
Abstract
Isocitrate dehydrogenase (IDH) mutations are beginning to drive decisions on therapy for glioma patients. Here we sought to determine the impact of adjuvant treatment in patients with IDH-mutant, 1p/19q non-codeleted secondary high-grade astrocytoma (sHGA) WHO grades III/IV. Clinical data of 109 sHGA patients grades III/IV, in addition to IDH mutation-, 1p/19q-codeletion- and MGMT-promoter methylation status—were retrospectively analyzed. Survival analysis in relation to adjuvant treatment modalities and molecular profiling were performed. Out of 109 patients, 88 patients (80.7 %) harbored IDH mutations, 30 patients had a 1p/19q-codeletion (27.5 %) and 69 patients (63.3 %) exhibited a methylated MGMT-promoter status. At a median follow-up of 9.8 years, 62 patients (57 %) died. The postsurgical treatment included: radio-chemotherapy (RT-CT; 54.5 %), RT alone (19.3 %), and CT alone (22.7 %). The median overall survival (OS) in the entire group was 3.4 years (1.9–6.7 years). Patients who received RT-CT had a significantly longer OS compared with those who underwent RT alone (6.5 vs. 1.2 years, HR 0.35, CI 0.32–0.51, p = 0.011). In the IDH-mutant 1p/19q non-codeleted sHGA subgroup the RT-CT cohort had a significantly longer OS in comparison to the RT cohort (6.4 vs. 1.2 years, HR 2.7, CI 1.1–6.5, p = 0.022). In the stepwise multivariable Cox model for OS of all 88 IDH-mutant sHGA patients, survival was strongly associated with only one factor, namely, adjuvant RT-CT at diagnosis of a sHGA. This retrospective long-term study demonstrates that RT and CT (mostly PCV) significantly improves progression-free and overall survival in IDH-mutant secondary high-grade astrocytoma patients, regardless of 1p/19q-codeletion status.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Secondary high-grade astrocytomas (sHGA) are rare tumors which develop in younger patients through progression from diffuse astrocytoma or mixed oligoastrocytoma (WHO grade II). Typical for sHGA are isocitrate dehydrogenase (IDH) gene mutations which were first described in secondary glioblastomas [1] and subsequently found in ∼70–80 % of sHGA [2]. IDH-mutant gliomas are characterized by the subsequent acquisition of further lineage-defining mutations, in particular 1p/19q-codeletion and TERT-mutations in oligodendroglioma, while the astrocytoma pathway is driven by TP53 and ATRX mutations [3].
Until recently, sHGA were treated similar to primary HGG. However, there is increasing knowledge about the role of the aforementioned molecular markers and their associations with patients’ survival. Long-term follow-up data of the randomized phase III Radiation Therapy Oncology Group (RTOG) trial 9402 and the EORTC trial 26951 demonstrated an improved outcome—in both progression-free and overall survival—in 1p/19q-codeleted anaplastic oligodendroglial tumors when procarbazine, lomustine and vincristine chemotherapy (PCV) was added to RT [4, 5]. Moreover, the RTOG 9402 trial concluded that even patients with non-codeleted 1p/19q but IDH-mutant oligodendroglial tumors lived longer after radio-chemotherapy.
Still, there is a lack of data examining the efficacy of radio-chemotherapy (RT-CT) in comparison to radiotherapy alone (RT) in astrocytic gliomas and in sHGA, in particular. We, therefore, sought to evaluate the efficacy of chemotherapy (CT) and RT in patients with IDH-mutant, 1p/19q non-codeleted secondary high-grade astrocytomas.
Methods and patients
Patients and samples
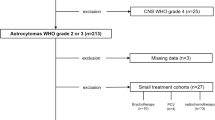
Samples and treatment data of 109 patients with astrocytomas grades III and IV, developed from histologically confirmed low-grade astrocytoma grade II (LGA) were available. All patients were treated for their sHGA between 1995 and 2013 in our institution. The diagnosis of sHGA and LGA was performed in our institute of neuropathology, according to the latest WHO classification for brain tumors [6]. Due to the fact that our patient collection extends across two WHO classifications, all pathological samples of our populations were recently reviewed by our senior neuropathologist (K.D.G.). All patients were at least 18 years of age at diagnosis and provided written consent for molecular analysis of tumor tissue and clinical data collection. Some of the clinical data of 64 individuals in this patients’ cohort have been published previously [7]. The study was approved by the local ethics committee.
