
Abstract
Glioblastoma (GBM) is the most common and aggressive primary brain tumor in adults with average disease relapse at 9 months and median survival rarely extending beyond 15 months. Brain tumor stem cells (BTSCs) have been implicated in not only initiating GBM but also conferring resistance to therapy. However, it is not clear whether the BTSC population that initiates tumor growth is also responsible for GBM recurrence. In this study, we have developed a novel in vitro treatment model to profile the evolution of primary treatment-naïve GBM BTSCs through chemoradiotherapy. We report that our in vitro model enriched for a CD15+/CD133− BTSC population, mirroring the phenotype of BTSCs in recurrent GBM. We also show that in vitro treatment increased stem cell gene expression as well as self-renewal capacity of primary GBMs. In addition, the chemoradiotherapy-refractory gene signature obtained from gene expression profiling identified a hyper-aggressive subtype of glioma. The delivery of in vitro chemoradiotherapy to primary GBM BTSCs models several aspects of recurrent GBM biology, and could be used as a discovery and drug-screening platform to uncover new biological drivers and therapeutic targets in GBM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM), the most common primary brain tumor in adults, is a highly aggressive astrocytic tumor (WHO grade IV), with uniformly fatal prognosis [1]. GBM is pathologically characterized by nuclear pleomorphism, microvascular proliferation and necrosis, and displays great inter-tumoral cellular heterogeneity [2–4]. Despite multimodal therapy, consisting of surgical resection followed by chemo and radiotherapy, patients typically experience tumor relapse at 9 months and median survival remains around 15 months [5]. At the genetic level, this heterogeneity has been classified into molecular subgroups, based on differential transcriptome profiling of hundreds of GBMs by the TCGA [1, 6]. At the cellular level, this heterogeneity can be explained by the existence of multiple cellular subpopulations of cancer cells that have acquired stem cell properties, variably labeled in the literature as brain tumor stem cells (BTSCs) or glioblastoma initiating cells (GICs) [7, 8]. Since cell surface markers allow sorting of bulk GBM into cellular subpopulations, much research has focused on the application of proteins such as CD133 [8], CD15 [9], integrin alpha6 [10] and L1CAM [11] to define functional BTSC subgroups. In addition, intracellular proteins such as RNA binding protein Musashi-1 [12], transcription factor Sox2 [13] and polycomb repressor Bmi1 [14] that have a characterized functional role in driving normal neural stem cell (NSC) self-renewal, have also been investigated as putative BTSC markers [15, 16]. Although recent data suggests that CD133+ GBM BTSCs are chemo- [17] and radioresistant [18], no study has prospectively identified whether such BTSCs are causal of tumor relapse, and whether the same BTSC populations that drive tumor initiation also drive recurrence.
In vitro functional assays of BTSC self-renewal have been shown to correlate with patient survival [19] and are used to study resistance of GBM BTSCs to temozolomide (TMZ), the chemotherapeutic used to treat GBM, or to radiation [18, 20]. However, the combinatorial effect of TMZ and radiation on BTSC populations in GBM has not been clearly studied and whether this leads to selection of subclonal population from which recurrence may arise remains to be explored.
In this study, we have developed a novel in vitro BTSC model of GBM recurrence to profile the evolution of BTSC populations through therapy. We investigated the effect of in vitro chemoradiotherapy on primary human GBMs (P-GBM) harvested from patients at initial diagnosis, identifying and characterizing the treatment-refractory BTSC population. We then compared it to the BTSC profile of clinically-treated recurrent human GBM specimens (R-GBM). By comparing in vitro treated P-GBM to R-GBM patient samples, we could (1) determine if our in vitro treatment protocol resulted in similar clonal evolution as seen in patients, and (2) establish a treatment-naïve versus post-treatment GBM differential BTSC profile through the application of stem cell assays and marker expression. Lastly, a global gene expression analysis comparing P-GBMs to corresponding in vitro generated treatment-refractory BTSCs identified signaling networks underlying therapy resistance.
