
Abstract
Tanycytic ependymoma is a rare subtybe of ependymoma with a predilection for the spinal cord and intracranial tanycytic ependymoma is thus extremely rare. Most studies on intracranial tanycytic ependymomas included only one or two cases. Here we report nine patients with pathologically confirmed intracranial tanycytic ependymomas. The clinical characteristics, including radiological and histological examination, operative records, and prognoses were reviewed. The case series included six male and three female patients with an average age of 19.3 years. Tumors were located in the lateral ventricle (3/9), the fourth ventricle (2/9), and the supratentorial extraventricle (4/9). Gross total resection (GTR) of the tumor was achieves in seven cases, and subtotal resection (STR) was achieved in the other two cases. One patient died 21 months after discharge. The left eight patients showed improved symptoms after surgery, and no tumor recurrence was found in these cases during the follow-up. It seems that intracranial tanycytic ependymoma has the best long-term prognosis compared to the other two subtypes of ependymoma. According to our experience, we recommend surgery including GTR and STR followed by radiotherapy for patients with intracranial tanycytic ependymomas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The 2016 World Organization Classification (WHO) of tumors of the central nervous system (CNS) grades ependymal tumor as subependymoma (grade I), myxopapillary ependymoma (grade I), ependymoma (grade II), ependymoma with RELA fusion–positive (grade II or III) and anaplastic ependymoma (grade III) [1]. WHO grade II ependymomas are further divided into three major subtypes: papillary, clear cell, and tanycytic ependymoma (TE) [1].
Ependymomas are slow-growing tumors, accounting for 3–7% of all brain tumors and 3–9% of all neuroepithelial tumors [2, 3]. TE is the rarest variant of ependymal tumor [4]. In 1978, Friede and Pollak described the pathologic features of TE for the first time [5]. TE exists predominantly in the spinal cord [4]. Therefore, intracranial TEs are extremely rare and merely 20 cases have been reported up to date in the English literature (Table 1) [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. Unfortunately, these studies had too few cases to show general characteristics of intracranial TEs. Here, we report a series of nine patients with histologically confirmed intracranial TEs and their long-term outcomes in a single center. To the best of our knowledge, this study is the largest case series of intracranial TE. We believe the clinical, radiological and pathological features of intracranial TE we showed here aim for better differential diagnosis in future.
Materials and methods
The study included nine consecutive cases of intracranial TE that were pathologically diagnosed in Beijing Tian Tan Hospital between August 2011 and May 2016 (Table 2). The first intracranial TE patient just admitted to our hospital at August 08, 2011. All patients had their primary tumor surgically resected in our centre.
The relevant clinical data (including presentation, radiological imaging, treatment, and follow-up outcomes) were collected through a chart review and telephone interviews with an approval of the institutional review board. The diagnosis of TE was pathologically confirmed by the Department of Neuropathology at Beijing Neurosurgical Institute using the 2016 WHO classification of tumors of the central nervous system.
The estimation of the extent of tumor removal was acquired from the operation records and postoperative magnetic resonance imaging (MRI). Gross total resection (GTR) and subtotal resection (STR) were defined as total and subtotal macroscopic removal, respectively. The Karnofsky Performance Scale (KPS) was applied to assess the neurological function of the patients. These assessments were performed before surgery, at discharge, and at the last follow-up.
Results
The clinic characteristics and treatment of the nine patients (including five children) of intracranial TE were detailed in Table 2. The average age at the time of diagnosis was 19.3 years (range from 8 to 39 years). The sex ratio (male: female) was 2:1. The durations of symptoms ranged from 10 days to 3 years. The initial symptom was related to the tumor-occupying effect and location, with headache (4/9) and nausea (3/9) being the most common symptoms. The preoperative KPS scores were 90 for all the patients.
