
Abstract
Due to the limitation of the theoretical specific capacity of traditional cathode materials, the key to improve the energy density of lithium-ion batteries (LIBs) is to develop high-performance cathode materials. As a promising cathode material for LIBs, Fe-containing lithium-rich manganese-based materials possess high specific capacity and high working voltage. At present, the research on the synthesis and electrochemical reaction mechanism of this kind of material is still imperfect. Here, a facile oxalate co-precipitation method is employed to prepare Li1.2Fe0.16Ni0.24Mn0.4O2 using different lithium sources. The effects of lithium source and electrochemical window on the specific capacity and cyclic stability of the materials are also systematically investigated. When applied in LIBs, the prepared material using LiNO3 as lithium source presents a micro-nano hierarchical structure and exhibits a high first discharge specific capacity of 337.7 mAh g−1 and a high reversible capacity of 208.4 mAh g−1 after 60 cycles at 0.1 C in the voltage range of 1.5 − 4.8 V. The results of this study can provide more selectivity for the lithium source of the electrode material during the preparation process, and select a suitable voltage test range for cathode materials to achieve specific properties. The outstanding performances make this oxalate co-precipitation process a prospective method to prepare high-capacity cathode materials for LIBs.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the development of economy and the continuous progress of industrialization, energy shortage has become a key issue. Lithium-ion batteries (LIBs) have drawn ever-increasing attention as electrochemical energy-storage applications because of the safety and reliability, small size, light weight, environmental friendliness, high energy density, and high output voltage (Zhao et al. 2019a; Zhou et al. 2018; Peng et al. 2019). In order to adapt the highly increasing demands dictated by portable electronics, electric mobility, and electrical grid markets, LIBs technology is continuously developing in recent years. Nevertheless, considering the future applications in power battery fields, the mass production with high energy density is crucial to the battery industry. Despite enormous efforts, the electrochemical performances of most traditional cathode materials are still limited by their low theoretical specific capacity. Preparation of high-capacity cathode materials with high voltage becomes today urgently needed for promoting the energy density of LIBs (Zhong et al. 2016; Zhao et al. 2015, 2018a).
Lithium, which exists in the cathode material of LIBs, plays a crucial role in battery performance (Lee et al. 2019). Li-rich manganese-based layered oxide, whose specific capacity is twice that of conventional layered LiCoO2 material, has been one of the hot spots in cathode material research for more than a decade. In particular, as an appealing cathode candidate for LIBs, Fe-containing Li-rich manganese-based oxides (FLMO) have driven a series of studies due to the environmental friendliness and low cost. However, low Coulombic efficiency, rapid capacity attenuation, and potential fade are still several technical barriers that need to be overcome before their commercialization. Our group has systematically studied the synthetic method and electrochemical properties of various types of FLMO (Zhao et al. 2017, 2018a, b, Zhao et al. 2019b, c). It is found that the preparation method and lithium source have great influence on the structure feature and electrochemical properties of FLMO. More importantly, the electrochemical reaction mechanism of FLMO in different voltage ranges during battery operation has not yet been clearly clarified. In order to facilitate their application, it is important and urgent to propose a rational preparation method and appropriate lithium source for FLMO cathode materials to further study their electrochemical behaviors in wide voltage window range (Li et al. 2012).
At present, there are abundant methods for preparing Li-rich materials, such as solid state method (West et al. 2012), co-precipitation method (Cui et al. 2019), freeze-drying method (Surace et al. 2015), sol-gel method (Sankar Devi et al. 2019), combustion method (Huang et al. 2019), and molten salt method (Zhao et al. 2019c). According to the reports, various Li-rich cathode materials can be successfully synthesized by an oxalate co-precipitation method. Compared with hydroxide co-precipitation or carbonate co-precipitation, oxalate co-precipitation is simple and easy to be used. It can obtain precursor materials without inert gas protection, and it is easy to realize electrode materials with micro-nano hierarchical structure, which is conducive to the electrochemical performance of Li-rich materials.
