
Abstract
In this work, MoS2-reduced graphene oxide (rGO) composites were prepared by a simple hydrothermal reaction and characterized by SEM, Raman, XRD, and electrochemical impedance spectroscopy (EIS). The obtained MoS2-rGO/GCE was used for electrochemical investigation of silver nanoparticles (AgNPs) by cyclic voltammetry (CV), differential pulse voltammetry (DPV), and chronocoulometry (CC). It was found that the MoS2-rGO/GCE exhibited good stability and could enhance current response of AgNP electrooxidation effectively. On the basis, a new electrochemical method was developed to detect AgNPs in aqueous solution. The proposed method exhibited satisfactory analytical performance for AgNP detection with the limit of detection (LOD) as 2.63 ng/L and the linear range from 5 to 120 ng/L. The determination assays in water samples by MoS2-rGO/GCE were also discussed. The assay recoveries in water samples were obtained as 94.9–99.4%. That suggested a promising application prospect in environmental analysis.
Graphical abstract
The MoS2-rGO/GCE enhanced the electrochemical response of AgNPs, which are attributed from the Ag absorbability of MoS2 and the electrochemical signal amplification of rGO. This finding resulted in a new electrochemical approach to simple detection of AgNPs in water

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Silver nanoparticles (AgNPs) were widely used in various applications because they can improve the performance of many consumer and industrial products, for example, in antibacterial medicine and devices, personal care products, water treatment, photocatalysis, and disinfection (Dankovich and Gray 2011; Rizzello and Pompa 2014; Yu et al. 2013). However, the growing use of AgNPs and releasing into environment has raised concerns about their potential implications to the environment and human health (Marambio-Jones and Hoek 2010; Loeschner et al. 2013). To address this issue, an urgent requirement has been the development of analytical techniques to detect the presence and the concentration of AgNPs in environment and biological tissues. Series of analytical techniques, such as ICP-MS (Artiaga et al. 2015; Paramelle et al. 2014), UV spectra (Chao et al. 2011), cloud point extraction (Alvaredo et al. 2013), chromatography (Cheng and Compton 2014), and electrochemical techniques (Zhou et al. 2011; Zhang et al. 2000) have been developed for detection and characterization of AgNPs. Among the above techniques, the electrochemical technique has been accepted as a powerful tool to detect AgNPs due to its portability, low cost, and real-time analysis. In particular, the analytical performance of electrochemical techniques always largely depends on electrode materials (Uslu and Ozkan 2007; Asadi et al. 2014). For that, novel electrode materials, especially with good adsorption performance for AgNPs and excellent electrochemical properties, are still expected in AgNP detection.
Molybdenum disulfide (MoS2) and related materials, which are considered promising electrode materials, have attracted a significant interest due to their unique 2D layered structure, good electrochemical properties, and outstanding semiconductor features (Matte et al. 2010). Due to the weak van der Waals forces, MoS2 materials exhibited typical 2D layered structure with high specific surface area (Li et al. 2014). The high surface area and sulfur element endue MoS2 materials promising adsorption ability for Ag by metal-sulfur covalent coordination (Pan et al. 2014). Zhu reported the adsorption and in situ reduction of Ag+ at MoS2 surface to form AgNPs-MoS2 composite by coordination effect (Zhao et al. 2015). Li also reported MoS2 nanosheets were used as a reaction substrate of AgNPs; the resultant AgNPs-MoS2 exhibited higher quality SERS activity (Xie et al. 2013). However, the aggregation of layered structure and the poor intrinsic conductivity of MoS2 still limit its further electrochemical application and required improvement (Huang et al. 2012). Composition with graphene is considered an effective approach to enhance the electrochemical properties and stability of semiconductor materials, due to the large specific surface area, good electric conductivity, and stable physicochemical properties (Zhao et al. 2015). Wang reported the layered MoS2 nanosheets supported on 3D graphene aerogel and the resultant MoS2-GA exhibited significant enhancement in electrochemical catalytic activity (David et al. 2014). Singh also reported the composition of exfoliated MoS2 and rGO flakes. The obtained MoS2-rGO was used for sodium-ion battery and exhibited enhanced electrochemical capacity and better Na+ ion cycling ability (Mohan et al. 2017).