Materials and methods
Dissociation and culture of GBM tissue
Human GBM brain tumors were obtained from consenting patients, as approved by the Hamilton Health Sciences/McMaster Health Sciences Research Ethics Board. Brief clinico-pathological information of each patient is included in Fig. 1a. Tumors were dissociated and cells resuspended in complete NSC medium as previously published [7, 8].

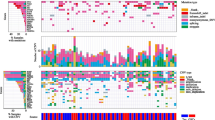
Inter-tumoral heterogeneity exists in BMI1 and SOX2 expression as well CD15 and CD133 cell surface expression in P-GBMs. a GBM patient demographics. RT: radiation, 60 Gy. TMZ: temozolomide. → adjuvant temozolomide. b BMI1 and c SOX2 gene expression level was determined in three different P-GBM samples (n = 2 for BT428, BT458 and BT465) with GAPDH expression as control. d Level of CD15 and CD133 cell surface protein expression was determined using flow cytometry in three P-GBM samples. The bar graph adjacent to the flow plots shows percent of CD15+ and CD133+ cells individually for each P-GBM sample. e Self-renewal capacity was determined using sphere formation assay with no significant difference in self-renewal between the three P-GBMs (n = 3, p = 0.5622). The bars represent mean ± sem
Real-time quantitative PCR
Total RNA was isolated using NorgenTotal RNA Purification kit. cDNA was synthesized by qScript cDNAsupermix (Quanta Biosciences) followed by real-time quantitative PCR using SsoAdvanced™ Universal SYBR®GreenSupermix (Bio-Rad). Samples were quantified using CFX Manager™ software. Data were presented as the ratio of the gene of interest to GAPDH. Primer sequences used for each gene are provided in Supp. Table S1.
Flow cytometry analysis
The percentage expression of CD133 and CD15 was determined on a MoFlo XDP flow cytometer (Beckman Coulter) using anti-CD133-APC (MiltenyiBiotec), anti-CD15-PE (Beckman Coulter) and matched isotype controls. Data was analyzed with Kaluza® Flow Analysis software.
Self-renewal assay
Once primary sphere formation was noted, spheres were dissociated to single cells and 200 cells/well were re-plated in 0.2 mL of complete NSC media in a 96 well plate as previously published [8]. The low cell density prevents cell aggregates from forming and allows for the formation of a sphere from a single cell. The spheres were counted after 7 days of incubation.
In vitro treatment
Single cells were plated at a density of 2 × 106 cells per mL of complete NSC media. The in vitro chemoradiotherapy protocol had three treatment groups. The radiation-only group received 1 Gy per day for five consecutive days. The combined chemoradiotherapy group Tx1 received 5 days of TMZ (Sigma-Aldrich) at 25 μM concurrently with 1 Gy per day of radiation and Tx2 received an additional 5 days of TMZ at 50 μM. The concentration of TMZ used in the study is adapted from clinically relevant doses received by GBM patients [21, 22]. Cells were treated with TMZ for 1 h, after which media was replaced with fresh complete NSC media and cells were immediately exposed to X-rays for a total dose of 1 Gy (Faxitron RX-650). Control cells received corresponding concentrations of DMSO for the same time periods. 1 week post-treatment, cells were analyzed using flow cytometry and stem cell assays, and RNA was extracted for RT-PCR and microarray analysis.
Illumina bead chip analysis
Briefly, 200 ng of RNA isolated using Qiagen RNeasy Micro Kit from the treated samples were labeled using the Illumina TotalPrep-96 RNA Amplification kit (Ambion). 750 ng cRNA was hybridized onto Human HT-12 V4 beadchips. BeadChips were stained as per Illumina protocol and scanned on the iScan (Illumina). Raw.IDAT files pre-processed using cubic spline normalization.
REMBRANDT data analysis
Publically available GBM sample data was downloaded from the Repository for Brain Neoplasia Data (REMBRANDT). The samples were clustered based on Control/Tx2 variable genes that were common to at least 2 of 3 GBM lines using non-negative matrix factorization (NMF) [23]. Survival analysis was completed in R and survival curves were graphed using Graphpad Prism 5.
