
Abstract
Despite accumulating knowledge regarding molecular backgrounds, the optimal management strategy for low-grade gliomas remains controversial. One reason is the marked heterogeneity in the clinical course. To establish an accurate subclassification of low-grade gliomas, we retrospectively evaluated isocitrate dehydrogenase-1 (IDH1) mutation in clinical specimens of diffuse astrocytomas (DA) and oligodendroglial tumors separately. No patients were treated with early radiotherapy, and modified PCV chemotherapy was used for postoperative residual tumors or recurrence in oligodendroglial tumors. Immunohistochemical evaluation of IDH status, p53 status, O6-methylguanine methyltransferase expression, and the MIB-1 index were performed. The 1p and 19q status was analyzed with fluorescence in situ hybridization. Ninety-four patients were followed for a median period of 8.5 years. For DAs, p53 was prognostic for progression- free survival (PFS) and IDH1 was significant for overall survival (OS) with multivariate analysis. In contrast, for oligodendroglial tumors, none of the parameters was significant for PFS or OS. Thus, the significance of IDH1 mutation is not clear in oligodendroglial tumors that are homogeneously indolent and chemosensitive. In contrast, DAs are heterogeneous tumors including some potentially malignant tumors that can be predicted by examining the IDH1 mutation status.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The therapeutic strategy for adult low-grade gliomas is still controversial, especially regarding postoperative early radiotherapy [1–3]. Heterogeneity in the natural history of the tumor makes application of a uniform treatment standard difficult [4, 5]. Some tumors remain dormant, whereas others rapidly transform into higher-grade gliomas [6]. Beyond the histological diagnosis, several molecular markers have been proposed as more precise predictors of outcome in patients with low-grade gliomas [5]. TP53 mutation is frequently observed in diffuse astrocytomas (DA), but its prognostic or predictive role is controversial and no consistent association with a response to therapy has been reported [7–9]. Methylation of the promoter region of O6-methylguanine methyltransferase (MGMT) predicts a favorable outcome in patients with anaplastic gliomas (WHO grade III) treated with DNA-damaging agents including radiotherapy [10], but its significance in low-grade gliomas (WHO grade II) remains unclear [11]. Combined deletion of chromosomes 1p and 19q (1p/19q co-deletion), which results from an unbalanced translocation, is common in oligodendroglial tumors [12]. 1p/19q co-deletion is a prognostic and predictive marker for anaplastic oligodendrogliomas but not established as a prognostic marker in grade II oligodendrogliomas [13, 14].
A somatic mutation in the gene encoding isocitrate dehydrogenase 1 (IDH1) is frequently observed in WHO grade II and III gliomas [15, 16]. This genetic abnormality occurs earlier than TP53 mutation or 1p/19q deletion in low-grade gliomas (WHO grade II) [17]. The most frequent IDH1 mutation (G395A) leads to the replacement of arginine with histidine at codon 132 (R132H), which is in the enzymatic active site [18]. This IDH1 mutation reduces production of α-ketoglutarate (α-KG) from isocitrate and also converts α-KG to 2-hydroxyglutarate (2-HG) [19]. The change in the metabolites induces extensive DNA hypermethylation by suppressing ten-eleven translocated (TET) function [20, 21]. The IDH1 mutation is associated with a good prognosis in high-grade gliomas [15, 22–24]. Because this gene mutation is not usually observed in primary glioblastoma which contains hypomethylated DNA, the mutation may be useful for excluding low-grade gliomas with a potential for rapid malignant transformation. However, inconsistent results have been reported regarding the prognostic value of IDH1 mutation in low-grade gliomas [24–27], partly because most previous studies included patients with heterogeneous histological types who underwent different treatments. We studied the prognostic value of the IDH1 mutation separately in WHO grade II DA and oligodendroglial tumors that were homogeneously treated without early radiotherapy.
