxin-activated neuronal apoptosis are essential mechanisms involved in the pathogenesis of various neurodegenerative diseases (Yuan and Yankner 2000). Apoptotic neuronal cells, characterized by cytoplasmic atrophy, mitochondrial fission, nuclear shrinkage and fragmentation with condensation of the chromatin, DNA damage, and impairment of DNA repair (Yuan and Yankner 2000), are initially developed from activation of intracellular pro-apoptotic molecules, such as poly (ADP-ribose) polymerase (PARP), which is a key protein involved in the process of apoptotic neuron death (Cheung and Slack 2004), and the regulation of extracellular signal-regulated kinases (ERKs) and Akt/glycogen synthase kinase-3 (GSK-3) is associated with cell survival and proliferation (Cheung and Slack 2004). In addition, down-regulation of B cell lymphoma-extra large (Bcl-xL), an essential mitochondrial transmembrane molecule that protects cells from DNA damage-induced apoptosis, is also an important event resulting in neuronal cell apoptosis (Jonas 2006).
Differentiated PC12 cells have been widely used to study morphological, biochemical, and molecular changes occurring in neuronal cell death (Mills et al. 1995). 1-Methyl-4-phenylpyridine ion (MPP+) is a commonly used neurotoxin to produce neuronal cell apoptosis and neurodegenerative models (Segura Aguilar and Kostrzewa 2004). In the present study, we first determined the effects of PF on cell viability, nuclear and mitochondrial apoptotic alternations, and intracellular level of calcium in differentiated PC12 cells exposed to MPP+. Then, we further examined the effects of PF on the apoptotic protein PARP and anti-apoptotic molecules Bcl-xL, ERKs, and Akt/GSK-3.
Materials and Methods
Cell Culture
Differentiated PC12 cells were used in the experiments. Cells were obtained from ATCC (CRL-1721; passages <10) and grown on a collagen I-coated flask as monolayer cultures in Dulbecco’s modified eagle medium (DMEM) supplemented with 10 % horse serum (HS), 5 % fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 mg/ml), under a humidified atmosphere containing 5 % CO2 and 95 % air at 37 °C. Cells were differentiated by adding 50 ng/ml nerve growth factor (NGF) in the culture medium for 48 h. The culture medium was replaced every 2–3 days. Culture cells were transferred to collagen I-coated 35-mm-diameter 6-well plates when the density reached 70–80 % confluence. Treatment experiments were performed when the density reached 70 % confluence in the plates.
Experimental Design
For PF dose–curve experiment, cells were treated with a range of PF doses from 25 to 400 μM for 3 h and then exposed to 4 mM MPP+ for 24 h. Cell viability was measured (see below) and an optimal dose of 200 μM PF was chosen for further experiments. To detect cellular apoptotic alternations, cells were treated with 200 μM PF for 3 h, followed by exposure to 4 mM MPP+ for 12 h. Cells were then collected and measured by flow cytometry. In a separate experiment, after cells were sequentially administered with 200 μM PF for 3 h and 4 mM MPP+ for 24 h, culture medium was collected for measurement of lactate dehydrogenase (LDH), a biomarker of neuronal cell death (Silva et al. 2006), and cells were collected for measurement of mitochondrial membrane potential (Δψm), a key parameter of mitochondrial permeability used as an indicator of neuronal cell health (Haeberlein 2004) and intracellular calcium analysis. Cells were also collected and prepared for Western blotting analysis of apoptosis-modulating proteins (Bcl-xL and PARP).
To examine the effects of PF alone and cotreatment with MPP+ on ERKs and Akt/GSK-3, cells were treated with 200 μM PF alone and collected at 0, 10, 30, 60, and 180 min. In the cotreatment, cells were treated with 200 μM PF for 3 h and then exposed to MPP+; cells were also collected at 0, 10, 30, 60, and 180 min after exposure to MPP+. Cells collected were prepared for Western blotting analysis. In a separate experiment, cells were pretreated with 20 μM PD98059, an ERK kinase inhibitor (Katsanakis et al. 2002), or 10 μM LY294002, an inhibitor of Akt/GSK-3 pathway (Cho and Park 2008), for 30 min, followed by treatment with 200 μM PF for 3 h and exposure to 4 mM MPP+. Cells were collected at 3 and 24 h after MPP+ exposure for Western blotting analysis and measurement of cell viability, respectively.