The abundance, low cost, and environmental friendliness of iron make Li1.2Fe0.16Ni0.24Mn0.4O2 one of the most promising cathode material choices for next-generation LIBs (Zhao et al. 2019d; Yao et al. 2019). In this work, Li1.2Fe0.16Ni0.24Mn0.4O2 is prepared by a facile oxalate co-precipitation route using Na2C2O4 as a precipitant. Meantime, the electrochemical behaviors of the prepared materials with two different lithium sources have been synthetically investigated in different electrochemical window ranges to further explore its electrochemical reaction mechanism. It is expected that the study results will provide more useful knowledge for the Fe-containing Li-rich materials and lay a foundation for the further development of the field of battery electrode materials.
Experimental section
Materials synthesis
All the chemicals used in this work were of analytical grade and not further purified. Li1.2Fe0.16Ni0.24Mn0.4O2 powder was synthesized by a co-precipitation method using Na2C2O4 as a precipitant. Stoichiometric amounts of FeSO4·7H2O (0.02 mol), NiSO4·6H2O (0.03 mol), and MnSO4·H2O (0.05 mol) were dissolved in 200 mL deionized water to obtain a transparent solution, denoted as solution A. 5.36 g Na2C2O4 was dissolved in 200 mL deionized water to get a clear solution, denoted as solution B. For preparing the precursor material (Fe0.2Ni0.3Mn0.5)C2O4, solution B was introduced dropwise into solution A for 2 h with vigorous stirring in a flask with three necks at 70 °C. The mixed solution was continuously stirred for 3 h at 70 °C for co-precipitation reaction. The precursors were collected after centrifuging the reacted mixture and washed with deionized water and absolute ethanol for three times to remove residual sodium and sulfuric species. After dried in a vacuum oven at 100 °C overnight, the precursors were uniformly mixed with a certain amount of lithium sources (Li2CO3 or LiNO3). To compensate for the loss during high temperature calcination, the lithium source was added in excess of 5%. Afterward, the obtained powder was subjected to further calcination using a step procedure (300 °C for 2 h, 500 °C for 6 h, 800 °C for 12 h) to obtain the final products. The prepared materials using Li2CO3 or LiNO3 as lithium sources were named as P-Li2CO3 and P-LiNO3, respectively.
Material characterizations
The morphology of the prepared powders was observed by a field emission scanning electron microscope (SEM, FEI, Quanata 200f). Element species were detected with an energy dispersive X-ray detector. The crystal structure analysis of the prepared precursor and active materials was carried out using X-ray diffraction (XRD; Rigaku Ultima IV-185) with a Cu Kα radiation source. The data were collected with a step size of 8° min−1 in a 2θ range of 10−90°. The surface chemical state of the prepared P-LiNO3 material was detected by X-ray photoelectron spectroscopy (XPS, PHI Quantera).
Electrochemical tests
For fabrication of the electrode, the prepared Li1.2Fe0.16Ni0.24Mn0.4O2 powder as active material was uniformly mixed with acetylene black and polyvinylidene fluoride (PVDF) by a ratio of 80:10:10 using N-methyl-2-pyrrolidone (NMP) as solvent. The obtained uniform slurry was coated onto Al foil and dried overnight at 100 °C in a vacuum oven. The electrode was roll-pressed and punched into circular plates before use. Charge-discharge tests were done with 2025 coin-type cells, which were assembled in a glove box filled with argon using Li metal as the anode. The electrolyte solution was 1 M LiPF6 dissolved in ethyl carbonate (EC) and dimethyl carbonate (DMC) (1:1 by volume). Celgard 2400 membrane was employed as the separator. The cells were cycled in the voltage ranges of 2−4.8 V or 1.5−4.8 V at different rates (1 C = 200 mA g−1). Cyclic voltammograms of the prepared cathodes were performed on an electrochemical workstation (CHI660E, Chenhua, Shanghai, China) at room temperature in the voltage range of 2−5 V at a sweep rate of 0.1 mV s−1.