Methods
Patients and treatment
All patients were histologically confirmed to have WHO grade II DA, oligodendroglioma (O) or oligoastrocytoma (OA). Age, sex, neurological symptoms, tumor location, tumor size, pathological diagnosis, and extent of resection were retrospectively reviewed. The histological diagnosis was verified by a neuropathologist other than the initial diagnostician. In the Chiba University Hospital, all patients with low-grade gliomas are homogeneously treated with maximum surgical resection and without early radiotherapy. Patients with DA were carefully followed-up after maximal surgical resection without any additional treatment. Patients having O/OA with post-operative residual tumors were treated with a standard nitrosourea-based chemotherapy (PAV, a modified PCV). For this chemotherapy, lomustine (CCNU) was replaced with nimustine (ACNU; [1-(4-amino-2-methyl-5-pyrimidinyl)-methyl-(2-chloroethyl)-3-nitrosourea hydrochloride]) which is a water- and lipid-soluble nitrosourea derivative. Magnetic resonance imaging was performed at 2-month intervals in all patients. A salvage surgery was performed for most of the patients with recurrent tumor. This study was approved by the Ethics Committee of Chiba University Hospital, and patients who needed chemotherapy were required to provide written informed consent.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue sections were deparaffinized in xylene on microscopic slides. Antigen retrieval was performed by microwaving the sections in 10 mM citric acid buffer (H 7.2). The primary antibodies used in this study were: anti-human IDH1-R132H monoclonal antibody and anti-human IDH1-R132S monoclonal antibody (1:100, IBL Co., Ltd, Gunma, Japan), MIB-1 monoclonal antibody against the Ki-67 antigen (1:100, Immunotech, Westbrook, ME), anti-p53 monoclonal antibody DO-1 (1:100, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and anti-MGMT monoclonal antibody MT3.1 (1:200, Chemicon, Inc., Temecula, CA, USA). The samples were incubated with the primary antibody overnight, followed by incubation with a biotinylated secondary antibody (1:500, Dako, Tokyo, Japan). The bound antibodies were visualized using the avidin biotin peroxidase complex method and diaminobenzidine tetrachloride (Santa Cruz Biotechnology, Inc.). To evaluate IDH1 staining, strong cytoplasmic staining in any number of cells was scored as positive. For MIB-1, p53, and MGMT scoring, the positive cells in a 200 × field (minimum of 1000 nuclei) were counted, and the labeling index was expressed as the percent of labeled tumor cells. A MIB-1 labeling index ≥3 %, p53 protein accumulation ≥10 %, and MGMT protein expression ≥10 % were considered positive.
1p19q FISH
Chromosome 1p and 19q deletion analyses were performed using standard fluorescence in situ hybridization (FISH) of fixed cytogenetic preparation of fresh tumor tissues [23]. FISH probes for 1p were the target region of 1p36 with a control region of 1q25, and those for 19q were the control region of 19p13 with the target region of 19q13. The total number of signals was counted, and a ratio of 1p:1q or 19q:19p of <0.5 was diagnosed as a loss. Parallel loss of both arms was considered 1p19q co-deletion.
Statistical analysis
Progression-free survival (PFS) was calculated from the date of diagnosis until the first sign of radiological progression, death, or last follow-up. Overall survival (OS) was calculated from the date of diagnosis until the date of death or last follow-up. The Kaplan–Meier method was used to estimate survival rates and the log-rank test was applied to compare the survival differences using StatView software (SAS Institute Inc., Cary, NC). Cox’s proportional hazard regression model was used to perform multivariate analysis of the possible prognostic variables including age, Karnofsky performance status (KPS), extent of resection, 1p19q status, MIB-1 labeling index, and expression of p53, MGMT, and mutant IDH1 proteins (SPSS, Inc., Chicago, IL). A p value <0.05 was considered significant.
Results
Patient profile
A total of 94 patients with histologically proven WHO grade II low-grade gliomas were identified between 1996 and 2013 and retrospectively analyzed (Table 1). Thirty-eight patients had DA, 47 had O, and nine had OA. Fifty-four men and 40 women with a mean age of 41 years (range 22–84 years) were analyzed. The KPS score was 70 % or more in 74 patients (79 %). Forty-six patients (49 %) underwent gross total resection, 27 patients (29 %) underwent subtotal tumor resection, and the other 21 (22 %) underwent partial resection. IDH1 mutation was examined with immunohistochemistry using anti-IDH1-R132H antibody, which specifically recognizes IDH1 with the R132H mutation and not wild-type IDH1, and showed that 78 cases (87.6 %) among 89 cases examined had mutated IDH1. The negative cases were also confirmed as negative for anti-IDH1R132S antibody. The patients were followed up for a median period of 8.5 years and no patient was lost during the follow-up period.