Cell Viability Assessment
Cell viability was measured using a quantitative colorimetric assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma-Aldrich, USA) as reported previously (Mosmann 1983). Briefly, cells were seeded into collagen I-coated 96-well plates at a density of 1 × 104 per well. At the completion of the 24-h treatment, 10 μL MTT solution (5 mg/mL) was added. Following incubation for 4 h at 37 °C, 100 μL dimethyl sulfoxide (DMSO) was used to dissolve crystals. The absorbance was measured using a microplate reader (Bio-Rad, USA) and cell viability was expressed as a percentage of control value.
Released LDH Analysis
Released LDH in cultured medium was measured using an enzyme assay kit (In Vitro Toxicology Assay Kit, Sigma-Aldrich) as reported previously (Legrand et al. 1992). 30 μL cultured medium collected was incubated with 60 μL mixed assay solution for 30 min at room temperature. 10 μL HCl (1 N) was added to terminate the reaction. The absorbance was measured using a microplate reader (Bio-Rad, USA) at a wavelength of 540 nm.
Flow Cytometric Analysis of Apoptosis
Cellular apoptotic alterations were detected using flow cytometry with annexin V labeled with fluorescein isothiocyanate (FITC) together with propidium iodide (PI) which stains DNA (Vermes et al. 1995). The density of cells collected was adjusted to 1 × 106/ml. Cells were suspended in binding buffer containing annexin V-FITC (20 μg/mL) and PI (50 μg/mL) and incubated for 10 min at room temperature. The intensity of fluorescence was analyzed using a flow cytometer (FACSCanto II Analyzer, Beckman). Living cells (annexin V−/PI−, Q4), early/primary apoptotic cells (annexin V+/PI−, Q3), late/secondary apoptotic cells (annexin V+/PI+, Q2), and necrotic cells (annexin V−/PI+, Q1) were identified and their percentages were calculated.
Δψm Analysis
The molecular probe JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethyl benzimidazolyl carbocyanine iodide) was used to assess Δψm. JC-1 is a lipophilic, cationic dye that can selectively enter into mitochondria and reversibly change color from green to red as the membrane potential increases (Cossarizza et al. 1993). In healthy cells with high Δψm, JC-1 spontaneously forms complexes known as J-aggregates with intense red fluorescence. On the other hand, in apoptotic or unhealthy cells with low Δψm, JC-1 remains in the monomeric form, which shows only green fluorescence (Cossarizza et al. 1993). Cells collected were incubated with 2 μM JC-1 at 37 °C for 10 min (Sigma-Aldrich, USA). Following three washes with phosphate-buffered saline (PBS), changes in fluorescent color in the mitochondria were examined using a fluorescent microscope (Carl Ziess).
Intracellular Ca2+ Concentration Analysis
The Ca2+-sensitive probe Fluo-4-AM staining (Invitrogen, USA) was used to detect changes in intracellular concentration of Ca2+ as reported previously (Soletti et al. 2010). Cells collected were incubated with Fluo-4-AM at a final concentration of 5 μM for 1 h at 37 °C and then washed three times to remove excess probe. The fluorescence intensity was examined using a fluorescent microscope (Carl Ziess). The average of fluorescence intensity for each cells was calculated with Image J and used to determine intracellular Ca2+ concentration.
Western Blot Assay
Whole cell pellets were lysed by radio-Immunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, USA) containing 1 % protease inhibitor cocktail (Sigma-Aldrich, USA) and 2 % phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich, USA). The supernatant was collected and protein concentration was assessed using the Bradford method. Proteins were separated using a 10 % SDS-PAGE gel and electrophoretically transferred onto nitrocellulose membranes (Bio Basic, Inc.). The transferred membranes were then blotted with rabbit antibodies against rat total ERK1/2 (t-ERK1/2), phospho-ERK1/2 (p-ERK1/2), total Akt (t-Akt), phospho-Akt (p-Akt), total GSK-3β (t-GSK-3β, a subunit of GSK-3), phospho-GSK-3β (p-GSK-3β), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), Bcl-xL, cleaved PARP, and total PARP (Cell signaling, USA) at a concentration of 1:1,000 at 4 °C overnight, followed by co-incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz, USA). Chemiluminescence was detected using ECL detection kits (GE Healthcare, UK). The intensity of the bands was quantified by scanning densitometry using Quantity One 4.5.0 software.