IDH1/2 mutations and MGMT-promoter status assessment
Data of IDH1/2 mutations, O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation status MGMT-promoter methylation status, as well as 1p/19q status were available in all patients and were correlated to clinical data.
IDH1/2 mutations were assessed using direct DNA sequencing, as previously described [7]. To ensure a tumor cell content of at least 80 % for DNA extraction, control slides stained with hematoxylin and eosin were examined by our local neuropathologist. The MGMT-promoter status was determined by methylation-specific polymerase chain reaction (MsPCR), as described by Esteller et al. [8].
Detection of 1p/19q-codeletion
1p/19q-codeletion was detected by fluorescence-in situ-hybridization analysis on samples of paraffin-embedded tumor tissues, as reported previously [9], according to the guidelines of the European Confederation of Neuropathological Societies [10].
P53 immunohistochemistry
Expression of p53 was performed in 77 patients using immunohistochemical staining (IHC). As addressed by Kelley et al., a positive immunoreaction by IHC in more than 50 % of tumor cell nuclei was interpreted as being a presence of TP53 mutation [11].
Treatment/radiotherapy
The postoperative adjuvant therapy was RT alone, CT alone, or sequential radio- plus chemotherapy (RT-CT).
Radiotherapy was prescribed to 60 or 59.4 Gy in 1.8–2 Gy fractions to the surgical cavity and the contrast-enhancing tumor region visible in the post-operative T1 weighted MRI plus a margin of 20 mm in all directions, excluding anatomical borders like bone, tentorium or falx. Non-contrast-enhancing tumor areas detected in T2 or FLAIR were always included in the target volume, if necessary by enlarging the above mentioned margin. Target volumes were compromised in vicinity to organs at risk, e.g. brainstem or optical nerves. From 2010 on, the target volume concept was slightly changed and specified to the definition of a clinical target volume (CTV) consisting of the surgical cavity and contrast-enhancing tumor areas (“gross tumor volume, GTV”) plus 20 mm and a planning target volume (PTV) consisting of the CTV+ a further margin of 3–5 mm, thus enlarging the total distance from the GTV to the PTV to 23–25 mm.
Treatment/chemotherapy
The CT was applied with alkylating agents including: procarbazine/CCNU/vincristine (PCV), temozolomide (TMZ), or rarely nimustine (ACNU). Of 109 patients, 82 patients underwent chemotherapy after the malignant transformation of their low-grade astrocytoma (57 patients received sequentially RT-CT and 25 only CT). 57 patients received PCV (70 %) as adjuvant chemotherapy after malignant progression, 23 received TMZ and two nimustine. The interval between end of RT and initiation of CT was in average 4 weeks.
There were no specific molecular or clinical criteria for the treatment modalities. E.g. most patients who underwent RT alone were treated because this treatment strategy was favored for malignant glioma patients in the late 1990s. Other patients who underwent RT alone or received CT alone, had secondary anaplastic astrocytomas WHO Grade III which were treated following the recommendations of the NOA-4 trial (RT or CT alone) [12].
Additionally, it should be mentioned that 13 patients received an early RT for their low-grade tumor, which was a common treatment strategy in the 1990s. All of those patients were treated with CT alone after progression to a sHGA. On the other hand, none of the patients in the entire group underwent chemotherapy alone at the time of the LGA-diagnosis.
Consequently, we focused our outcome comparison only on patients treated with RT alone or with sequential RT-CT.
In this retrospective analysis, only patients with proven astrocytic tumor histology of their low-grade tumor were included. Therefore, we excluded patients with pure oligodendrogliomas. Furthermore, we excluded sHGA patients with progression from anaplastic astrocytomas, or those without available tissue for molecular analysis. In addition, patients lost to follow-up were not considered in our analysis.