Network analysis
Common Control/Tx2 variable genes to at least 2 of 3 GBM lines were mapped as genes onto nodes of the REACTOME functional interaction network [24, 25]. Markov clustering was used to subset the network and identify modules of interacting genes. Subsequently modules were annotated with significantly enriched pathways. All network analyses was carried out using Cytoscape (v2.8.2) and the Reactome FIs plugin (v2012).
Statistical analysis
All quantitative data presented are the mean ± SEM. Samples used and respective n values are listed in the figure legends. The level of significance was determined by Student’s two-tailed t test or ANOVA using GraphPad Prism 5 software.
Results
Inter-tumoral heterogeneity in stem marker expression in P-GBMs
We determined the gene and surface protein expression levels of known BTSC markers using RT-PCR and flow cytometry respectively, (Fig. 1b–d) in three P-GBM samples: BTs 428, 458 and 465. All P-GBMs express BMI1 (Fig. 1b) and SOX2 (Fig. 1c), genes known to play a role in maintaining GBM self-renewal. However, the expression was highly variable across individual patient P-GBMs with BT428 and BT458 expressing 4–5 fold higher levels of BMI1 and up to 10 times lower level of SOX2 as compared to BT465. Similarly, the cell surface expression of CD15 and CD133 varied considerably between P-GBM samples as BT428 and BT458 had higher numbers of CD133+ and CD15+ cells than BT465 (Fig. 1d). This variability was not unexpected considering the high inter-tumoral heterogeneity that exists in GBM [1]. Despite variable stem cell gene and protein expression levels, we found no significant difference in secondary sphere formation capacity between the three P-GBMs (Fig. 1e), illustrating the significance of functionally quantifying self-renewal capacity in addition to characterizing stem cell gene and protein expression. We also determined the MGMT promoter methylation status of our P-GBMs (Supp. Fig. S1). Both BT428 and BT465 harbor unmethylated MGMT promoter while BT458 has a partially methylated MGMT promoter. Based on the expression of 21 subtype specific genes as described in Verhaak et al. 2010 [1] we subtyped the GBM samples used in this study (Supp. Table S3) and found that BT428 and BT465 belonged to Proneural subtype while BT458 belonged to Classical subtype.
In vitro chemoradiotherapy increases the expression of genes potentially driving self-renewal
In order to study the effect of in vitro chemoradiotherapy on BMI1 and SOX2 expression, we designed an in vitro protocol combining TMZ and radiation treatment (Fig. 2a). The protocol was divided into three treatment groups: Rad, Tx1 and Tx2 (Fig. 2b). We found that in vitro chemoradiotherapy significantly increased the expression of BMI1 in both Tx1 and Tx2 group in all-P-GBMs tested by 2–3 fold except in BT465 Tx1 (Fig. 2c). Similarly, in vitro chemoradiotherapy significantly increased SOX2 expression in both Tx1 and Tx2 treatment groups in all P-GBMs (Fig. 2d). In contrast, radiotherapy alone induced variable changes in BMI1 and SOX2 expression across the P-GBM samples (Supp. Fig. S2) validating that combined in vitro chemoradiotherapy provides a better model to study therapy resistance in GBM.