DA
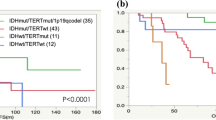
The 5- and 10-year PFS rate were 63.8 and 53.4 %, respectively, and the median PFS was 147 months. The OS rate was 77.3 % at 5 years and 68.5 % at 10 years of observation (Fig. 1a). Older patients (>50 years) had a significantly shorter PFS (p = 0.0017) and OS (p = 0.0002) (Fig. 1b). The KPS score was not significantly associated with PFS or OS. The tumor size (>5 cm) and extent of surgical resection were both significant for PFS (p = 0.0456 and 0.0060, respectively) but not OS (Fig. 1c, d). MIB-1 labeling index ≥3 % was observed in 12 cases (32 %), and was a significant factor both for PFS and OS (p = 0.0003 and 0.0072, respectively) (Fig. 1e). p53 protein expression was observed in 15 cases (48 %), and was a significant factor for PFS (p = 0.0278) (Fig. 1f). Mutant IDH1 protein was expressed in 27 cases (82 %) and was a significant prognostic factor for OS (p = 0.0030) (Fig. 1g). In contrast, among six patients with negative IDH-R132H staining, four experienced malignant progression and survived a significantly shorter time than those with positive staining. Multivariate analysis showed that age, KPS, tumor size, extent of surgery, and MIB-1 index were no longer significant prognostic factors, but p53 for PFS and IDH1 for OS remained independent prognostic factors (p = 0.0258 and 0.0108, respectively) (Table 2). A salvage second surgery was performed in 15 patients, and malignant transformation was observed in 13 patients (87 %).

Kaplan–Meier survival curves for the progression-free survival (PFS) and overall survival (OS) of the diffuse astrocytoma patients, comparing the potential prognostic factors. All patients (a), age (b) (blue line <50, red line ≥50), tumor size (c) (blue line <5 cm, red line ≥5 cm), extent of surgery (d) (blue line total removal, red non-total), MIB-1 labeling index (e) (blue line <3 %, red line ≥3 %), p53 protein expression (f) (blue line <10 %, red line ≥10 %), IDH1-R132H expression (g) (blue line negative, red line positive)
O/OA
The 5- and 10-year PFS rates were 72.3 and 46.9 %, respectively, and the median PFS was 100 months. The OS rate was 97.4 % at 5 years and 89.6 % at 10 years of observation (Fig. 2a). Age (Fig. 2b), KPS, and extent of surgery (Fig. 2c) did not significantly affect PFS or OS. A MIB-1 index ≥3 % was observed in 16 patients (28 %) who experienced recurrence significantly earlier than those with <3 % (p = 0.0049) (Fig. 2d). Such patients tended to survive for a shorter time, but the result was not statistically significant (p = 0.1316). A histopathological diagnosis of oligoastrocytoma was obtained in nine patients (16 %) who survived for a significantly shorter period than patients with pure O (p = 0.0238). 1p/19q co-deletion was observed in 37 of 52 patients (71 %) who were analyzed with FISH. Loss of only 1p or only 19q was observed in two patients. The median PFS for patients with 1p/19q co-deleted tumors was 96 months and that for non-deleted tumors was 57 months. A median OS was not reached for either group. We found no significant difference in PFS or OS between patients with 1p/19q co-deleted tumors and those without co-deletion (p = 0.1616 and 0.6835, respectively) (Fig. 2e). Patients with MGMT protein expression had a significantly shorter PFS than those without expression of the protein (Fig. 2f). Mutant IDH1 protein was expressed in 51 cases (91 %), and was not a significant factor for PFS or OS although the number of negative cases was small for a powerful statistical detection (Fig. 2g). The multivariate analysis showed that age, extent of resection, 1p19q status, and IDH1 were not significantly associated with the length of survivals. A salvage second surgery was performed in 16 patients, and malignant transformation was observed in seven cases (44 %). Six patients developed grade III anaplastic oligodendrogliomas, and one patient with OA developed glioblastoma.