Here, we achieved a simple route to synthesize MoS2-rGO composite by hydrothermal reaction. The as-prepared MoS2-rGO was used to modify glassy carbon electrode (GCE). Compared with single MoS2/GCE, MoS2-rGO/GCE exhibited enhanced cycling stability. Moreover, in the electroanalytical application of AgNPs, MoS2-rGO/GCE performed better electrochemical response currents than GCE, MoS2/GCE, or rGO/GCE. After the optimization of pH and enrichment time, the proposed electrode performed good linear relationship and satisfactory sensitivity for AgNPs. This study maybe provides a new opportunity for AgNPs detection.
Experimental
Reagents, instruments, and measurements
Ammonium heptamolybdate, potassium ferricyanide, potassium ferrocyanide, trisodium citrate, thiourea, ammonia, nitric acid, ethanol, sodium hydroxide, sodium dihydrogen, sodium dihydrogen phosphate, and disodium hydrogen phosphate were purchased from Chongqing chemical reagent factory. Silver nitrate and sodium chloride were purchased from Kelong chemical reagent factory. Gum acacia were purchased from Aladdin Chemicals Corporation. All the solutions were prepared and diluted by ultrapure water. AgNP standard solution was prepared by previous literature (Ferrari and Basko 2013) and was diluted to required concentration. Dynamic light scattering (DLS) (Fig. S1) and SEM (Fig. S2) results exhibited the particle size of AgNPs was uniform. The average size (Zd) and standard deviation (Σ) of AgNPs were measured by DLS as 5.26 nm and 0.87 nm, respectively. The polydisperse index (PDI) was calculated as 0.16, by the formula PDI=Σ2/Zd2. The obtained AgNP solutions were stored in dark place at 4 °C for future use.
Dynamic light scattering (DLS) and zeta potential of AgNPs were carried by a Zetasizer Nano-ZS90 instrument (Malvern, UK). Scanning electron microscopy (SEM) images were taken on a Hitachi model S-4800 field emission scanning microscope (Hitachi, Japan). XRD measurements were carried on XD-3 X-ray diffractometer (Puxi, Beijing, China). Raman spectra were obtained by a Renishaw inVia Raman microscope (Renishaw, UK).
Cyclic voltammetry (CV), chronocoulometry (CC), and differential pulse voltammetry (DPV) were performed on a CHI660e electrochemical workstation (Chenhua, China). Electrochemical impedance spectroscopy (EIS) measurements were performed on the IM6ex electrochemical workstation (ZAHNER, Germany). All the abovementioned electrochemical experiments were carried out with a conventional three-electrode cell at room temperature. The three-electrode cell was employed including a modified glassy carbon electrode (GCE) as the working electrode, a platinum wire electrode as the counter electrode, and an Ag/AgCl electrode as the reference electrode. Before LSV, DPV, or CV measurement, working electrode was immersed in electrolyte for a certain adsorption time.
Preparation of rGO, MoS2 and MoS2-rGO composite
The process of material preparation and electrode modification is briefly shown in Scheme 1. rGO and MoS2-rGO were prepared by hydrothermal reduction method based on previous reports with minor modification (David et al. 2014). Eighty-milligram graphene oxide (GO) powders were dispersed in 40 mL deionized water. The obtained GO dispersion was transferred into a Teflon reaction kettle and heated at 150 °C for 12 h. After cooling down and freeze drying, the rGO was obtained.