Statistical Analysis
One- or two-way variance analysis (ANOVA) was used to detect statistical significance, followed by post hoc multiple comparisons. Data are expressed as mean ± standard deviation (SD). Statistical significance was defined as P < 0.05.
Results
The Effects of PF Against Cell Damage and Apoptotic Alternations
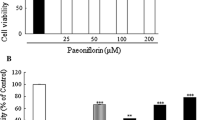
Exposure of cells to 4 mM MPP+ for 24 h resulted in approximately 42 % loss of cells; but, cells pretreated with PF in a range of 50–400 μM had a significant higher cell viability compared to cells treated with MPP+ alone (70–77 vs. 58 %, P < 0.05). The most significant effect was observed at a dose of 200 μM (Fig. 1a). This dose was then applied in further experiments.

Cells were treated with various doses of PF for 3 h, followed by exposure to 4 mM MPP+ for 24 h. Cell viability was examined using the MTT method, a LDH release was measured using an enzyme assay in culture medium collected from cells pretreated with 200 μM PF for 3 h and exposed to 4 mM MPP+ for 24 h, b Cellular apoptotic alterations were examined using flow cytometry in cells pretreated with 200 μM PF and exposed to MPP+ for 12 h, c Data are expressed as mean ± standard deviation (SD) (n = 3–6) and analyzed using one-way ANOVA.# P < 0.05 vs. control (CTRL), *P < 0.05 and **P < 0.01 vs. MPP+-exposed cells
MPP+ treatment also resulted in a 43 % increase of LDH release into cultured medium compared to untreated cells (P < 0.05); but, the addition of 200 μM PF strikingly inhibited the MPP+-induced LDH release (87 vs. 143 %, P < 0.01) (Fig. 1b).
Flow cytometry revealed that there was a significantly higher proportion of apoptotic cells (Q2 + Q3) in MPP+-exposed cells (16.8 %) than that in unexposed cells (6.2 %, P < 0.01), but pretreatment with PF significantly reduced the proportion of apoptotic cells to the level of the unexposed cells (6.1 %, P < 0.01) (Fig. 1c).
The Effects of PF Against Intracellular Ca2+ Overload and Mitochondrial Apoptotic Alteration
While intense red fluorescence indicative of healthy cells was present in untreated cells, a notable number of cells stained with intense green fluorescence indicative of mitochondrial apoptosis were observed in MPP+-exposed cells, but most of green fluorescence-stained cells disappeared when PF was added (Fig. 2).

Mitochondrial membrane potential (Δψm) was determined using the molecular probe JC-1 in cells pretreated with 200 μM PF and exposed to 4 mM MPP+ for 24 h. Red fluorescence indicates healthy cells with high Δψm, whereas green fluorescence indicates apoptotic or unhealthy cells with low Δψm. Scale bar 20 μM (Color figure online)
Exposure to the neurotoxin resulted in a 44 % increase of intracellular Ca2+ load compared to untreated cells (P < 0.05). Pretreatment with PF significantly reduced the MPP+-induced increase of Ca2+ load (112 vs. 144 %, P < 0.05) (Fig. 3).

Intracellular Ca2+ level was quantified using the probe Fluo-4-AM in cells pretreated with 200 μM PF and exposed to 4 mM MPP+ for 24 h. Scale bar 20 μM. Data are expressed as mean ± SD (n = 3) and analyzed using one-way ANOVA.# P < 0.05 vs. control (CTRL), *P < 0.05 vs. MPP+-exposed cells
The Effects of PF on the Expression of Cleaved PARP and Bcl-xL
MPP+ exposure resulted in a 60 % increase of the expression of cleaved PARP and a 21 % decrease of the expression of Bcl-xL compared to unexposed cells (P < 0.05). PF pretreatment markedly suppressed the MPP+-induced increase of the expression of cleaved PARP to 55 % and restored the expression of Bcl-xL to 94 % of untreated cells (P < 0.05) (Fig. 4).