Progression and survival
Progression-free survival (PFS) was defined as the time from first diagnosis of sHGA to tumor progression or end of follow-up. Overall survival (OS) was the time from the day of first confirmation of a sHGA to death or end of follow-up. All patient data were updated on January 09, 2015. The minimum follow up was 1 year after the diagnosis of a sHGA, developed from histologically confirmed LGA WHO II. Patients who were operated on a sHGA with less than one year of follow-up were excluded.
Statistical analysis
Kaplan–Meier survival curves were plotted and the log-rank test was used to investigate survival differences among groups. The prognostic significance of the aforementioned molecular marker for PFS and OS were first analyzed by univariate analysis. The Fisher’s exact test was used to test for association of clinical variables and molecular markers. Cox proportional hazards modeling was performed, adjusting for predictors associated with survival on univariate analysis. A p value <0.05 was considered statistically significant.
Results
The clinical and molecular characteristics of the 109 patients—in relation to their treatment—are summarized in Table 1. The median OS from the diagnosis of LGA to last follow-up in the entire group was 8 years (1.4–27.3 years). It was 8.9 years (1.65–27.3) in the IDH-mutant tumor group and 4.9 years (1.4–19.4) in the IDH wild-type group. The interval between LGA diagnosis and sHGA for all 88 IDH-mutant LGA patients (time to malignant transformation) was 4.8 years (0.6–18.6 years). The median time until malignant transformation of LGA patients with IDH mutation and early RT was 6.7 years (1.1–18.6 years), which was significantly longer than in IDH-mutant LGA patients who did not undergo early RT with 4.1 years (0.7–17.4 years, p < 0.05).
The median age at sHGA diagnosis was 42.5 years (23.4–76.8 years). 56 % of the patients were male (n = 61). 88 patients harbored IDH1 or -2 mutations (80.7 %; IDH1, n = 87; IDH2, n = 1), 30 patients had a 1p/19q-codeletion (27.5 %), and 69 patients (63.3 %) exhibited a methylated MGMT-promoter status. Of 77 patients, a p53 overexpression was present in 32 cases (41.5 %). The initial histology of the low-grade tumor was diffuse astrocytoma grade II in 74 patients (67.8 %) and mixed oligo-astrocytoma grade II in 35 patients (32.1 %). The median follow-up of this cohort from the initial diagnosis was 9.8 years, and 62 patients (57 %) had died.
Of 88 patients with an IDH mutation, 30 patients had 1p/19-codeleted tumors (34.1 %) and 58 a methylated MGMT-promoter status (66 %) (The clinical and molecular characteristics of the 88 patients with an IDH mutation are summarized in Table S1, suppl. Data). The median OS of all patients in the entire group was 3.4 years (1.9–6.7 years), 5.3 years (1.9–17.4 years) in the IDH-mutant group, and 1.3 years (0.3–6.8 years) in the IDH wild-type group (p = 0.12). The median PFS of all patients was 2.0 years (1.4–2.6 years), it was 2.2 years (1.7–2.5 years) in the IDH-mutant cohort, compared with 1.0 years in the IDH wild-type group (0.3–1.4 years). The longest survival was seen in IDH-mutant, 1p/19q-codeleted sHGA patients with a methylated MGMT-promoter (n = 21). These patients had a median OS of 5.7 years (3.3–18 years).
The postsurgical treatment of the 88 IDH-mutant sHGA included: RT alone in 17 patients (19.3 %), a sequential RT-CT in 48 patients (54.5 %) and CT alone in 20 patients (22.7 %). Three patients in the IDH-mutant group did not receive any adjuvant treatment after the initial diagnosis due to poor neurological function after surgery.
The OS in patients who received RT-CT was 6.5 years, which is significantly longer compared with those who underwent RT alone (1.2 years, hazard ratio 0.35, 95 % CI 0.32–0.51. p = 0.011; Fig. 1). Moreover, the PFS in the RT-CT group was 3.4 years, which was significantly longer compared with 0.9 years in the group treated with RT alone (p = 0.007).