The brief schematic graph for the process of material preparation and electrode modification
MoS2-rGO was synthesized by the hydrothermal reaction of (NH4)6Mo7O24 and CH4N2S on rGO surface. Similar to the above procedure, 80 mg GO dispersion was transferred into a Teflon reaction kettle and heated at 150 °C for 12 h. Then, 200 mg (NH4)6Mo7O24 and 2g CH4N2S were added in dispersion. Then, the mixed dispersion was transferred into another Teflon reaction bottle and heated at 180 °C for 24 h. After the bottle cooled to room temperature, the product was washed by water and ethanol to remove impurities. After the final drying, MoS2-rGO composite was obtained.
For comparison, pure MoS2 without rGO modification was also prepared by similar procedures. Two-hundred-milligram (NH4)6Mo7O24 and 2g CH4N2S were dispersed in 40 mL deionized water. The dispersion was transferred into Teflon reaction bottle and heated at 180 °C for 24 h. After the cooling to room temperature, purification, and drying, pure MoS2 was obtained.
Preparation of MoS2-rGO modified glassy carbon electrodes
Prior to modification, the GCE (Φ = 3 mm) was polished carefully on a polishing cloth with 0.05 mm alumina powder and rinsed with ultrapure water. Before use, the GCE was treated by sonication in 50 wt% ethanol aqueous solution. Ten-milligram MoS2-rGO composite was dispersed in 10 mL ultrapure water to form MoS2-rGO dispersion (1 mg/mL). Five microliters of MoS2-rGO dispersion was drop-casted onto GCE surface. After the GCE dried in air, MoS2-rGO/GCE was obtained. For comparison, MoS2/GCE and rGO/GCE were prepared with similar steps by drop-casting same amount of MoS2 dispersion or rGO dispersion as the modifier of GCE, respectively.
Results and discussion
Characterization of materials
The obtained material was characterized by SEM. Figure 1A and B show the SEM images of rGO and MoS2-rGO, respectively. In Fig. 1A, rGO exhibited a typical crumpled and wrinkled flake-like structure. After the growth of MoS2, MoS2-rGO remained original flake structure and assembled with MoS2 nanosheets on rGO surface. The morphology suggested the successful composition of MoS2-rGO. At higher magnification rate, MoS2-rGO displayed scattered flowerlike architectures (Fig. 1C and D). The scattered flowerlike architectures indicated rGO surface enhanced the dispersibility of MoS2 nanosheets effectively.

SEM images of rGO (A) and MoS2-rGO (B, C, and D)
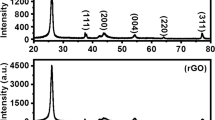
Figure 2A displays the Raman spectrum of rGO and MoS2-rGO composite. For rGO, two characteristic peaks, which were corresponding to the D band and G band of carbon materials, appeared at 1328 cm−1 and 1598 cm−1 respectively. For MoS2-rGO, the two peaks still existed without any peak shift after the composition of MoS2. That indicated MoS2-rGO maintained the carbon structure of rGO. In general, the intensity ratio value of D band to G band (ID/IG) is an indicator of surface defect in carbon materials (Zheng et al. 2014). The intensity ratio value of D band to G band (ID/IG) for MoS2-rGO (1.13) was higher than that for pure rGO (0.97), demonstrating that the introduction of MoS2 nanosheets increased the carbon defects of rGO surface. Moreover, the emergence of two new characteristic peaks at 371 cm−1 and 406 cm−1 were associated with the in-plane E12g and out-of-plane A1g vibrational modes of hexagonal MoS2, respectively (Lee et al. 2010). The result showed MoS2 maintained original structure after the modification with rGO. The XRD patterns of rGO, MoS2, and MoS2-rGO are shown in Fig. 2B. The diffraction peak at around 23.5° could be assigned to graphene (002) diffraction peak for rGO (Liu et al. 2012). After the composition, MoS2-rGO maintained the (002) peak of rGO. Compared with the simulation XRD pattern (JCPDS 37-1492), the prominent peaks of MoS2-rGO, including (002) at 14°, (100) at 33°, (103) at 39°, and (106) at 59°, matched well with the hexagonal structure of MoS2. That indicated the crystal structure of MoS2 was not changed in composition process.