Effects of 3-h pretreatment with 200 μM PF on the expression of cleaved PARP, and Bcl-xL in cells exposed to 4 mM MPP+ for 24 h using Western blotting analysis. Data are expressed as mean ± SD (n = 3) and analyzed using one-way ANOVA, # P < 0.05 vs. control, *P < 0.05 vs. MPP+-exposed cells
The Effects of PF on the Expression of ERKs and AKT/GSK3β
While MPP+ exposure for 30 and 60 min significantly inhibited the expression of p-ERK1/2, but not t-ERK1/2, PF treatment alone enhanced the expression of p-ERK1/2, with a significant effect observed at 30 and 60 min (P < 0.001). PF pretreatment also strikingly reversed the MPP+ suppression of p-ERK1/2 expression at 10 and 30 min (Fig. 5a, b). The blocker PD98059 treatment alone and in combination with PF, MPP+, or both completely inhibited the expression of p-ERK1/2, but did not affect the level of t-ERK1/2 (Fig. 5c).

Effects of PF treatment alone and cotreatment with MPP+ as well as the addition of the ERK blocker PD98059 on the expression of p-ERKs and ERKs using Western blotting analysis. a, b Cells were treated with 200 μM PF alone and collected at 0, 10, 30, 60, and 180 min. In cotreatment, following pretreatment with 200 μM PF for 3 h, cells were collected at 0, 10, 30, 60, and 180 min after MPP+ exposure. c Cells were pretreated with 20 μM PD98059 for 30 min, followed by treatment with 200 μM PF and 4 mM MPP+ for 3 h each. In b, data are expressed as mean ± SD (n = 3) and analyzed using two-way ANOVA, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. control within the same time point
The expression of p-Akt and p-GSK-3β as well as t-Akt and t-GSK-3β was unaltered over time in cells exposed to MPP+ compared to unexposed cells. Both PF alone and plus MPP+ produced a time-dependent increase of the expression of p-Akt and p-GSK-3β, but did not affect the expression of t-Akt and t-GSK-3β (Fig. 6a–d). The significant effect on p-Akt was observed at 10 min through 180 min in PF alone and at 30 min through 180 min in PF plus MPP+ (P < 0.05), whereas the significant effect on p-GSK-3β was observed at 60 and 180 min in PF alone and 30 min through 180 min in PF plus MPP+ (P < 0.05). The addition of blocker LY294002 alone and plus PF, MPP+, or both completely inhibited the expression of p-Akt and p-GSK-3β, but did not change the expression of t-Akt and t-GSK-3β (Fig. 6e).

Effects of PF treatment alone and cotreatment with MPP+ as well as the addition of the Akt/GSK3 blocker LY294002 on the expression of p-Akt, Akt, p-GSK3β, and GSK3β using Western blotting analysis. a–d Cells were treated with 200 μM PF alone and collected at 0, 10, 30, 60, and 180 min. In cotreatment, following pretreatment with 200 μM PF for 3 h, cells were collected at 0, 10, 30, 60, and 180 min after MPP+ exposure. e Cells were pretreated with 10 μM LY294002 for 30 min, followed by treatment with 200 μM PF and 4 mM MPP+ for 3 h each. In b, d, data are expressed as mean ± SD (n = 3) and analyzed using two-way ANOVA, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. control within the same time point
The Effects of the Blockers on PF Protection of Cell Viability
Pretreatment with either PD98059 or LY294002 alone did not affect cell viability compared to untreated cells and cells exposed to MPP+, but significantly diminished the potency of PF in improving viability of cells exposed to MPP+ (P < 0.01) (Fig. 7).