Kaplan–Meier survival curves for the progression-free survival (PFS) and overall survival (OS) of the patients with oligodendroglial tumors, comparing the potential prognostic factors: All patients (a), age (b) (blue line <50, red line ≥50), extent of surgery (c) (blue line total removal, red non-total), MIB-1 labeling index (d) (blue line <3 %, red line ≥3 %), 1p/19q co-deletion (e) (blue line positive, red line negative), MGMT protein expression (f) (blue line <10 %, red line ≥10 %), IDH1-R132H expression (g) (blue line negative, red line positive)
Discussion
As shown with IDH1-R132H immunostaining, the present study demonstrated that patients with low-grade gliomas had a high rate of IDH1 mutation (87 %), including 82 % of DA and 91 % of O/OA. Patients with IDH1-mutated DA lived significantly longer than those with wild-type IDH1 tumors, whereas the survival times of patients with O/OA were not different regardless of IDH1 status when not treated with early radiotherapy. Thus, IDH1 mutation is a prognostic factor for DA but not for low-grade oligodendroglial tumors. The influence of the IDH1 mutation on survival of patients with low-grade gliomas is unclear due to inconsistent results reported [24–30]. Sanson et al. described an association of IDH1 mutations with improved survival in patients with low-grade gliomas [24]. Houillier et al. also showed that the IDH1 mutation is a significant marker of a positive prognosis in patients with low-grade gliomas, whereas its impact on the course of untreated tumors is limited [25]. Sabha et al. showed that patients with low-grade diffuse gliomas with the IDH1 mutation survived significantly longer than those without the mutation in a cohort not treated with radiation therapy [26]. In contrast, Kim et al. reported no significant influence of IDH1 mutations on survival in more than 300 low-grade glioma patients who were heterogeneously treated [27]. Mukasa et al. and Boots-Sprenger et al. also reported that IDH1 mutation is prognostic only for patients with grade III gliomas and not for those with grade II gliomas treated with early radiotherapy [28, 29]. Although these studies included both astrocytic tumors and oligodendroglial tumors, the prognostic significance of the IDH1 mutation in astrocytic tumors and oligodendroglial tumors should be evaluated separately because these tumors are completely distinct both genetically and clinically [5]. Although many reports included heterogeneously treated patients, separate analysis of each treatment modality or at least a description of the rate of each modality is desirable. In addition, the method of evaluation was also different among studies including direct sequencing or immunohistochemistry.
Several important reports have separately analyzed the influence of the status of IDH1 according to histological types [31–37]. Dubbink et al. showed that the presence of the IDH1 mutation in DA is associated with a significantly improved OS in a cohort not treated with early radiotherapy [32]. Hartmann et al. showed that patients with IDH1-mutated DA survive longer than those with a non-IDH1 mutated tumor when not treated with early radiotherapy [33]. In contrast, Ahmadi et al. reported no association between the IDH1 mutation status and PFS or OS in DA in which the patient cohort was heterogeneously treated with early radiotherapy in 38 % of the patients [34]. These previous reports suggest that the IDH1 mutation is a prognostic marker in DA when no additional genotoxic therapies such as radiation therapy are performed, and that radiation therapy may affect the clinical course of patients with wild type IDH1 DA, providing a better outcome [35, 36]. In contrast, because clinical trials have failed to demonstrate a survival benefit with early radiation therapy in DA as a whole, patients with “true DA” harboring mutated IDH1 have a disease that is basically dormant and not responsive to radiation therapy.
Regarding oligodendroglial tumors, few studies have dealt primarily with oligodendroglial tumors concerning the influence of the IDH1 mutation on patient survival, and the results have been inconsistent [38, 39]. Many studies of low-grade gliomas showing a positive correlation between the IDH1 mutation and a favorable survival have included many patients with oligodendroglial tumors, suggesting that a significant contribution of the IDH1 mutation exists regarding the prognosis of oligodendroglial tumors [24, 25, 40, 41]. In contrast, Lee et al. showed a negative result for an IDH1 contribution to the survival of patients with oligodendrogliomas, most of whom were not treated with early radiotherapy [38]. In a study of patients who were homogeneously not treated with early radiotherapy, Hartmann et al. reported a result similar to our present study showing that oligodendroglial tumors have similar prognosis regardless of the IDH1 mutational status [33].