In vitro chemoradiotherapy increases the expression of BMI1 and SOX2 in P-GBMs. a Timeline of the in vitro chemoradiotherapy protocol. Red arrows show the time frame during which radiation and TMZ was delivered to cells in vitro. b Four treatment groups were studied, labeled as control, Rad, Tx1 and Tx2. The treatment received by each group is described in the table. c BMI1 expression level was determined in Tx1 and Tx2 groups and compared to control samples (n = 3). Both Tx1 and Tx2 show significantly higher expression of BMI1 except in BT465 Tx1 group (BT428, p < 0.05; BT458, p < 0.0001; BT465 Tx2, p < 0.05). d SOX2 expression level was determined in Tx1 and Tx2 groups and compared to control samples (n = 3). Both Tx1 and Tx2 show significantly higher expression of SOX2 in all P-GBMs (BT428, p < 0.05; BT458, p < 0.0001; BT465 Tx2, p < 0.05). The bars represent mean ± sem. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001
In vitro chemoradiotherapy enriches for the CD15+/CD133− cell population, mirroring the BTSC marker profile of R-GBMs
In addition to changes in BMI1 and SOX2 gene expression, we wanted to study the evolution of the CD15/CD133 flow profile of P-GBMs over the course of in vitro chemoradiotherapy (Fig. 3a). Radiation alone increased the percentage of CD133+ cell population in all three P-GBMs, supporting previous studies identifying CD133+ BTSCs as radioresistant [18]. In both BT428 and BT458, the increase in the CD133+ cell population following in vitro chemoradiotherapy was characteristically restricted to the CD15−/CD133+ subpopulation. Only BT465 showed an increase in CD15+ cell population in response to radiation alone.

In vitro chemoradiotherapy enriches for CD15+/CD133− cell population in P-GBM and increases secondary sphere formation capacity, a profile similar to that of R-GBMs. a Flow cytometry analysis was done on P-GBMs through the course of the in vitro chemoradiotherapy for all treatment groups. Rad group is enriched for CD133+/CD15− cell population in both BT428 and BT458, while both Tx1 and Tx2 of all P-GBMs have enriched CD15+ cell population as compared to controls. The bar graphs adjacent to the flow plots show the percent of CD15+ and CD133+ cells individually for each P-GBM. b Flow cytometry analysis of R-GBM for CD15 and CD133 levels show a CD15 high and CD133 low profile for all three R-GBMs. The bar graph adjacent to the flow plots shows the percent of CD15+ and CD133+ cells individually for each R-GBM sample. c Control and Tx2 treatment cells from each P-GBM sample were plated for secondary sphere formation assay. Self-renewal capacity of Tx2 treatment group for all three P-GBMs is increased as compared to DMSO controls (n = 3, p < 0.001). d Secondary sphere formation capacity is significantly higher in R-GBMs (BT241, BT566 and BT618) as compared to P-GBMs (BT428, BT458 and BT465) (p = 0.0002). The bars represent mean ± sem. ***p < 0.001
In contrast to flow profiles of P-GBMs post-radiation, combined chemoradiotherapy enriched exclusively for the CD15+/CD133− cell population (Fig. 3a). Both BT428 and BT458 expressed higher level of CD15+ in control groups and showed dramatic increases in the CD15+/CD133− population in Tx1 and Tx2 treatment groups. On the other hand, BT465 with its low fraction of CD15+ cells showed only a slight increase in the CD15+ cell population after treatment. All P-GBMs showed significant decrease in CD133+/CD15− subpopulation in the Tx2 group as compared to control.
In order to investigate the relevance of CD15+/CD133− subpopulation enrichment in P-GBMs in response to in vitro chemoradiotherapy, we profiled R-GBMs for CD15 and CD133 cell surface expression levels. Intriguingly, R-GBMs were also characterized by a CD15high/CD133low subpopulation (Fig. 3b). In fact, both BT241 and BT566 had threefold higher levels of CD15+ cells than CD133+ cells, while BT618 exclusively presented a CD15+ cell population with less than 0.5 % of cells expressing CD133. This characteristic CD15high/CD133lowflow profile of R-GBMs was very similar to profiles obtained by in vitro chemoradiotherapy of P-GBMs (Fig. 3a, Tx2). Hence, in vitro chemoradiotherapy in Tx2 treatment group of P-GBMs generated flow profiles similar to those of patient-derived R-GBMs.