Kaplan–Meier estimates of overall survival (OS) in relation to treatment [radio-chemotherapy (RT-CT) or radiotherapy (RT) or chemotherapy (CT)] in all 88 IDH-mutant secondary high-grade astrocytomas. The OS in patients who received RT-CT was significantly longer compared with those who underwent RT alone (1.2 years, hazard ratio 0.35, 95 % CI 0.32–0.51. p = 0.011; median OS RT-CT group: 6.5 years, median OS RT group: 1.2 years)
Among sHGA patients with an IDH mutation but without a 1p/19q-codeletion and with any adjuvant treatment (n = 55), there was a significantly improved OS in the RT-CT cohort (n = 33) in comparison to patients who received RT alone (n = 10, 6.4 vs. 1.2 years, hazard ratio 2.7, 95 % CI 1.1–6.5, p = 0.022). Furthermore, a trend toward improved OS was seen in the RT-CT group when compared with those who underwent CT alone (n = 12, 6.4 vs. 1.6 years, hazard ratio 2. 95 % CI 0.8–4, p = 0.097; Fig. 2). In other words, patients who received RT alone had a 2.7 fold higher likelihood of dying in comparison to those who received RT-CT. Also, the median PFS of the RT-CT cohort was significantly longer than in the RT group (3.12 vs. 0.5 years, p = 0.005; Fig. 3). There were significantly more 5- and 10-year survivors after RT plus CT than with RT alone in the IDH-mutant cohort (58 and 39 vs. 29 and 29 %; p < 0.05; Figs. 1 and 2).

Kaplan–Meier estimates of overall survival (OS) in relation to treatment [Radiochemotherapy (RT-CT) or Radiotherapy (RT) or chemotherapy (CT)] in IDH-mutant and 1p/19q non-codeleted patients. The curves estimate a significantly improved OS in the RT-CT cohort (n = 33) in comparison to patients who received RT alone (n = 10, hazard ratio 2.7, 95 % CI 1.1–6.5, p = 0.022, median OS 6.4 versus 1.2 years)
All patients with 1p/19q-codeleted sHGA (n = 30; all of them oligoastrocytomas) harbored an IDH mutation; they had a median OS of 5.7 years (2.8–8.5 years), which was non-significantly longer than the OS of patients with IDH-mutant non-codeleted tumors (3 years, range 0.8–5.3 years, p = 0.30). Of 30 1p/19q codeleted sHGA patients, 7 received RT and 15 RT-CT; all of those PCV.
After adjustment of median OS in relation to treatment, the median OS in patients with codeleted IDH-mutant sHGA after RT-CT (n = 15) was not reached in comparison with non-codeleted IDH-mutant sHGA patients (n = 33, median OS 6.5 years, 1.9–11 years); however, this was not significant: p = 0.79.
The median OS of sHGA patients with a methylated MGMT-promoter who received RT-CT (n = 39) was not significantly different in comparison to patients with a methylated MGMT-promoter who received RT or CT alone (n = 66; 8.2 vs. 9.43 years, p = 0.089). However, the interaction of IDH and methylated MGMT-promoter as a parameter in the multivariate analysis was proven to be significant (p = 0.019, data not shown). Remarkably, patients with an unmethylated MGMT-promoter had a similarly long OS after RT-CT compared to those who received RT alone (n = 19, 9.46 years, p = 0.801).
Of the 32 patients with p53 overexpression, 27 patients harbored an additional IDH mutation (84.3 %) and 18 patients a methylated MGMT-promoter (56.2 %). The median OS of the IDH-mutant sHGA patients who presented a p53 overexpression was 3.2 years (0.3–7 years). Their median OS was non-significantly longer than the median OS of patients with IDH-mutant and codeleted tumors without p53 over expression (n = 30, median OS 5.7 years, p = 0.58).
In the stepwise multivariable Cox model for OS of all 88 IDH-mutant sHGA patients, which incorporated the variables of age, gender, 1p/19q-codeletion, MGMT-promoter methylation status, and treatment modalities (RT alone vs. RT-CT vs. CT alone), survival was strongly associated with only one factor, namely, RT-CT as adjuvant treatment at diagnosis of a sHGA (HR 2.6; 95 % CI 1.2–5.5; p = 0.014; Table 2).