Radiological features
The tumors were located in the lateral ventricle (3/9), fourth ventricle (2/9), frontal lobe (3/9), and parieto-occipital (1/9). T1-weighted imaging of MRI revealed hypointense signal in six patients, iso- to hypo-intense signal in two patients and isointense signal in one patient. T2-weighted imaging showed hyperintense signal in six cases, iso- to hyper-intense in two cases and isointense signal in one case. On contrast-enhanced T1-weighted images, three cases showed markedly heterogeneous enhancement, four cases showed moderately heterogeneous enhancement, one case showed mild heterogeneous enhancement, and the last one had markedly homogeneous enhancement. Unfortunately, only three cases (33.3%) were diagnosed as ependymoma based on the preoperative MRI. The other six patients were misdiagnosed with glioma (4/6), subependymoma (1/6) or hemangioblastoma (1/6) (Fig. 1).

Case 3: Preoperative MRI revealed a cystosolid lesion, which was misdiagnosed as astrocytoma. Plain T1WI (a, b) and enhanced T1WI signals (c–e) showing a lesion in the left frontal and basal ganglia. Postoperative CT and MRI (f–j) revealed gross total resection of the tumor. Pathological examination (k–o) revealed a spindle cell neoplasm of moderate cellularity arranged in interlacing fascicles. The tumor cells had discreetly elongated bipolar nuclei (k). The immunohistochemical examinations showed that the tumor cells were strongly positive for GFAP (l), EMA (m) and Nestin (n), and 3% of cells are positive for Ki-67 (o). The pathological diagnosis was consistent with TE. Original magnification in (k–o) is ×200
Intraoperative findings
All tumors were removed through craniotomy under operative microscope. Intraoperatively, the lesions typically appeared grayish red, smooth-surfaced, mildly vascular, and rubbery. Most of the masses were well circumscribed from the brain tissues, which made it facility for operators to expose and dissect. GTR and STR were achieved in seven and two patients, respectively. Failure of GTR occurred in two tumors located in the fourth ventricle.
Pathological examination
Microscopically, all tumors showed typical histological indications of TE according to the WHO classification. Tanycytes (spindle cells with oval and elongated nuclei) were observed by hematoxylin and eosin staining in all nine cases. Immunohistochemistry showed expression of glial fibrillary acidic protein (GFAP), epithelial membrane antigen (EMA), and Nestin in tumor cells. The Ki-67 positive cells in all the lesions were less than 5%, suggesting a low proliferation capacity. Ultrastructural examination demonstrated sarciniform spindle cells with deficient extracellular matrix. Abundant slim surface microvilli, and microvilli-lined lumina were discovered at the intercellular junctions (Fig. 2).

Case 7: Preoperative CT (a, b) showing a partly calcified mass in the right lateral ventricle. MRI showing hypointense T1 (c) and isointense T2 (d) signals as well as heterogeneous enhancement (e–g). Postoperative MRI (h–j) revealing gross total resection of the tumor. Pathological examination revealed the diagnosis of TE (k) with positivity for GFAP (l), EMA (m) and Nestin (n), and 2% of cells are positive for Ki-67 (o). Electron micrograph showing intercellular junctions and numerous slender surface microvilli (p). Original magnification in (k–o) is ×200, p is ×6000
Long-term outcomes
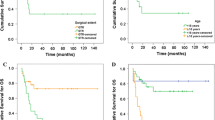
All patients were still alive except for one who died 21 months after the first surgery. The deceased underwent second surgery 16 months after the first one in our hospital. The follow-up time for the patients who were still alive was 8–65 months, with an average of 33.0 months. At present, all the eight patients have recovered, and their postoperative KPS scores were all reached 100. They live a normal life, including working or studying normally. Currently, there is no evidence of tumor recurrence according to the last MRI. None of the eight patients underwent second surgery and seven of them underwent further adjuvant therapy. External radiation therapy was administered to six of them. Also, one patient underwent the proton therapy in Germany. We have drawn a survival curve to compare our nine patients with previous reports (available data from Table 1) (Fig. 3). Our study includes complete patients follow-up data, with longer follow-up duration than previous reported. There was no significant difference in overall survival rate between the two groups, indicating favorable prognosis of intracranial TE.