Raman spectra (A) and XRD patterns (B) of rGO and MoS2-rGO
Electrochemical characterization of electrodes
The electrochemical characterization of electrodes (GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE) in electrolyte containing 1.0×10−3 M [Fe(CN)6]3−/[Fe(CN)6]/4− was investigated by EIS, CV, and CC. EIS experiment was a powerful tool to study the electrode interface properties. The EIS results (Nyquist plots, −Z″ vs. Z′) for four electrodes are shown in Fig. 3A. For electrode/electrolyte system, EIS always showed a typical curve containing an obvious semicircle in the high frequency region and an approximate straight line in the low frequency region (Lisdat and Schäfer 2008; Chang and Park 2010). The diameters of the semicircle corresponded to the interfacial charge-transfer resistance (Rct) which usually represented the resistance of electrochemical reactions on the electrode and was often called the Faraday resistance. The interfacial charge-transfer resistance (Rct) values of GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE could be achieved as 79.48 Ω, 166.5 Ω, 677 Ω, and 233.7 Ω by simulation, respectively. The highest Rct values of MoS2/GCE indicated the poor electric conductivity of single MoS2. Compared with MoS2/GCE, MoS2-rGO/GCE exhibited lower Rct value. That phenomenon meant rGO had excellent electric conductivity and could promote the charge transfer ability of MoS2. This conclusion was consistent with the CV results (Fig. 3B). A pair of electrochemical peaks, which were corresponding to the redox of [Fe(CN)6]3−/[Fe(CN)6]/4−, were presented at all four electrodes’ CV results (Fig. 3B). The area under the CV curve at MoS2-rGO/GCE was found to be larger than that at other three electrode. This observation indicated that the hybrid material showed good capacitive behavior due to a synergistic contribution of both MoS2 and rGO (Kamila et al. 2017; Joseph et al. 2018). Although capacitive current seems dominant for the four electrodes, the oxidation and reduction peaks at MoS2-rGO/GCE (Ipa=102 mA, Ipc=107 mA) exhibited higher currents than those at GCE (Ipa=67 mA, Ipc=63 mA) and MoS2/GCE (Ipa=82 mA, Ipc=85 mA). This indicated that the combination of rGO could promote the charge transfer ability and then enhance the electrochemical response currents effectively. The electrode stability was further discussed in Fig. 3C. With the three continuous CV tests, the electrochemical behavior of MoS2/GCE changed greatly, including the decrease of response currents and the visible shift of peak potentials. On the contrary, there were no obvious potential or current changes occurred in three continuous CV tests for MoS2-rGO/GCE. That indicated MoS2-rGO/GCE exhibited better electrochemical stability than MoS2/GCE and the stability which might attribute to the composition of rGO. The CC results of GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE are displayed in Fig. 3D. The highest slope value of MoS2-rGO/GCE indicated the satisfied electrode performance in the electrochemical redox of [Fe(CN)6]3−/[Fe(CN)6]/4−. Both the CV and CC results showed that the MoS2-rGO exhibited the biggest capacitive current than rGO or MoS2. That was attributed to synergistic contribution of the pseudocapacitance from MoS2 and the double layer capacitance from rGO (Kamila et al. 2017; Joseph et al. 2018). Furthermore, the electrochemical active surface area (A) of the electrodes could be calculated by the CC results of K3Fe(CN)6 (Fig. 3D) with Anson equation (Cheng et al. 2013): Q=2nFAC0D1/2π−1/2t1/2+Qdl+Qads, where F is the Faraday constant (F=96500 C·mol−1), n is the number of electron transferred (n=1), D is the diffusion coefficient of K3Fe(CN)6 (D=6.3×10−6 cm2/s), t is the reaction time, Qdl is the charge of electric double-layer capacitor, and Qads is the adsorption charge. In the equation, Q and t1/2 exhibited linear relationship with the linear slope of 2nFAC0D1/2π−1/2. Based on the linear slope value of Q~t1/2 curves, the electrochemical surface area (A) of GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE could be calculated as 0.0103 cm2, 0.0118 cm2, 0.0166 cm2, and 0.03778 cm2, respectively. The highest electrochemical surface area of MoS2-rGO/GCE would benefit the enrichment and adsorption of analytes.