Effects of the addition of the ERK and Akt/GSK3 blockers on the potency of PF in improving cell viability. Cells were pretreated with 20 μM PD98059 a or 10 μM LY294002 b for 30 min, followed by treatment with 200 μM PF for 3 h and exposure to 4 mM MPP+ for 24 h. Cells were then collected for measurement of cell viability using the MTT method. Data are expressed as mean ± SD (n = 6) and analyzed using one-way ANOVA, ## P < 0.01, ### P < 0.001 vs. control (CTRL); **P < 0.01 vs. MPP+; ^^P < 0.01 vs. PF + MPP+
Discussion
Consistent with previous studies (Cao et al. 2010; Wankun et al. 2011), the present study clearly demonstrated that PF possesses robust neuroprotective effects, as evidenced by the fact that PF significantly ameliorated the MPP+-induced decrease of cell viability and cellular apoptotic alternations. The robust protective effect of PF is also reflected in its suppression of the neurotoxin-induced over-release of LDH. Increased release of LDH has been observed in cultured neuronal cells exposed to neurotoxins (Cao et al. 2010) and brain injury (Zandbergen et al. 2001). Therefore, the suppression of LDH release seems to be a consequence of the protective effects of PF against neurotoxicity and neural injury.
Mitochondria play a central part in neurodegeneration and neuronal death (Naoi et al. 2009). A key feature of mitochondrial apoptosis is disruption of the membrane potential mainly caused by increased membrane permeability (Mayer and Oberbauer 2003). Our study revealed that PF protected MPP+-induced mitochondrial apoptotic alternations by improving Δψm, a surrogate for the membrane integrity (Cossarizza et al. 1993), suggesting that PF could maintain mitochondrial membrane integrity by preventing the membrane permeability from neurotoxicity. Our study further revealed that exposure of cells to the neurotoxin resulted in intracellular calcium overflow; pretreatment with PF remarkably blocked the calcium influx. Similar results also have been extensively observed in other in vitro and animal experiments (Cao et al. 2010; Sugaya et al. 1991). It is well known that excessive cytosolic calcium causes a wide range of subcellular pathological responses, in particular dysfunction of mitochondrial membrane permeability (Giorgi et al. 2012). It appears that the effect of PF in maintaining mitochondrial membrane integrity is related to its capacity of regulating cytosolic calcium homeostasis. On the other hand, Bcl-xL is an essential member of the B cell lymphoma-2 (Bcl-2) family that is located on the outer membrane of mitochondria and regulates the mitochondrial membrane permeability (Chan and Yu 2004). The present study showed that the activity of Bcl-xL was significantly down-regulated by exposure to MPP+, but was almost completely restored by pretreatment with PF. Previous studies have shown that PF has remarkable effects in modulating other members of the Bcl-2 family (Li et al. 2007). Therefore, the neuroprotective effects of PF are at least partly attributed to its maintenance of mitochondrial membrane integrity by improving intracellular calcium homeostasis and up-regulating the activity of mitochondria-dependent anti-apoptotic molecules.
PARP is a family of proteins mainly involved in DNA repair and programmed cell death with activation of executory caspases (Piskunova et al. 2008). Our present study found that exposure to the neurotoxin significantly increased the expression of cleaved PARP. Pretreatment with PF strikingly suppressed the increment of cleaved PARP expression. On the other hand, ERKs are key molecules in the mitogen-activated protein kinase (MAPK)-ERK pathways that execute signal transduction from the neuronal surface into the nucleus (Sweatt 2001). Phospho activation of ERKs results in their nuclear translocation and phosphorylation of numerous substrates, promoting proliferation and inhibiting pro-apoptotic signals (Sweatt 2001). In the present study, we demonstrated that both PF treatment alone and cotreatment with MPP+ produced rapid phosphorylation of ERKs within 1 h, whereas MPP+ down-regulated p-ERK levels. However, PF enhancement of the expression of p-ERK was completely abolished in the presence of the ERK blocker PD98059. The presence of PD98059 also eradicated the effect of PF in protecting cell viability from MPP+ toxicity. These results clearly indicate that the protective effects of PF against neurotoxicity are closely associated with its blockade of apoptotic signal transduction from the cytosol to the nucleus by up-regulating activity of MAPK/ERK pathway.