The survival-curve for our series and previous reports
Discussion
Epidemiology and clinical features
The Neurosurgery Department of Beijing Tian Tan Hospital performs brain tumor surgeries to about 7000 patients every year, among which, there are generally 300 cases of relapse. Hence, approximately 60,000 patients with primary brain tumors had their first surgery in Beijing Tian Tan Hospital from January 2008 to January 2017. We had reviewed the records of Beijing Tian Tan Hospital and identified nine patients with pathologically confirmed intracranial TE among a total of 1787 ependymoma patients. The proportion of intracranial TEs among ependymoma in our hospital was only 5‰. Intracranial TE was so rare that only 29 cases (including the current study) were reported. The ages of these patients ranged from 2 to 59 years (median, 30.5 years) when diagnosed. Similar to ependymal tumors, which predominantly affect children and young adults, intracranial TE also happened to young people [21]. Actually, 15 of the 29 cases were diagnosed below 25 years old. The mean ages at the time of the diagnosis in male and female patients were 27.4 and 29.5 years, respectively. Obvious gender difference was found, with male patients seemed more likely to be affected (male:female = 3.8:1). However, in our case series, the mean age at diagnosis was 19.3 years and the sex ratio (male:female) was 2:1. This may because the difference of race or due to relatively low number of cases included.
Locations varied among the cases, companied with corresponding symptoms in these patients. The most frequent affected location was supratentorial extraventricle (51.7%, 15/29), followed by intraventricle (41.1%, 12/29) and cerebellopontine angle (6.9%, 2/29). Similar to other intracranial tumors, the clinical symptoms of intracranial TE included headache, nausea, and vomiting, usually due to increased intracranial pressure. Particularly, headache and seizure were the most common initial symptoms. In addition, TE was occasionally found in supratentorial extraventricle locations and can present with seizures [15]. It was noteworthy that 10/15 (66.7%) patients with supratentorial extraventricle TEs suffered from seizures but only 1/12 (8.3%) patients with intraventricle TEs had seizures.
Pathogenesis
TE is a sort of WHO grade II tumor with a relatively better prognosis than other grade II ependymomas [4, 11, 16, 22]. The diagnosis and treatment of intracranial TE is critical to achieve a satisfying prognosis. The cell of origin for this neoplasm is still elusive. However, recent evidence shows that they may originate from radial glias such as the tanycytes of ependyma and the initial cells of neurogliocytes [4, 20]. The tanycyte is implicated in two CNS tumors, namely, tanycytic ependymoma and astroblastoma [3]. Tanycytes are elongated spindly bipolar cells which generally present in the circumventricular organs especially in the third ventricle and central canal of spinal cord [18].
The electron microscopy shows ultrastructural features of TE including distinct intercellular junctions, abundant slender microvilli, and microvilli-lined lumina [4]. Ependymomas normally have strong GFAP reactivity and variable staining for S-100, vimentin and EMA [3]. Similarly, intracranial TEs are also positive for GFAP in most cases, including ours. However, the expression of Ki-67 in intracranial TE is usually lower than that of the other subtypes of WHO grade II ependymomas, and the Ki-67 index can be as low as 1% [16]. Our results of immunochemistry were consistent with the previous reports.
Radiological features and diagnosis
MRI plays important role in differential diagnosis and surgical plan of CNS tumors. In most of our cases showed hypointense to isointense on T1WI and isointense to hyperintense on T2WI, and contrast MRI sequences show moderately to markedly heterogeneous enhancement. Furthermore, boundaries of those lesions are relatively clear. There were a variant of diagnoses suggested by the imaging features, but these were not confirmed by the pathologic diagnosis. As intracranial TE is associated with no specific imaging features, the confirmative diagnosis depends on pathologic characteristics. The electron microscopy can contribute to diagnosis and differential diagnosis. Several cases were diagnosed as astrocytoma or subependymoma or hemangioblastoma by preoperative imaging examination, which, however, were pathologically confirmed as misdiagnosis. In pathological diagnosis, it is essential for pathologist to improve the understanding of the histological features of this tumor.