EIS spectra (A), cyclic voltammetries (B), and chronocoulometries (D) for GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE in [Fe(CN)6]3−/[Fe(CN)6]/4−solution; CV stability tests (C) for MoS2/GCE and MoS2-rGO/GCE
Electrochemical behaviors of AgNPs at MoS2-rGO/GCE
Figure 3A displays the CV results of AgNPs at GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE. At GCE, there is no obvious response current of AgNPs. At rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE, the oxidation peak of AgNPs appeared significantly. Compared with rGO/GCE or MoS2/GCE, AgNPs at MoS2-rGO/GCE showed higher oxidation current. The results indicated that MoS2-rGO/GCE exhibited enhanced electrochemical responsibility of oxidation of AgNPs and could be employed as a potential sensing platform of AgNPs. The CC result of blank solution and CC results of AgNPs at four electrodes are obtained in Fig. 4B. The absorption amount of AgNPs in electrochemical reaction could be calculated by CC results and Anson equation (Duan and Huang 2016). The absorption amounts of AgNPs of GCE, MoS2/GCE, rGO/GCE, and MoS2-rGO/GCE were obtained as 6.73×10−12 mol, 2.96×10−11 mol, 3.54×10−11 mol, and 6.35×10−11 mol, respectively. The highest absorption mount indicated MoS2-rGO/GCE exhibited excellent adsorption ability to AgNPs than GCE, rGO/GCE, or MoS2/GCE. That might attribute to the Ag-S coordination effect and the high electrochemical surface area.

CV (A) and CC (B) results for GCE, rGO/GCE, MoS2/GCE, and MoS2-rGO/GCE in AgNP solution
The effect of pH of electrolyte was also discussed in obtained experiment. Figure 5A displays the oxidation peak current values of AgNPs at different pH in the range from 2.77 to 7.71. While pH increased from 2.77 to 4.76, the peak current of AgNPs increased that might be attributed to the electrostatic adsorption between MoS2-rGO and AgNPs; while pH increased from 4.76 to 7.71, the peak current of AgNPs decreased. At electrolyte pH of 4.76, the oxidation peak current achieved the highest value. In order to further explore the influence of pH on electrochemical detection, the zeta potentials of AgNPs and MoS2-rGO with different electrolyte pH were measured (Fig. S3). At electrolyte pH of 4.76, the zeta potential of AgNPs and MoS2-rGO exhibited highest difference value. The large zeta potential difference at pH 4.76 indicated the electrostatic repulsion between MoS2-rGO and AgNPs was minimal, which was beneficial for electrostatic adsorption and electrochemical detection towards AgNPs (Cepriá et al. 2016). Therefore, pH 4.76 was selected as the optimum pH in this work. As an important factor for electrochemical signal amplification, adsorption time was also discussed in obtained detection. Before each LSV measurement, MoS2-rGO/GCE as working electrode was immersed in electrolyte for a certain adsorption time from 0.5 to 8 h, in order to enrich AgNPs onto the electrode surface. After the adsorption procedure, the LSV measurement was carried from −0.4 to 0.6 V, with the scan rate at 50 mV/s. Figure 5B displays the reaction charge values of AgNPs with different adsorption time from 0.5 to 8 h (reaction charge value could be obtained by LSV result). Similar to previous works, the reaction charge increased linearly with the adsorption time approximately (Duan and Huang 2016; Stuart et al. 2014). Considering the detection time and sensitivity in the actual detection, 3 h was selected as the optimized adsorption time for AgNP detection in this work (Cepriá et al. 2016).