After addition of p53 overexpression as a variable to the stepwise multivariable Cox model for OS (n = 62), RT-CT continued to be the only significant factor which was associated with a prolonged survival (p = 0.010).
Of the 17 HGA patients with RT alone after malignant progression, eight patients underwent a second surgery at recurrence followed with PCV in three cases, TMZ in one case, Gliadel wafer in two cases and no CT in two cases. The remaining five patients did not receive further therapy due to bad neurological conditions upon massive tumor progression, whereas four patients with a secondary anaplastic astrocytoma are still progression-free at follow-up.
Discussion
This retrospective long-term study shows that the addition of alkylating agents (mainly PCV) to RT significantly improves progression-free and overall survival in IDH-mutant secondary high-grade astrocytoma patients, regardless of 1p/19-codeletion status. In contrast, neither the methylation of MGMT-promoter nor TP53 overexpression demonstrates a significant treatment-related increase in patients’ survival in this study.
The RTOG 9402 trial demonstrated an improved outcome in 1p/19q-codeleted anaplastic oligodendroglial tumor patients when PCV was added to RT [4,]. The median OS of 1p/19q non-codeleted IDH-mutant sHGA patients who received RT-CT or RT in our study was comparable with the reported OS in the RTOG 9402 trial (Table 3) [4, 5]. These results implicate that IDH-mutant sHGA patients might show the same clinical benefit after sequential CT–RT like patients with de novo anaplastic oligodendroglial gliomas. As opposed to the EORTC 26951 trial, MGMT-promoter methylation failed to show a significant association with patients’ outcome in our study as a solely parameter. However, the combination of IDH mutation and methylated MGMT-promoter was superior to MGMT-promoter methylation alone in predicting survival of sHGA patients in our study. This is in accordance to recently published data [13–15].
Further on, multivariate analyses identified only one significant favorable prognostic variable in the IDH-mutant patients’ cohort: the use of RT-CT. In this cohort, the risk of dying was increased by a factor of 2.7 when patients underwent only RT compared to RT-CT. None of the other investigated factors—such as 1p/19q-codeletion, MGMT-promoter methylation, p53 overexpression, age or gender—had a significant influence on patients’ survival as a solely parameter. Additionally, these data are in concordance with the results of the RTOG 9402 and EORTC 26951 trials [4, 5].
In general, IDH-mutant gliomas, regardless of their WHO grade, are markedly different in their clinical presentation, genetic alterations, and overall natural history when compared with IDH wild-type gliomas. As reported previously, post-malignant progression survival among patients with IDH-mutant low-grade astrocytomas is improved compared to patients with IDH wild-type astrocytomas [7, 16]. In this regard, our recent study may provide so far a strong evidence for differential responsiveness and outcomes in relation to different treatment modalities (RT vs. RT-CT) in IDH-mutant sHGA patients. This observation supports the hypothesis that IDH mutation might not only be a prognostic but also a predictive marker for survival in sHGA patients.
Further typical anatomical and radiological characteristics of IDH-mutant gliomas are the frequent location in the frontal lobe, the bi-hemispheric involvement, and the relatively low enhancement in comparison to IDH wild-type high-grade gliomas [9]. IDH-mutant gliomas are more amenable to surgical resection of enhancing tumor; a fact which might contribute to the better prognosis observed in this patient population [17]. Furthermore, an increased radiosensitivity of IDH-mutant gliomas has been reported. This effect was clinically translated as improved survival [16]. Furthermore, recent in vitro results [18] and clinical data from EORTC trial 26951 [19] support the hypothesis that IDH-mutant gliomas are highly sensitive to RT. This sensitivity might be due the fact that wild-type IDH enzymes protect cells against gamma irradiation by maintaining NADPH levels that buffer against irradiation-induced reactive oxygen species, thus protecting cells from apoptosis [20, 21]. Furthermore, a recently published study demonstrated that IDH-mutant glioblastomas have a more pronounced radiographic response to chemo-radiation therapy by serial quantitative MR volumetry than do their IDH wild-type counterparts [22].