Results and discussion
Structural characterization
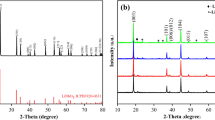
X-ray diffraction is generally employed to detect the crystal structure of the powders. The resulted diffraction peaks of the prepared precursor and Li-rich materials in this work are shown in Fig. 1. For comparison, the standard peak positions of layered phase are also displayed in the last XRD spectrum. The diffraction peaks of the precursor indicate that oxalate products can be successfully synthesized through the method in this work (Linjing Zhang et al. 2013). By contrast, the typical characteristic peaks of P-Li2CO3 and P-LiNO3 are very similar, which are well consistent with the diffraction peaks of lithium-rich manganese-based materials. Obviously, the existence of several low-intensity diffraction peaks between 20 and 23° is a feature of Li2MnO3-phase like structure, which can be attributed to monoclinic cell C2/m (Jeom-Soo Kim et al. 2004). Except for those peaks in 20−23°, all the other diffraction peaks are ascribed to the standard hexagonal α-NaFeO2-type phase with a space group of R3m. Therefore, no matter which lithium source (Li2CO3 or LiNO3) is used, the oxalate co-precipitation method can successfully synthesize lithium-rich manganese-based cathode materials. Lattice parameters of these two materials have been analyzed and compared in detail according to the results of XRD patterns. The results have been listed in Table S1 in the supporting information. It can be seen that P-LiNO3 displays higher c value, V value, and I(003)/I(104), indicating it has wider interlayer spacing and larger volume for Li+ extraction/insertion.

XRD patterns of the precursor and prepared P-Li2CO3 and P-LiNO3 materials
SEM images and EDS
SEM images in Fig. 2a, b reveal the particle morphology and size of the prepared precursor. The precursor presents a dense and sharp angular state with a particle size of ~ 10 μm. The morphologies of P-Li2CO3 and P-LiNO3 are displayed in Fig. 2c, d and Fig. 2e, f, respectively. Obviously, two Li-rich material samples prepared from different lithium sources are both composed of micron-sized particles. The morphology of P-Li2CO3 may be more likely to be micron bulk materials. However, P-LiNO3 material in Fig. 2e–f displays some micron-sized particles composed of many small spherical-like particles, which means that it has a hierarchical structure. Generally speaking, the diffusion distance and diffusion time of lithium ions in small particles are short, which is conducive to improving the rate capability of the cathode materials (Yamada et al. 2001; Sun et al. 2019). The micron-level overall structure is beneficial to reduce side reactions between the electrode and the electrolyte, and maintains the cycle stability of the electrode materials. In addition, the particle size distribution of lithium-rich material prepared with LiNO3 as lithium source is more uniform. More pores with even distribution are also observed in Fig. 2e, f. Therefore, it is speculated that P-LiNO3 has a higher specific surface area, which is conducive to the electrolyte infiltration into the material, so P-LiNO3 is expected to exhibit better electrochemical performances (Zhao et al. 2013).

SEM images of a, b the precursor; c, d P-Li2CO3; and e, f P-LiNO3. g EDS image of P-LiNO3
The EDS results and elemental mapping images of P-LiNO3 are illustrated in Fig. 2 g and Fig. S1, respectively. In addition to the inability to detect Li due to its low energy density, other expected elements including Fe, Mn, Ni, and O, are all distributed evenly in the prepared material (Yang et al. 2014), indicating that Fe-containing Li-rich material is successfully synthesized using the facile oxalate co-precipitation method in this work. According to our previous work (Zhao et al. 2017) and literature investigation (Cao et al. 2017), the use of different lithium sources mainly affects the morphology and electrochemical properties of the final cathode materials (Bao Zhang et al. 2014). However, the type of lithium source cannot affect the stoichiometric ratio. Both LiNO3 and Li2CO3 can be used as lithium source to synthesize lithium-rich manganese-based cathode materials with stoichiometric ratio. However, the addition amount of lithium source can affect the proportion of lithium element in the final cathode material, which is due to the loss of lithium source in the high temperature calcination process. Therefore, in this work, 5% excess of lithium source (Li2CO3 and LiNO3) was added to synthesize Li-rich cathode materials.