The Akt-GSK-3 pathway is a ubiquitous intracellular signal transduction system diversely involved in regulating cell survival, proliferation, energy metabolism, neurogeneration, and neuroprotection (Liang and Slingerland 2003). Phosphorylated Akt (p-Akt), an activated form, inactivates its major downstream molecule GSK-3 by phosphorylating it, while GSK-3 activation induces apoptosis by disrupting cell cycle and glucose metabolism (Liang and Slingerland 2003). The current study found that while the activity of both p-Akt and p-GSK-3β, a subunit of p-GSK3, was unaltered in exposure to MPP+, PF treatment alone and in combination with MPP+ rapidly and significantly increased p-Akt and p-GSK-3β levels in a time-dependent manner without changing the expression of Akt and GSK-3β. Pretreatment with LY294002, an inhibitor of Akt-GSK-3 pathway, completely eliminated p-Akt and p-GSK-3β activity, but did not affect the expression of t-Akt and t-GSK-3β. The addition of the inhibitor antagonized the effect of PF in improving cell viability. These data indicate that PF is capable of enhancing the activation of Akt and in turn inactivating GSK3.
In summary, the present study demonstrates that the neuroprotective effects of PF are related to its modulation of multiple anti-apoptotic and apoptotic pathways, including blockade of intracellular calcium overload, prevention of mitochondrial membrane integrity, inhibition of activity of apoptotic molecules, and up-regulation of the activity of anti-apoptotic molecules associated with cell survival and proliferation. The study provides evidence supporting PF as a potential therapeutic agent used for the treatment of neurodegenerative diseases and neural injury.
References
Cao BY, Yang YP, Luo WF, Mao CJ, Han R, Sun X, Cheng J, Liu CF (2010) Paeoniflorin, a potent natural compound, protects PC12 cells from MPP+ and acidic damage via autophagic pathway. J Ethnopharmacol 131(1):122–129
Chan SL, Yu VC (2004) Proteins of the bcl-2 family in apoptosis signalling: from mechanistic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol 31(3):119–128
Cheung EC, Slack RS (2004) Emerging role for ERK as a key regulator of neuronal apoptosis. Sci STKE 2004 (251):PE45
Cho JY, Park J (2008) Contribution of natural inhibitors to the understanding of the PI3K/PDK1/PKB pathway in the insulin-mediated intracellular signaling cascade. Int J Mol Sci 9(11):2217–2230
Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C (1993) A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun 197(1):40–45
Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, Wieckowski MR, Pinton P (2012) Mitochondrial Ca(2+) and apoptosis. Cell Calcium 52(1):36–43
Haeberlein SL (2004) Mitochondrial function in apoptotic neuronal cell death. Neurochem Res 29(3):521–530
Jonas E (2006) BCL-xL regulates synaptic plasticity. Mol Interv 6(4):208–222
Katsanakis KD, Owen C, Zoumpourlis V (2002) JNK and ERK signaling pathways in multistage mouse carcinogenesis: studies in the inhibition of signaling cascades as a means to understand their in vivo biological role. Anticancer Res 22(2A):755–759
Legrand C, Bour JM, Jacob C, Capiaumont J, Martial A, Marc A, Wudtke M, Kretzmer G, Demangel C, Duval D et al (1992) Lactate dehydrogenase (LDH) activity of the cultured eukaryotic cells as marker of the number of dead cells in the medium [corrected]. J Biotechnol 25(3):231–243
Li CR, Zhou Z, Zhu D, Sun YN, Dai JM, Wang SQ (2007) Protective effect of paeoniflorin on irradiation-induced cell damage involved in modulation of reactive oxygen species and the mitogen-activated protein kinases. Int J Biochem Cell Biol 39(2):426–438
Liang J, Slingerland JM (2003) Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2(4):339–345