We acknowledge that our retrospective study has several limitations including: The sample size for each treatment modality was small; the non-randomized allocation of treatment and the administration of different alkylating agents (CCNU, TMZ or rarely ACNU). A further limitation is that TP53 mutations are identified indirectly by p53 overexpression rather than sequencing techniques. In addition, the fact that out of 13 of 25 patients received an early RT after the diagnosis of a low-grade astrocytoma—all treated with CT alone after malignant progression—is one of the limitations which led us to focus our outcome comparison only on patients treated with RT alone or RT-CT. It should be mentioned, that in our cohort of patients with sHGA there was no suggestion for improved outcome after early RT compared to those patients who received RT at their HGA diagnosis.
On the other hand, our study is the first study which demonstrates a treatment-related survival improvement in IDH-mutant secondary high-grade astrocytoma patients—different from the RTOG 9402 and EORTC 26951 trials which focused on anaplastic oligodendroglial tumors. An additional strength of the current study is the long-term follow-up (median of 9.8 years) in this rare patient subpopulation. However, the results of the ongoing CATNON (NCT00626990), multicenter phase III trial will shed more light on the best treatment concepts of affected patients. This trial will help to determine the predictive value of IDH mutation in anaplastic gliomas.
In summary, sHGA are heterogeneous tumors with a variable response to treatment. This clinical variability underlines the urgent need for marker that can reliably guide clinical decision-making. Our long-term follow-up data have suggested that patients with IDH-mutant sHGA should be treated with addition of alkylating chemotherapy to radiation, irrespective 1p/19q deletion status. Radio-chemotherapy (mostly PCV) has significantly improved progression-free and overall survival in our patients cohort compared with radiotherapy or chemotherapy alone. On the other hand, MGMT-promoter methylation status was merely in combination with IDH mutation a marker for outcome in relation to radio-chemotherapy.
References
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. doi:10.1126/science.1164382
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. doi:10.1056/NEJMoa0808710
Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, Dias-Santagata D, Ellisen LW, Borger DR, Fendt SM, Vander Heiden MG, Batchelor TT, Iafrate AJ, Cahill DP, Chi AS (2014) Targetable signaling pathway mutations are associated with malignant phenotype in idh-mutant gliomas. Clin Cancer Res 20:2898–2909. doi:10.1158/1078-0432.CCR-13-3052
Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, Buckner JC, Fink KL, Souhami L, Laperriere NJ, Huse JT, Mehta MP, Curran WJ (2014) Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol 32:783–790. doi:10.1200/JCO.2013.49.3726
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Enting RH, French PJ, Dinjens WN, Vecht CJ, Allgeier A, Lacombe D, Gorlia T, Hoang-Xuan K (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31:344–350. doi:10.1200/JCO.2012.43.2229
Louis DN, Ohgaki H, Wiestler O, Cavenee W, Burger P, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumors of the central nervous system. IARC Press, Lyon
Juratli TA, Kirsch M, Geiger K, Klink B, Leipnitz E, Pinzer T, Soucek S, Schrok E, Schackert G, Krex D (2012) The prognostic value of IDH mutations and MGMT promoter status in secondary high-grade gliomas. J Neurooncol. doi:10.1007/s11060-012-0977-2
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343:1350–1354. doi:10.1056/NEJM200011093431901
Juratli TA, Engellandt K, Lautenschlaeger T, Geiger KD, von Kummer R, Cerhova J, Chakravarti A, Krex D, Schackert G (2013) Is there pseudoprogression in secondary glioblastomas? Int J Radiat Oncol Biol Phys 87:1094–1099. doi:10.1016/j.ijrobp.2013.09.036