XPS analysis
XPS is usually used to analyze the solid surface of oxide material to preliminary evaluate the chemical or elemental composition. Figure 3 displays the typical XPS spectra and fitting curves of Ni 2p, Fe 2p, and Mn 2p to investigate the surface chemical state of the prepared P-LiNO3 material in this work. The XPS overall spectrum in Fig. 3a shows the binding energy characteristics of the oxide material. In Fig. 3b, nickel shows similar characteristics to Ni2+ valence state in lithium-rich materials Li[Li1/3–2x/3NixMn2/3–x/3]O2, which is attributed to the binding energy of 854.9 eV at Ni 2p3/2 position (Tran et al. 2006). There are two characteristic main peaks of Fe 2p3/2 and Fe 2p1/2 in the Fe 2p spectrum, which are in good agreement with Fe3+ in Fe2O3 (Yamashita and Hayes 2008). The binding energy positions are 711.2 eV and 724.4 eV, respectively. Moreover, the two satellite peaks observed in Fig. 3c further confirm the presence of Fe3+. In Fig. 3c, the binding energy of the two main peaks, Mn 2p3/2 (642.4 eV) and Mn 2p1/2 (654 eV), are well consistent with the binding energy values of the stable Mn4+ state in Li-rich materials (Liu et al. 2014). Generally, tetravalent manganese ions can play a key role in stabilizing the structure in P-LiNO3 material.

XPS spectra and fitting curves of the prepared P-LiNO3 material. a The overall spectrum. b Ni 2p. c Fe 2p. d Mn 2p
Charge and discharge capacities
The effect of different lithium sources on the charge/discharge specific capacities at a low rate is clearly studied. Figure 4a and b show the first charge/discharge curves of P-Li2CO3 and P-LiNO3 at 0.1 C in the voltage range of 2−4.8 V. The first charge curve and first discharge curve of these two materials show similar characteristics. The first charge curve contains two distinct voltage platforms with 4.5 V as the boundary, and the voltage in the first discharge curve shows a steady downward trend. In the voltage range of 2−4.8 V, the first charge specific capacity (320.9 mAh g−1) of P-Li2CO3 is obviously higher than that (305 mAh g−1) of P-LiNO3. However, the first charge specific capacity (152.2 mAh g−1) of P-Li2CO3 below 4.5 V is lower than that (162.5 mAh g−1) of P-LiNO3 below 4.5 V. Generally speaking, under the voltage of 4.5 V or below, lithium ions are mainly extracted from the lithium layer of lithium-rich materials. When the voltage is higher than 4.5 V, lithium ions is easy to escape from the transition metal layer, accompanied by the release of oxygen, resulting in a high irreversible capacity loss (Armstrong et al. 2006). The resulting oxygen vacancy may migrate to the adjacent transition metal site, leading to the crystal structure transition from layered to spinel phase (Armstrong et al. 2006). In other words, the extraction of lithium ions above 4.5 V easily leads to the instability of the crystal structure of Li-rich material. The above data indicate that compared with P-Li2CO3, more lithium ions can be extracted from lithium layer and less lithium ions can be extracted from transition metal layer in P-LiNO3 during charge process. Therefore, P-LiNO3 is speculated to have more stable crystal structure. The discharge specific capacity of P-LiNO3 in the first cycle is 195.6 mAh g−1, which is slightly higher than that (173.4 mAh g−1) of P-Li2CO3. The higher discharge specific capacity of P-LiNO3 further confirms the above conclusion.