Optimization of pH value (A) and adsorption time (B) for AgNPs determination with working potential at 0.27 V
Analytical performance and electrochemical detection
Under the optimal experimental conditions (electrolyte pH=4.76, adsorption time is 3h), the analytical performances for AgNPs of the proposed electrode were evaluated by DPV. Figure 6A displays the DPV results of AgNPs with different concentrations in the range from 5 ng/L to 120 ng/L. As observed in Fig. 6B, the oxidation peak currents (Ipa) exhibited good linear relationship with the AgNP concentrations (c). The linear regression equation between Ipa and c was defined as Ipa (μA)=1.501+0.0709c (ng/L) (R2=0.9952, n=12). Based on the above equation, the AgNP detection method was established with the limit of detection (LOD) as 2.63 ng/L and linear range as 5–120 ng/L. Compared with other electrochemical detection techniques in AgNP detection, our electrode exhibited satisfactory analytical performances, including lower working potential and LOD (Table 1).

DPV curves for different concentrations of AgNPs in MoS2-rGO/GCE (A); the linear relationship between AgNP concentrations and peak current values (B)
To examine the applicability of the proposed method, the detection assays for AgNPs in lake water samples were performed. Prior to testing, the lake water samples were not subjected to any further treatment, except for a simple filtration with filter paper. Lake water samples were added to the electrolyte using the standard addition method. The measurement results are presented briefly in Table 2 and Fig. S4. The detection method for AgNPs exhibited a satisfactory result with the recoveries range from 94.9 to 99.4%. The recoveries demonstrate that the proposed method was potentially applicable for determination of AgNPs in environmental samples. Furthermore, the stability and reproducibility were also considered. The relative standard deviation (RSD) of determination results for the seven MoS2-rGO/GCE electrodes, which were fabricated under same condition, was calculated as 2.6%. Meanwhile, the RSD of determination results for MoS2-rGO/GCE in six repetitive tests was 4.3%. That indicated MoS2-rGO/GCE exhibited satisfactory stability and reproducibility.
The interference of common ions in AgNPs detection was also evaluated. It was found that 1 M of Na+, K+, and PO43−; 1 mM of Ca2+, Mg2+, and NH4+; 0.1 mM of SO42− and NO3−; and 100 μg/L of Cu2+, Pb2+, Zn2+, and Cd2+ did not interfere with the electrochemical responses of 100 ng/L of AgNPs (RSD below ±5%). That indicated that MoS2-rGO/GCE exhibited high anti-interference capacity in AgNP determination.
Conclusion
The main feature of this work was to propose an electrochemical method to detect AgNPs in aqueous solution by MoS2-rGO composite. MoS2-rGO/GCE exhibited satisfactory performance not only on the good electric conductivity, but also on the electrochemical detection of AgNPs. Due to the Ag-S coordination effect, MoS2-rGO composite could promote the adsorption and electrochemical response of AgNPs at electrode surface. The obtained method exhibited outstanding electroanalytical performance for AgNPs, including with a mild applied potential and a superior LOD, which is expected to be applied in further environmental analysis.