In vitro chemoradiotherapy increases self-renewal capacity of P-GBMs
Although we see an increased expression of both BMI1 and SOX2 genes as well as an enrichment of the CD15+ cell population in response to in vitro chemoradiotherapy, we wanted to determine if the changes had a functional consequence on regulating self-renewal capacity of P-GBMs. Control and Tx2 treatment cells from each GBM sample were plated for secondary sphere formation assay 1-week post treatment. We find that in vitro chemoradiotherapy increases the secondary sphere formation capacity of Tx2 group compared to DMSO controls (Fig. 3c). In fact, R-GBMs also exhibit higher self-renewal than P-GBMs (Fig. 3d), again validating that our in vitro chemoradiotherapy protocol is mimicking BTSC biology of R-GBMs. We tested whether in vitro chemoradiotherapy refractory cells were more resistant to subsequent exposure to TMZ and radiation. We found that BT458 cells with previous chemoradiotherapy treatment presented higher cell survival when challenged with subsequent chemoradiotherapy and had significantly increased sphere formation capacity as compared to challenged DMSO treated control cells (Supp. Fig. S3). This suggests that cell survival in response to in vitro chemoradiotherapy is not stochastic and rather cells with acquired resistance to TMZ and radiation survive the treatment.
Gene expression profiling of treated P-GBMs reveals patterns of acquired resistance and identifies hyper-aggressive brain tumors
Our observations that acquired resistance to in vitro chemoradiotherapy appeared to model the biology of R-GBMs prompted us to do comprehensive global gene expression profiling of control and treated P-GBMs. For each of the 3 P-GBMs we identified the top 250 variably expressed genes between the control and Tx2 samples, which represented our best in vitro model of untreated and clinically treated GBM, and used these genes to cluster control, radiation, Tx1 and Tx2 treated GBMs (Fig. 4a). Analysis of the resulting clusters revealed a similar pattern of resistance acquisition in each GBM sample. Radiation-only treated samples were generally more similar to control GBM cultures, whereas both chemoradiotherapy-treated cultures (Tx1 and Tx2) were highly similar to each other. Importantly these data suggest that the radiation-only GBM group represent an intermediary step that occurs during the acquisition of chemoradiotherapy resistance by GBM (Fig. 4a).

Transcriptional analysis of in vitro treated P-GBM cultures identifies pathways associated with resistance and hyper-aggressive subtypes of brain tumors. a Hierarchical clustering of various GBM cultures based on the top 250 variably expressed genes in Control and Tx2 treated cultures. b Venn-diagram of overlapping Control/Tx2 variable genes among the three treated GBM cultures. c, d Survival analysis of the 3 brain tumor subtypes identified by NMF clustering of the REMBRANDT database using Control/Tx2 variable genes common to at least 2 of the 3 treatment experiments. e Network analysis of Control/Tx2 variable genes common to at least 2 of the 3 treatment experiments
We next examined the 250-control/Tx2 variable genes for overlap between the three GBMs. 62 genes were common to at least 2 of the 3 GBMs (Fig. 4b), which we hypothesized represents a signature of chemoradiotherapy resistance in brain tumors (henceforth called 62-RS). We completed unsupervised clustering using NMF of brain tumors that comprised the repository for brain neoplasia data (REMBRANDT, n = 286) based on the expression of the 62-RS genes (Fig. 4c). This analysis revealed that REMBRANDT samples optimally stratified into 3 classes of brain tumors based on the expression of 62-RS genes. Intriguingly, patients whose tumors were assigned to class 2 experienced dramatically poorer survival than class 1 and 3 tumors. (Class 1 vs. 2, HR 0.4, *p < 0.0001, Class 3 vs. 2, HR 0.059, *p = 0.0004) (Fig. 4d). Therefore, the 62-RS identified using our in vitro chemoradiotherapy model of brain tumor could also be used to identify patients with ultra-high risk brain tumors.