Treatments and outcomes
Currently, complete surgical resection remains the mainstay of therapy for patients with intracranial TE [23, 24]. GTR should always be attempted with microsurgical techniques if a diagnosis of ependymoma is suggested by intraoperative pathology. Most of the tumors did not adhere to the brain tissue, and GTR was feasible with delicate microsurgical techniques. However, it can be difficult to remove the entire tumor if it is closely adherent to brain tissue or is rich in vascular supply. Since tanycytic ependymoma is benign histologically and generally well demarcated, a good prognosis can be anticipated after complete removal. In reviewing the therapeutic strategy for intracranial TE, it was noted that most scholars are supportive of adjuvant radiotherapy following surgical resection, as this is expected to reduce the recurrent rate. Some other scholars argue that radiotherapy may not be mandatory following complete resection, especially for younger patients, and that adjuvant radiotherapy may be given in case of incomplete resection or recurrence [22,23,24]. What we recommend is to give postoperative radiotherapy regardless of whether it is a complete or incomplete resection, as this might be favorable for patient prognosis.
The prognosis of TE is related to the age of patient, the location of tumor and the extent of surgical resection. A high Ki-67 index is also associated with recurrence but is not closely related to outcomes [4]. Comparing to other subtypes of ependymomas, intracranial tumors have a low Ki-67 index [4], suggesting a better prognosis. Indeed, it has been reported that TEs have a higher 5- and 10-year survival rate than other subtypes of ependymoma [4, 11].
Conclusions
The current case series and literature review suggest that intracranial TE should be taken into consideration when intracranial lesions are detected in young patients, especially when the tumors show enhancement on MRI. TE is the rarest variant of ependymoma and its diagnosis largely depends on histopathological examination. Neurosurgeons should be aware of this lesion entity. Fortunately, intracranial TE usually has a favorable prognosis, and complete surgical resection is expected whenever possible. After surgery, follow-up imaging examination is helpful for monitor.
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. doi:10.1007/s00401-016-1545-1
Lopez G, McLendon RE, Peters KB (2015) Supratentorial tanycytic ependymoma in an adult male: case report and review of literature. Case Rep Oncol 8:159–163. doi:10.1159/000380906
Ragel BT, Townsend JJ, Arthur AS, Couldwell WT (2005) Intraventricular tanycytic ependymoma: case report and review of the literature. J Neurooncol 71:189–193. doi:10.1007/s11060-004-1371-5
Agarwal S, Stevenson ME, Sughrue ME, Wartchow EP, Mierau GW, Fung KM (2014) Features of intraventricular tanycytic ependymoma: report of a case and review of literature. Int J Clin Exp Pathol 7:3399–3407
Friede RL, Pollak A (1978) The cytogenetic basis for classifying ependymomas. J Neuropathol Exp Neurol 37:103–118
Langford LA, Barre GM (1997) Tanycytic ependymoma. Ultrastruct Pathol 21:135–142
Daneyemez M, Can C, Izci Y, Beduk A, Timuckaynak E (1999) The tanycytic ependymoma of the lateral ventricle: case report. Minim Invasive Neurosurg 42:201–203. doi:10.1055/s-2008-1053399
Hayashi S, Kameyama S, Fukuda M, Takahashi H (2000) Ganglioglioma with a tanycytic ependymoma as the glial component. Acta Neuropathol 99:310–316
Richards AL, Rosenfeld JV, Gonzales MF, Ashley D, Mc Lean C (2004) Supratentorial tanycytic ependymoma. J Clin Neurosci 11:928–930. doi:10.1016/j.jocn.2004.03.022