References
Alvaredo J, Bayón M, Bettmer J (2013) Speciation of silver nanoparticles and silver(I) by reversed-phase liquid chromatography coupled to ICPMS. Anal Chem 85:1316–1321
Anson FC (1966) Innovations in the study of adsorbed reactants by chronocoulometry. Anal Chem 38:54–57
Artiaga G, Ramos K, Ramos L, Cámara C, Gómez-Gómez M (2015) Migration and characterisation of nanosilver from food containers by AF4-ICP-MS. Food Chem 166:76–85
Asadi M, Kumar B, Behranginia A, Rosen BA et al (2014) Robust carbon dioxide reduction on molybdenum disulphide edges. Nat Comm 5:4470
Cepriá G, Córdova W, JiménezLamana J, Laborda F, Castillo JR (2014) Silver nanoparticle detection and characterization in silver colloidal products using screen printed electrodes. Anal Methods 6:3072–3078
Cepriá G, Pardo J, Lopez A, Peña E, Castillo JR (2016) Selectivity of Silver nanoparticle sensors: discrimination between silver nanoparticles and Ag+. Sens Actuator B 230:25–30
Chang B, Park S (2010) Electrochemical impedance spectroscopy. Ann Rev Anal Chem 3:207–229
Chao J, Liu J, Yu S, Feng Y, Tan Z, Liu R, Yin Y (2011) Speciation analysis of silver nanoparticles and silver ions in antibacterial products and environmental waters via cloud point extraction-based separation. Anal Chem 83:6875–6882
Cheng W, Compton RG (2014) Electrochemical detection of nanoparticles by ‘nano-impact’ methods, TRAC-Trend. Anal Chem 58:79–89
Cheng W, Stuart EJE, Tschulik K, Cullen JT, Compton RG (2013) A disposable sticky electrode for the detection of commercial silver NPs in seawater. Nanotechnol 24:505501
Dankovich TA, Gray DG (2011) Bactericidal paper impregnated with silver nanoparticles for point-of-use water treatment. Environ Sci Technol 45:1992–1998
David L, Bhandavat R, Singh G (2014) MoS2/graphene composite paper for sodium-ion battery electrodes. ACS Nano 8:1759–1770
Duan S, Huang Y (2016) Polyethylenimine-carbon nanotubes composite as an electrochemical sensing platform for silver nanoparticles. Talanta 160:607–613
Ferrari AC, Basko DM (2013) Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat Nanotech 8:235–246
Goda T, Ambrosi A, Miyahara Y, Pumera M (2014) Simultaneous electrochemical detection of silver and molybdenum nanoparticles. Chemelectrochem 1:529–531
Huang X, Qi X, Boey F, Zhang H (2012) Graphene-based composites. Chem Soc Rev 41:666–686
Joseph N, Shafi PM, Bose AC (2018) Metallic MoS2 anchored on reduced graphene oxide sheets with edge orientation, and its electrochemical investigation on energy storage application. ChemistrySelect 3(42):11993–12000
Kamila S, Mohanty B, Samantara AK (2017) Highly active 2D layered MoS2-rGO hybrids for energy conversion and storage applications. Sci Rep 7(1):1–13
Lee K, Lee K, Lee J, Ahn B, Woo S (2010) Electrochemical oxygen reduction on nitrogen doped graphene sheets in acid media. Electro Comm 12:1052–1055
Li W, Guo M, Zhang Y (2014) Edge-specific Au/Ag functionalization-induced conductive paths in armchair MoS2 nanoribbons. Chem Mater 26:5625–5631
Li XT, Batchelor-McAuley C, Compton RG (2019) Silver nanoparticle detection in real world environments via particle impact electrochemistry. ACS Sensors 4:464–470
Liu S, Zhang X, Shao H, Xu J, Chen F, Feng Y (2012) Preparation of MoS2 nanofibers by electrospinning. Mater Lett 73:223–225
Lisdat F, Schäfer DD (2008) The use of electrochemical impedance spectroscopy for biosensing. Anal Bioanal Chem 391:1555–1567
Loeschner K, Navratilova J, Købler C, Mølhave K, Wagner S, Kammer F, Larsen E (2013) Detection and characterization of silver nanoparticles in chicken meat by asymmetric flow field flow fractionation with detection by conventional or single particle ICP-MS. Anal Bioanal Chem 405:8185–8195