To identify the biological programs measured by the 62-RS, we generated a protein interaction network (Fig. 4e) comprising of protein products and their interaction partners using the Reactome database. Clustering of the interaction network suggested that the 62-RS interacted in 6 sub-networks, or modules, each of which was associated with distinct biological processes (Supp. Table S4, Supp. Fig. S4). Although no pathways were significantly enriched in module 0 (FDR < 0.05), module 0 comprised many genes involved in inflammation including CCL2, JUN, MGP, and LMO2. Module 1 was likely associated with proliferation, as it displayed enrichment in many proliferation-associated pathways, including mitosis as well as PLK1 and FOXM1 signaling. Module 2 was enriched in pathways associated with cell adhesion and angiogenesis, including focal adhesion, signaling through VEGFR, and the PDGFR signaling pathway. Although module 3 was only significantly enriched in a p53 related signaling pathway, it also comprised many genes involved in cellular proliferation including CDC20, CCNB1 and CCNB2. Module 4 was also not significantly enriched in any pathways, but is likely associated with hypoxia as it contains HIF1A, ADM, and CA9, which are all either functionally involved, or markers of hypoxia in tumor cells. Finally, module 5 was enriched in pathways associated with chromosome stability and maintenance. Hence, together these data reveal that brain tumor resistance to chemoradiotherapy is multifactorial, and likely results in changes to multiple biological pathways, including proliferation, chromosome stability, angiogenesis, inflammation, hypoxia and cell adhesion.
Discussion
GBM is characterized by both genetic and cellular heterogeneity, which together drive the hierarchical organization of the tumor, its clonal evolution and subsequent therapeutic resistance. Tumor evolution over the course of disease progression and in response to therapy may generate genetically and functionally distinct BTSC clones throughout tumor progression [26, 27]. These chemo- and radio-resistant BTSC clones might substantially contribute to tumor recurrence. Adding to the complexity, this evolution is dynamic in spatial organization within the tumor mass, as well as temporally throughout the disease course. Ideally, sampling different parts of same tumor tissue or sampling tissue at different time-points might help in identifying the BTSC clonal subpopulation. Unfortunately, in case of GBM, re-sampling post-treatment or at the time of relapse may encounter problems: risk of infection, risk of neurological deficits and psychological deficits like depression [28, 29]. Therefore, matched R-GBM is a rare specimen, as patients do not always undergo additional surgical resections for their relapsed tumor. In the present study, we have developed a benchtop model to study GBM recurrence and identify the evading treatment-refractory population in an individual patient with P-GBM (Fig. 5). This may constitute a unique model for studying the later stages of tumor progression from P-GBM to R-GBM. This in vitro model will be crucial for the investigation of genetic, epigenetic and cellular alterations, for the study of proliferation, migration and tumor recurrence and to define potential molecular markers for tumor progression. Ultimately, it will accelerate the development of personalized therapeutic strategies targeting BTSCs driving recurrent GBM.

In vitro chemoradiotherapy of P-GBM models the biology of R-GBM. In vitro treatment of BTSC enriched P-GBMs with radiation and TMZ leads to increase in expression of BMI1 and SOX2, enriches for CD15+ cell population and increases self-renewal capacity of P-GBMs. The global gene expression profile identifies a Tx2 specific 62-gene signature that clusters the REMBRANDT glioma dataset into an ultra-high risk brain tumor subgroup
The upregulation of stem cell genes, BMI1 and SOX2 is not a surprising finding. Previously, Bmi1 protein was shown to interact with DNA damage response machinery and confer radioresistance to irradiated GBM cells [30]. Additionally, loss of Sox2 is shown to delay tumor progression in GBMs through polo-like kinase 1 (PLK1), which possibly drives radio-resistance in GBM [31, 32]. Interestingly, our data identified PLK1 pathway as significantly variable signaling event in our Tx2 treatment group (Supp. Table S4). The involvement of these signaling pathways is not entirely novel; however it further validates this unique in vitro model to study GBM recurrence. Further studies are warranted to specifically elucidate the role of Bmi1 and Sox2 in GBM therapy resistance and tumor relapse.
The enrichment of CD15+/CD133− subpopulation in P-GBM after chemoradiotherapy is an exciting finding, which was further corroborated by the presence of a predominant CD15high/CD133low subpopulation in R-GBM. Our findings add to the growing body of evidence that there is a hierarchy of self-renewing BTSCs, playing different roles in tumor initiation, progression and recurrence. While BTSCs with neural stem cell marker CD133 expression represent the tumor-initiating subpopulation, cells with neural progenitor cell marker CD15 may represent BTSCs driving tumor recurrence. Further studies investigating the molecular heterogeneity and defining the cellular hierarchy of BTSCs will lead to better understanding of the disease process.