Liu DZ, Zhu J, Jin DZ, Zhang LM, Ji XQ, Ye Y, Tang CP, Zhu XZ (2007) Behavioral recovery following sub-chronic paeoniflorin administration in the striatal 6-OHDA lesion rodent model of Parkinson’s disease. J Ethnopharmacol 112(2):327–332
Mayer B, Oberbauer R (2003) Mitochondrial regulation of apoptosis. News Physiol Sci 18:89–94
Mills JC, Wang S, Erecinska M, Pittman RN (1995) Use of cultured neurons and neuronal cell lines to study morphological, biochemical, and molecular changes occurring in cell death. Methods Cell Biol 46:217–242
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1–2):55–63
Naoi M, Maruyama W, Yi H, Inaba K, Akao Y, Shamoto-Nagai M (2009) Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J Neural Transm 116(11):1371–1381
Piskunova TS, Yurova MN, Ovsyannikov AI, Semenchenko AV, Zabezhinski MA, Popovich IG, Wang ZQ, Anisimov VN (2008) Deficiency in poly(ADP-ribose) polymerase-1 (PARP-1) accelerates aging and spontaneous carcinogenesis in mice. Curr Gerontol Geriatr Res:754190
Segura Aguilar J, Kostrzewa RM (2004) Neurotoxins and neurotoxic species implicated in neurodegeneration. Neurotox Res 6(7–8):615–630
Silva RF, Falcao AS, Fernandes A, Gordo AC, Brito MA, Brites D (2006) Dissociated primary nerve cell cultures as models for assessment of neurotoxicity. Toxicol Lett 163(1):1–9
Soletti RC, del Barrio L, Daffre S, Miranda A, Borges HL, Moura-Neto V, Lopez MG, Gabilan NH (2010) Peptide gomesin triggers cell death through L-type channel calcium influx, MAPK/ERK, PKC and PI3K signaling and generation of reactive oxygen species. Chem Biol Interact 186(2):135–143
Sugaya A, Suzuki T, Sugaya E, Yuyama N, Yasuda K, Tsuda T (1991) Inhibitory effect of peony root extract on pentylenetetrazol-induced EEG power spectrum changes and extracellular calcium concentration changes in rat cerebral cortex. J Ethnopharmacol 33(1–2):159–167
Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76(1):1–10
Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C (1995) A novel assay for apoptosis. flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods 184(1):39–51
Wankun X, Wenzhen Y, Min Z, Weiyan Z, Huan C, Wei D, Lvzhen H, Xu Y, Xiaoxin L (2011) Protective effect of paeoniflorin against oxidative stress in human retinal pigment epithelium in vitro. Mol Vis 17:3512–3522
Wu CF (1985) A review on the pharmacology of Paeonia lactiflora and its chemical components. Zhong Yao Tong Bao 10(6):43–45
Wu SH, Wu DG, Chen YW (2010) Chemical constituents and bioactivities of plants from the genus Paeonia. Chem Biodivers 7(1):90–104
Xiao L, Wang YZ, Liu J, Luo XT, Ye Y, Zhu XZ (2005) Effects of paeoniflorin on the cerebral infarction, behavioral and cognitive impairments at the chronic stage of transient middle cerebral artery occlusion in rats. Life Sci 78(4):413–420
Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407(6805):802–809
Zandbergen EG, de Haan RJ, Hijdra A (2001) Systematic review of prediction of poor outcome in anoxic-ischaemic coma with biochemical markers of brain damage. Intensive Care Med 27(10):1661–1667
Zhang XJ, Chen HL, Li Z, Zhang HQ, Xu HX, Sung JJ, Bian ZX (2009) Analgesic effect of paeoniflorin in rats with neonatal maternal separation-induced visceral hyperalgesia is mediated through adenosine A(1) receptor by inhibiting the extracellular signal-regulated protein kinase (ERK) pathway. Pharmacol Biochem Behav 94(1):88–97
Acknowledgments
This study was supported by the Health and Health Service Research Fund (HHSRF) (09101141) and the HKU intramural seed funds (201011159095, 201102160005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, D., Wong, H.K., Feng, YB. et al. Paeoniflorin, a Natural Neuroprotective Agent, Modulates Multiple Anti-Apoptotic and Pro-apoptotic Pathways in Differentiated PC12 Cells. Cell Mol Neurobiol 33, 521–529 (2013). https://doi.org/10.1007/s10571-013-9914-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-013-9914-y