Woehrer A, Sander P, Haberler C, Kern S, Maier H, Preusser M, Hartmann C, Kros JM, Hainfellner JA, Societies RCotECoN (2011) FISH-based detection of 1p 19q codeletion in oligodendroglial tumors: procedures and protocols for neuropathological practice—a publication under the auspices of the Research Committee of the European Confederation of Neuropathological Societies (Euro-CNS). Clin Neuropathol 30:47–55
Kelley TW, Tubbs RR, Prayson RA (2005) Molecular diagnostic techniques for the clinical evaluation of gliomas. Diagn Mol Pathol 14:1–8
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, Sabel MC, Koeppen S, Ketter R, Meyermann R, Rapp M, Meisner C, Kortmann RD, Pietsch T, Wiestler OD, Ernemann U, Bamberg M, Reifenberger G, von Deimling A, Weller M (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880. doi:10.1200/JCO.2009.23.6497
Molenaar RJ, Verbaan D, Lamba S, Zanon C, Jeuken JW, Boots-Sprenger SH, Wesseling P, Hulsebos TJ, Troost D, van Tilborg AA, Leenstra S, Vandertop WP, Bardelli A, van Noorden CJ, Bleeker FE (2014) The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. doi:10.1093/neuonc/nou005
SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z, LanXiao W, Fei Y (2012) IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci 103:269–273. doi:10.1111/j.1349-7006.2011.02134.x
Wick W, Meisner C, Hentschel B, Platten M, Schilling A, Wiestler B, Sabel MC, Koeppen S, Ketter R, Weiler M, Tabatabai G, von Deimling A, Gramatzki D, Westphal M, Schackert G, Loeffler M, Simon M, Reifenberger G, Weller M (2013) Prognostic or predictive value of MGMT promoter methylation in gliomas depends on IDH1 mutation. Neurology 81:1515–1522. doi:10.1212/WNL.0b013e3182a95680
Juratli TA, Kirsch M, Robel K, Soucek S, Geiger K, von Kummer R, Schackert G, Krex D (2012) IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol 108:403–410. doi:10.1007/s11060-012-0844-1
Beiko J, Suki D, Hess KR, Fox BD, Cheung V, Cabral M, Shonka N, Gilbert MR, Sawaya R, Prabhu SS, Weinberg J, Lang FF, Aldape KD, Sulman EP, Rao G, McCutcheon IE, Cahill DP (2014) IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro Oncol 16:81–91. doi:10.1093/neuonc/not159
Li S, Chou AP, Chen W, Chen R, Deng Y, Phillips HS, Selfridge J, Zurayk M, Lou JJ, Everson RG, Wu KC, Faull KF, Cloughesy T, Liau LM, Lai A (2013) Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol 15:57–68. doi:10.1093/neuonc/nos261
van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, Frenay M, Tijssen CC, Lacombe D, Idbaih A, van Marion R, Kros JM, Dinjens WN, Gorlia T, Sanson M (2010) IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res 16:1597–1604. doi:10.1158/1078-0432.CCR-09-2902
Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W, Troost D, Vandertop WP, Bardelli A, Van Noorden CJ (2010) The prognostic IDH1(R132) mutation is associated with reduced NADP+ -dependent IDH activity in glioblastoma. Acta Neuropathol 119:487–494. doi:10.1007/s00401-010-0645-6
Kim SY, Lee SM, Tak JK, Choi KS, Kwon TK, Park JW (2007) Regulation of singlet oxygen-induced apoptosis by cytosolic NADP+ -dependent isocitrate dehydrogenase. Mol Cell Biochem 302:27–34. doi:10.1007/s11010-007-9421-x
Tran AN, Lai A, Li S, Pope WB, Teixeira S, Harris RJ, Woodworth DC, Nghiemphu PL, Cloughesy TF, Ellingson BM (2014) Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol 16:414–420. doi:10.1093/neuonc/not198
Acknowledgments
The authors thank sincerely Mrs. Silke Soucek for her support by the statistical analyses. The study was presented at the 19th Annual scientific meeting of the Society for Neuro-Oncology in Miami.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
The authors did not receive any funding that support this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1: The life table curve showed a significantly prolonged median progression free survival in the radio-chemotherapy cohort (RT-CT) in comparison with the radiotherapy (RT) group (3.4 vs. 0.9 years). p = 0.007 (Wilcoxon-Test).
Rights and permissions
About this article

Cite this article
Juratli, T.A., Lautenschläger, T., Geiger, K.D. et al. Radio-chemotherapy improves survival in IDH-mutant, 1p/19q non-codeleted secondary high-grade astrocytoma patients. J Neurooncol 124, 197–205 (2015). https://doi.org/10.1007/s11060-015-1822-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1822-1