Charge-discharge curves at different cycles of a, c P-Li2CO3 and b, d P-LiNO3 at 0.1 C in the voltage range of 2−4.8 V
The charge/discharge curves of P-LiNO3 and P-Li2CO3 at different cycles are compared in Fig. 4c, d. After 100 cycles at 0.1 C, both the charge specific capacity and discharge specific capacity are attenuated to a certain extent. The reversible discharge specific capacity at 100th cycle of P-LiNO3 is about 140.6 mAh g−1, while the reversible discharge specific capacity of P-Li2CO3 is only 119.6 mAh g−1. The result proves that the as-prepared P-LiNO3 material exhibits higher reversible specific capacity. In addition, both of the two materials show voltage drop with cycling, which is one of the key bottlenecks in the commercialization of lithium-rich materials.
Cycling stability
Fig. 5a compares the cycling performances and Coulombic efficiencies of the prepared P-Li2CO3 and P-LiNO3 materials in the voltage range of 2−4.8 V. Obviously, the Coulombic efficiencies of P-Li2CO3 and P-LiNO3 at different cycles are all nearly 100%. In the initial cycles, the Coulombic efficiency of P-Li2CO3 has a little turbulence, and then return to stability. However, the Coulombic efficiency of P-LiNO3 is relatively stable during the 100 cycles. In addition, the first Coulombic efficiency of P-LiNO3 is 64.1%, which is significantly higher than that (54.0%) of P-Li2CO3. In terms of cycle stability, the specific capacities of the two materials in the voltage range of 2−4.8 V gradually decrease as the cycle progresses. Moreover, the charge/discharge specific capacity of P-LiNO3 is always higher than that of P-Li2CO3 at the same cycle. In brief, the capacity retention of P-LiNO3 is 72%, which is higher than that (69%) of P-Li2CO3, indicating a better cycling stability of P-LiNO3.

a Cycling performances and Coulombic efficiencies of P-Li2CO3 and P-LiNO3 at 0.1 C in the voltage range of 2−4.8 V. b Cycling performance and c charge-discharge curves of P-LiNO3 at 0.1 C in the voltage range of 1.5 − 4.8 V
The cycling stability of P-LiNO3 is also investigated at 0.1 C in the voltage range of 1.5−4.8 V. The cycling performance and charge-discharge curves of P-LiNO3 at different cycles are clearly shown in Fig. 5b, c. The first charge specific capacity and discharge specific capacity of the material are 351 mAh g−1 and 337.7 mAh g−1 respectively, and the first Coulombic efficiency is as high as 96.2%. Similarly, as the cycle progresses, the charge/discharge specific capacity gradually decreases. After 100 cycles, the remaining reversible discharge specific capacity is 173.8 mAh g−1. Obviously, the charge/discharge specific capacity in a wide electrochemical window range of 1.5−4.8 V is always higher than the charge/discharge specific capacity tested in a narrow electrochemical window range of 2−4.8 V. Despite the higher specific capacity, the capacity retention after 100 cycles in the voltage range of 1.5−4.8 V is only 51.5%, which indicates that the prepared material has a faster specific capacity decay rate in a wide voltage range. As can be seen in Fig. 5c, the decrease of discharge specific capacity is not only due to the voltage range above 2 V, but also related to the voltage range below 2 V. The electrochemical reaction below 2 V is considered to be the redox process between Fe3+ and Fe2+. The cycling performance and charge-discharge curves of P-Li2CO3 in the voltage range of 1.5−4.8 V have been illustrated in Fig. S2. It can be seen that the discharge capacities of P-Li2CO3 are lower than those of P-LiNO3. Moreover, the platform below 2 V gradually disappears with the cycle, resulting in a sharp decline in discharge specific capacity. The higher specific capacities of P-LiNO3 than those of P-Li2CO3 in the voltage range of 1.5–4.8 V may be due to the wider interlayer spacing of P-LiNO3, which is conducive to Li+ insertion/extraction. In addition, the electrochemical reaction of P-LiNO3 below 2 V is more stable than that of P-Li2CO3. By comparing the specific capacity in different electrochemical windows, it can be concluded that a wide voltage range is conducive to the improvement of charge and discharge specific capacities, but is not conducive to maintaining cycle stability.