Marambio-Jones C, Hoek EMV (2010) A review of the antibacterial effects of silver nanomaterials and potential implications for human health and the environment. J Nanoparticle Res 12:1531–1551
Matte H, Gomathi A, Manna A et al (2010) MoS2 and WS2 analogues of graphene. Angew Chem Int Ed 49:4059–4062
Mohan Y, Raju K, Sambasivudu K, Singh S, Sreedhar B (2017) Preparation of acacia-stabilized silver nanoparticles: a green approach. J Appl Polym Sci 106:3375–3381
Paramelle D, Sadovoy A, Gorelik S, Free P, Hobbley J, Fernig DG (2014) A rapid method to estimate the concentration of citrate capped silver nanoparticles from UV-visible light spectra. Analyst 139:4855–4861
Pan L, Liu Y, Xie X, Zhu X (2014) Coordination-driven hierarchical assembly of silver nanoparticles on MoS2 nanosheets for improved lithium storage. Chem-Asian J 9:1519–1524
Rizzello L, Pompa P (2014) Nanosilver-based antibacterial drugs and devices: mechanisms, methodological drawbacks, and guidelines. Chem Soc Rev 43:1501–1518
Sadik O, Du N, Kariuki V, Okello V, Bushlyar V (2014) Current and emerging technologies for the characterization of nanomaterials. ACS Sustain Chem Eng 2:1707–1716
Stuart E, Tschulik K, Lowinsohn D, Cullen JT, Compton RG (2014) Gold electrodes from recordable CDs for the sensitive, semi-quantitative detection of commercial silver nanoparticles in seawater media. Sens Actuator B 195:223–229
Uslu B, Ozkan SA (2007) Electroanalytical application of carbon based electrodes to the pharmaceuticals. Anal Lett 40:817–853
Vidal JC, Torrendo D, Menés S, Fuente A, Castillo JR (2020) Voltammetric sensing of silver nanoparticles on electrodes modified with selective ligands by using covalent and electropolymerization procedures. Discrimination between silver (I) and metallic silver. Microchim Acta 187:1-11
Xie J, Zhang J, Li S, Grote F, Zhang X et al (2013) Controllable disorder engineering in oxygen-incorporated MoS2 Ultrathin nanosheets for efficient hydrogen evolution. J Am Chem Soc 135:17881–17888
Yu S, Yin Y, Liu J (2013) Silver nanoparticles in the environment. Environ Sci Processes Impacts 15:78-92
Zhang S, Wright G, Yang Y (2000) Materials and techniques for electrochemical biosensor design and construction. Biosens Bioelectron 15:273–282
Zhao J, Zhang Z, Yang S, Zheng H, Li Y (2015) Facile synthesis of MoS2 nanosheet-silver nanoparticles composite for surface enhanced Raman scattering and electrochemical activity. J Alloy Compd 559:87–91
Zhou Y, Rees N, Compton RG (2011) The electrochemical detection and characterization of silver nanoparticles in aqueous solution. Angew Chem Int Ed 50:4219–4221
Zhao Y, Xie X, Zhang J, Liu H, Ahn H, Sun K, Wang G (2015) MoS2 nanosheets supported on 3D graphene aerogel as a highly efficient catalyst for hydrogen evolution. Chem-Eur J 21:15908–15913
Zheng X, Xu J, Yan K, Wang H, Wang Z, Yang S (2014) Space-confined growth of MoS2 nanosheets within graphite: the layered hybrid of MoS2 and graphene as an active catalyst for hydrogen evolution reaction. Chem Mater 26:2344–2353
Funding
Open Foundation of Hubei Key Laboratory for Processing and Transformation of Agricultural Products (No. 2020HBSQGDKFB08), National Key Research and Development Program of China (No. 2019YFC1606000), National Natural Science Foundation of China (No. 32001772), and University Research Project of Wuhan Polytechnic University (No. 04).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, J., Duan, S., Yue, R. et al. High sensitive electrochemical detection of silver nanoparticles based on a MoS2/graphene composite. J Nanopart Res 24, 92 (2022). https://doi.org/10.1007/s11051-022-05454-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-022-05454-y