Ito T, Ozaki Y, Nakagawara J, Nakamura H, Tanaka S, Nagashima K (2005) A case of cervicomedullary junction tanycytic ependymoma associated with marked cyst formation. Brain Tumor Pathol 22:29–33. doi:10.1007/s10014-005-0174-5
Zhang S, Wang X, Zhang Z, Chen Y (2008) Tanycytic ependymoma arising from the right lateral ventricle: a case report and review of the literature. Neuropathology 28:427–432. doi:10.1111/j.1440-1789.2007.00857.x
Du J, Zhou X-j, Tang Q-q, Ma H-h, Zhou H-b, Wang J-d, Lu Z-f, Yin H-l (2009) Tanycytic ependymoma: two case reports and review of the literature. Comp Clin Pathol 18:449–453. doi:10.1007/s00580-008-0780-9
Reis F, Schwingel R, de Morais FC, de Souza Queiroz L (2011) Supratentorial tanycytic ependymoma: an uncommon fibrillary ependymoma variant. Arq Neuropsiquiatr 69:723
Arvanitis LD, Gattuso P, Nag S (2013) A 40-year-old male with an intraventricular tumor. Combined tanycytic ependymoma and subependymoma. Brain Pathol 23:359–360. doi:10.1111/bpa.12054
Rigante L, Novello M, Massimi L, Caldarelli M (2013) A cortical cystic epileptogenic lesion: tanycytic ependymoma. Acta Neurol Belg 113:523–525. doi:10.1007/s13760-012-0157-3
Kambe A, Kurosaki M, Watanabe T, Nakazato Y (2014) Pediatric supratentorial cortical tanycytic ependymoma associated with absence seizures. Clin Neuropathol 33:308–310. doi:10.5414/NP300726
Shukla S, Malhotra KP, Awasthi NP, Husain N, Singh SK (2014) intraventricular tanycytic ependymoma: an uncommon fibrillary variant. Neurol India 62:200–201. doi:10.4103/0028-3886.132401
Liu Z, Li J, Liu Z, Wang Q, Famer P, Mehta A, Chalif D, Wang Y, Li JY (2014) Supratentorial cortical ependymoma: case series and review of the literature. Neuropathology 34:243–252. doi:10.1111/neup.12087
Kuga Y, Ohnishi H, Kodama Y, Takakura S, Hayashi M, Yagi R, Fukutome K, Matsushima K, Okamoto K, Taomoto K, Takahashi H (2014) Cerebral and spinal cord tanycytic ependymomas in a young adult with a mutation in the NF2 gene. Neuropathology 34:406–413. doi:10.1111/neup.12109
Divito A, Keller JT, Hagen M, Zuccarello M (2014) Vestibular schwannoma or tanycytic ependymoma: immunohistologic staining reveals. Surg Neurol Int 5:158. doi:10.4103/2152-7806.144595
Massimino M, Buttarelli FR, Antonelli M, Gandola L, Modena P, Giangaspero F (2009) Intracranial ependymoma: factors affecting outcome. Future Oncol 5:207–216. doi:10.2217/14796694.5.2.207
Krisht KM, Schmidt MH (2013) Tanycytic ependymoma: a challenging histological diagnosis. Case Rep Neurol Med 2013:170791. doi:10.1155/2013/170791
Cepeda S, Hernandez-Lain A, Munarriz PM, Martinez Gonzalez MA, Lagares A (2014) Spinal tanycytic ependymoma associated with neurofibromatosis type 2. Clin Neuropathol 33:311–314. doi:10.5414/NP300704
Radhakrishnan N, Nair NS, Hingwala DR, Kapilamoorthy TR, Radhakrishnan VV (2012) Tanycytic ependymoma of filum terminale: a case report. Clin Neurol Neurosurg 114:169–171. doi:10.1016/j.clineuro.2011.09.017
Acknowledgements
The authors thank all the patients who trusted them and all the physicians and staff who helped them in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflict of interest concerning the materials or methods used in this study or the findings described in this paper. No benefits in any form have been or will be received from any commercial party related directly or indirectly to the subject of this manuscript.
Ethical approval
This retrospective study was approved by Institutional Review Board of Beijing Tian Tan Hospital, Capital Medical University.
Rights and permissions
About this article

Cite this article
Tao, X., Dong, J., Hou, Z. et al. The clinical features and surgical outcomes of intracranial tanycytic ependymomas: a single-institutional experience. J Neurooncol 134, 339–347 (2017). https://doi.org/10.1007/s11060-017-2531-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-017-2531-8