The prognostic or predictive role of 1p/19q loss is well defined for anaplastic oligodendrogliomas [42, 43], whereas the prognostic relevance is less well defined for low-grade oligodendroglial tumors [44–48]. Although some reports have shown that patients with low-grade oligodendroglial tumors harboring 1p/19q deletion survive longer than those without these deletions, Weller et al. reported that 1p/19q deletion loses its prognostic impact when the tumors are not treated with radiation therapy or chemotherapy after surgery [48]. Thus, we speculate that radiation therapy will negatively modify the survival of patients with oligodendroglial tumors without 1p/19q loss, whereas radiation therapy may be partially effective for patients with IDH1-wild type DA. Both 1p/19q deletion and IDH1 mutation are candidate predictive markers for response to radiation therapy.
The limitations of this study are the small number of patients, especially in the IDH1 mutation-negative cases, and the retrospective nature of the study design. However, our results suggest that the prognostic value of IDH1 mutation for DA should be validated in future prospective study.
References
Karim AB, Afra D, Cornu P et al (2002) Randomized trial on the efficacy of radiotherapy for cerebral low-grade gliomas in the adult: European Organization for Research and Treatment of Cancer Study 22845 with Medical Research Council Study BRO4: an interim analysis. Int J Radiat Oncol Biol Phys 52:316–324
Shaw E, Arusell R, Scheithauer B et al (2002) Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Group study. J Clin Oncol 20:2267–2276
van den Bent MJ, Afra D, de Witte O et al (2005) EORTC Radiotherapy and Brain Tumor Groups and the UK Medical Research Council. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 366:985–990
Papagikos MA, Shaw EG, Stieber VW (2005) Lessons from randomized clinical trials in adult low grade glioma. Lancet Oncol 6:240–244
Behin A, Hoang-Xuan K, Carpentier AF, Delattre J-Y (2003) Primary brain tumors in adults. Lancet 361:323–331
Wessels PH, Weber WE, Raven G et al (2003) Supratentorial grade II astrocytoma: biological features and clinical course. Lancet Neurol 2:395–403
Okamoto Y, Di Patre PL, Burkhard C et al (2004) Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol 108:49–56
Peraud A, Kreth FW, Wiestler OD, Kleihues P, Reulen HJ (2002) Prognostic impact of TP53 mutations and P53 protein overexpression in supratentorial WHO grade II astrocytomas and oligoastrocytomas. Clin Cancer Res 8:1117–1124
Ständer M, Peraud A, Leroch B, Kreth FW (2004) Prognostic impact of TP53 mutation status for adult patients with supratentorial World Health Organization Grade II astrocytoma or oligoastrocytoma: a long-term analysis. Cancer 101:1028–1035
Weller M, Stupp R, Reifenberger G et al (2010) MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol 6:39–51
Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T (2003) Promoter hypermethylation of the DNA repair gene O6-methylguanine- DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low-grade diffuse astrocytomas. Brain Pathol 13:176–184
Jenkins RB, Blair H, Ballman KV et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861
Cairncross JG, Ueki K, Zlatescu MC et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479
Weller M, Berger H, Hartmann C et al (2007) German Glioma Network. Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin Cancer Res 13:6933–6937
Parsons DW, Jones S, Zhang X et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL, Xiong Y (2013) Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 32:663–669
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M et al (2009) Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27:5743–5750
Van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoom MJB et al (2010) IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res 16:1597–1604
Sanson M, Marie Y, Paris S, Idbaih A et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Houillier C, Wang X, Kaloshi G et al (2010) IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 75:1560–1566
Sabha N, Knobbe CB, Maganti M et al (2014) Analysis of IDH mutation, 1p/19q deletion, and PTEN loss delineates prognosis in clinical low-grade diffuse gliomas. Neuro Oncol 16:914–923
Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Sure U et al (2010) Molecular classification of low-grade diffuse gliomas. Am J Pathol 177:2708–2714
Mukasa A, Takayanagi S, Saito K, Shibahara J, Tabei Y et al (2012) Significance of IDH mutations varies with tumor histology, grade, and genetics in Japanese glioma patients. Cancer Sci 103:587–592
Boots-Sprenger SHE, Sijben A, Rijntjes J et al (2013) Significance of complete 1p/19q co-deletion, IDH1 mutation and MGMT promoter methylation in gliomas: use with caution. Mod Pathol 26:922–929
Leu S, von Felten S, Frank S, Vassella E, Vajtai I et al (2013) IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro Oncol 15:469–479
Okita Y, Narita Y, Miyakita Y, Ohno M, Matsushita Y et al (2012) IDH1/2 mutation is a prognostic marker for survival and predicts response to chemotherapy for grade II gliomas concomitantly treated with radiation therapy. Int J Oncol 41:1325–1336
Dubbink HJ, Taal W, van Marion R et al (2009) IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology 73:1792–1795
Hartmann C, Hentschel B, Tatagiba M et al (2011) Molecular markers in low-grade gliomas: predictive or prognostic? Clin Cancer Res 17:4588–4599
Ahmadi R, Stockhammer F, Becker N, Hohlen K, Misch M, Christians A, Dictus C et al (2012) No prognostic value of IDH1 mutations in a series of 100 WHO grade II astrocytomas. J Neurooncol 109:15–22
Juratli TA, Kirsch M, Robel K et al (2012) IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol 108:403–410
Dahlrot RH, Kristensen BW, Hjelmborg J, Herrstedt J, Hansen S (2013) A population-based study of low-grade gliomas and mutated isocitrate dehydrogenase 1 (IDH1). J Neurooncol 114:309–317
Thon N, Eigenbrod S, Kreth S et al (2012) IDH1 mutations in grade II astrocytomas are associated with unfavorable progression-free survival and prolonged postrecurrence survival. Cancer 118:452–460
Lee D, Suh YL, Kang SY, Park TI, Jeong JY, Kim SH (2013) IDH1 mutations in oligodendroglial tumors: comparative analysis of direct sequencing, pyrosequencing, immunohistochemistry, nested PCR and PNA-mediated clamping PCR. Brain Pathol 23:285–293
Myung JK, Cho HJ, Park CK, Kim SK, Phi JH, Park SH (2012) IDH1 mutation of gliomas with long-term survival analysis. Oncol Rep 28:1639–1644
Figarella-Branger D, Bouvier C, de Paula AM et al (2012) Molecular genetics of adult grade II gliomas: towards a comprehensive tumor classification system. J Neurooncol 110:205–213
Metellus P, Coulibaly B, Colin C et al (2010) Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol 120:719–729
Caincross G, Berkey B, Shaw E et al (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma; Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714
van den Bent MJ, Carpentier AF, Brandes AA et al (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organization for Research and Treatment of Cancer phase III trial. J Clin Oncol 24:2715–2722
Iwadate Y, Matsutani T, Hasegawa Y, Shinozaki N, Higuchi Y, Saeki N (2011) Favorable long-term outcome of low-grade oligodendrogliomas irrespective of 1p/19q status when treated without radiotherapy. J Neurooncol 102:443–449
Kanner AA, Staugaitis SM, Castilla EA et al (2006) The impact of genotype on outcome in oligodendroglioma: validation of the loss of chromosome arm 1p as an important factor in clinical decision making. J Neurosurg 104:542–550
Mariani L, Deiana G, Vassella E et al (2006) Loss of heterozygosity 1p36 and 19q13 is a prognostic factor for overall survival in patients with diffuse WHO grade 2 gliomas treated without chemotherapy. J Clin Oncol 24:4758–4763
Jaeckle KA, Ballman KV, Rao RD, Jenkins RB, Buckner JC (2006) Current strategies in treatment of oligodendroglioma: evolution of molecular signature of response. J Clin Oncol 24:1246–1252
Weller M, Berger H, Hartmann C et al (2007) Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin Cancer Res 13:6933–6937
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no financial disclosures from the authors.
Rights and permissions
About this article

Cite this article
Iwadate, Y., Matsutani, T., Hirono, S. et al. IDH1 mutation is prognostic for diffuse astrocytoma but not low-grade oligodendrogliomas in patients not treated with early radiotherapy. J Neurooncol 124, 493–500 (2015). https://doi.org/10.1007/s11060-015-1863-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1863-5