Although previous studies have been conducted to examine the resistance of GBMs to TMZ and radiation, none looked at the combinatorial effects of chemoradiotherapy in modulating therapy resistance and tumor relapse, despite the fact that most GBM patients undergo combined therapy. Our work suggests that studying a therapy module in isolation may not inform on the complexity of the tumor recurrence. This is illustrated by the CD15/CD133 flow profile where radiation alone led to an enrichment of the CD133+/CD15− population; if taken in isolation, this data would warrant attempted targeting of the CD133+ population in P-GBM, even if this cell population is not driving tumor recurrence after subsequent chemotherapy. Our data suggests that combined chemoradiotherapy further uniquely modulates GBM subpopulations, as in our model it leads to a subsequent enrichment of the CD15+/CD133− subpopulation. Similarly, the gene expression of stem cell genes BMI1 and SOX2 was only significantly increased after combined chemoradiotherapy, demonstrating the need to model disease progression through combined therapies with concurrent gene expression profiling, to define the state of therapy resistance. Only in a combined treatment model can treatment-refractory cell populations and gene signatures be identified, which can then lead to development of informed combinatorial therapies not only in GBM, but also in other malignancies that exhibit heterogeneity.
References
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Cancer Genome Atlas Research N (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. doi:10.1016/j.ccr.2009.12.020
Wechsler-Reya R, Scott MP (2001) The developmental biology of brain tumors. Annu Rev Neurosci 24:385–428. doi:10.1146/annurev.neuro.24.1.385
Zhu Y, Parada LF (2002) The molecular and genetic basis of neurological tumours. Nat Rev Cancer 2:616–626. doi:10.1038/nrc866
Huse JT, Holland EC (2010) Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer 10:319–331. doi:10.1038/nrc2818
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, EuropeanOrganisation for R, Treatment of Cancer Brain T, Radiotherapy G, National Cancer Institute of Canada Clinical Trials G (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. doi:10.1056/NEJMoa043330
Cancer Genome Atlas Research N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. doi:10.1038/nature07385
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. doi:10.1038/nature03128
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828
Son MJ, Woolard K, Nam DH, Lee J, Fine HA (2009) SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 4:440–452. doi:10.1016/j.stem.2009.03.003
Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, Rich JN (2010) Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 6:421–432. doi:10.1016/j.stem.2010.02.018
Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, Hjelmeland AB, Rich JN (2008) Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res 68:6043–6048. doi:10.1158/0008-5472.CAN-08-1079
Kaneko Y, Sakakibara S, Imai T, Suzuki A, Nakamura Y, Sawamoto K, Ogawa Y, Toyama Y, Miyata T, Okano H (2000) Musashi1: an evolutionally conserved marker for CNS progenitor cells including neural stem cells. Dev Neurosci 22:139–153
Graham V, Khudyakov J, Ellis P, Pevny L (2003) SOX2 functions to maintain neural progenitor identity. Neuron 39:749–765
Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S (2007) shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 1:87–99. doi:10.1016/j.stem.2007.04.001
Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G (2009) BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci 29:8884–8896. doi:10.1523/JNEUROSCI.0968-09.2009
Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, Curry WT, Martuza RL, Rivera MN, Rossetti N, Kasif S, Beik S, Kadri S, Tirosh I, Wortman I, Shalek AK, Rozenblatt-Rosen O, Regev A, Louis DN, Bernstein BE (2014) Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157:580–594. doi:10.1016/j.cell.2014.02.030
Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular cancer 5:67. doi:10.1186/1476-4598-5-67
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. doi:10.1038/nature05236
Venugopal C, Li N, Wang X, Manoranjan B, Hawkins C, Gunnarsson T, Hollenberg R, Klurfan P, Murty N, Kwiecien J, Farrokhyar F, Provias JP, Wynder C, Singh SK (2012) Bmi1 marks intermediate precursors during differentiation of human brain tumor initiating cells. Stem Cell Res 8:141–153. doi:10.1016/j.scr.2011.09.008
Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, Proescholdt M, Brawanski A, Bogdahn U, Trampe-Kieslich A, Giebel B, Wischhusen J, Reifenberger G, Hau P, Beier CP (2008) Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res 68:5706–5715. doi:10.1158/0008-5472.CAN-07-6878
Hammond LA, Eckardt JR, Baker SD, Eckhardt SG, Dugan M, Forral K, Reidenberg P, Statkevich P, Weiss GR, Rinaldi DA, Von Hoff DD, Rowinsky EK (1999) Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J Clin Oncol 17:2604–2613
Barazzuol L, Jena R, Burnet NG, Jeynes JC, Merchant MJ, Kirkby KJ, Kirkby NF (2012) In vitro evaluation of combined temozolomide and radiotherapy using X rays and high-linear energy transfer radiation for glioblastoma. Radiat Res 177:651–662
Brunet JP, Tamayo P, Golub TR, Mesirov JP (2004) Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci USA 101:4164–4169. doi:10.1073/pnas.0308531101
Wu G, Stein L (2012) A network module-based method for identifying cancer prognostic signatures. Genome Biol 13:R112. doi:10.1186/gb-2012-13-12-r112
Wu G, Feng X, Stein L (2010) A human functional protein interaction network and its application to cancer data analysis. Genome Biol 11:R53. doi:10.1186/gb-2010-11-5-r53
Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE (2011) Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469:362–367. doi:10.1038/nature09733
Dalerba P, Cho RW, Clarke MF (2007) Cancer stem cells: models and concepts. Annu Rev Med 58:267–284. doi:10.1146/annurev.med.58.062105.204854
Barami K, Fernandes R (2012) Incidence, risk factors and management of delayed wound dehiscence after craniotomy for tumor resection. J Clin Neurosci 19:854–857. doi:10.1016/j.jocn.2011.09.025
Chang SM, Parney IF, McDermott M, Barker FG 2nd, Schmidt MH, Huang W, Laws ER Jr, Lillehei KO, Bernstein M, Brem H, Sloan AE, Berger M, Glioma Outcomes I (2003) Perioperative complications and neurological outcomes of first and second craniotomies among patients enrolled in the Glioma Outcome Project. J Neurosurg 98:1175–1181. doi:10.3171/jns.2003.98.6.1175
Facchino S, Abdouh M, Chatoo W, Bernier G (2010) BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci 30:10096–10111. doi:10.1523/JNEUROSCI.1634-10.2010
Tandle AT, Kramp T, Kil WJ, Halthore A, Gehlhaus K, Shankavaram U, Tofilon PJ, Caplen NJ, Camphausen K (2013) Inhibition of polo-like kinase 1 in glioblastoma multiforme induces mitotic catastrophe and enhances radiosensitisation. Eur J Cancer 49:3020–3028. doi:10.1016/j.ejca.2013.05.013
Lee C, Fotovati A, Triscott J, Chen J, Venugopal C, Singhal A, Dunham C, Kerr JM, Verreault M, Yip S, Wakimoto H, Jones C, Jayanthan A, Narendran A, Singh SK, Dunn SE (2012) Polo-like kinase 1 inhibition kills glioblastoma multiforme brain tumor cells in part through loss of SOX2 and delays tumor progression in mice. Stem Cells 30:1064–1075. doi:10.1002/stem.1081
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Stem Cell Network Undergraduate Co-op Award and Canadian Institute of Health Research Canada Graduate Scholarship – Master’s Award (to M.A.Q.); Stem Cell Network Stem Cell Drug Discovery Award, Terry Fox Foundation New Investigator Award, Ontario Institute for Cancer Research Cancer Stem Cell Program, and Canada Research Chair Award (to S.K.S.).
Conflict of Interest
The authors declare no conflict of interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qazi, M.A., Vora, P., Venugopal, C. et al. A novel stem cell culture model of recurrent glioblastoma. J Neurooncol 126, 57–67 (2016). https://doi.org/10.1007/s11060-015-1951-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1951-6