Cyclic voltammograms
Cyclic voltammetry (CV) was performed on P-Li2CO3 and P-LiNO3 to evaluate the redox potential of the transition metal ions during electrochemical reaction. Figure 6 shows the CV curves of P-Li2CO3 (the first three cycles) and P-LiNO3 (the first five cycles) between 2 and 5 V at a scan rate of 0.1 mV s−1. The peaks in the CV curves correspond to oxidation or reduction reactions of transition metal ions that occur during lithium extraction or insertion in the crystal lattice of the prepared material. In contrast, the CV curve characteristics of P-Li2CO3 and P-LiNO3 are similar. The initial anode peak of about 4.2 V in the first cycle is related to the oxidation of Ni from Ni2+ to Ni4+, but Mn always maintains the oxidation state of 4+ during the first charging process. The second anode peak at a higher potential of 4.6–4.7 V is mainly derived from an irreversible electrochemical activation reaction, which strips Li2O from the Li2MnO3 phase to form an electrochemically active MnO2 component (Armstrong et al. 2006). Obviously, the intensity of the first anode peak of P-LiNO3 is higher than that of P-Li2CO3, while the intensity of the second anode peak of P-LiNO3 is lower than that of P-Li2CO3, which is well consistent with the length of charging platform shown in Fig. 4a, b. During the first discharge process, P-Li2CO3 shows only a significant reduction peak around 3.8 V, which is attributed to the reduction reaction of Ni4+ to Ni2+. By comparison, P-LiNO3 shows two distinct reduction peaks at around 3.2 V and 3.8 V. The peak at 3.2 V appear in the CV curve of P-LiNO3 is associated with the reduction reaction of Mn4+ to Mn3+ (Donghan Kim et al. 2010; Fu et al. 2014; Ates et al. 2014), which is derived from the electrochemical activation reaction of Mn4+ above 4.5 V in the first charging process. The peak at 3.8 V is related to the reduction process of Ni4+ to Ni2+. It is worth noting that the peak intensity at 3.2 V does not decrease with the cycle, which is one of the reasons why the discharge specific capacity of P-LiNO3 is higher than that of P-Li2CO3. In addition, the CV curves of P-LiNO3 in the subsequent cycles have better overlap, showing smaller polarization and faster lithium extraction/insertion dynamics. The reduction peak of 3.2 V also exists in P-Li2CO3 sample, but the peak intensity is not obvious. This peak usually starts to appear in the first discharge process of lithium-rich cathode. The difference of the peak intensity in the two samples is related to the characteristics of the material itself.

CV curves of a P-Li2CO3 and b P-LiNO3 in the voltage range of 2−5 V
Universality of the method
In order to verify the universality of the method employed in this work, two kinds of other lithium-rich materials (Li1.2Co0.13Ni0.13Mn0.54O2 and Li1.2Ni0.2Mn0.6O2) were prepared by the same oxalate co-precipitation method, and their electrochemical properties were also tested. Figure 7 displays the electrochemical performances of these two Li-rich cathode materials. In Fig. 7a, the prepared Li1.2Co0.13Ni0.13Mn0.54O2 material exhibits super high first charge specific capacity of 431.3 mAh g−1 at 0.1 C, even though the Coulombic efficiencies at different cycles are not very ideal. Figure 7b shows the typical charge-discharge curves at different cycles of Li1.2Co0.13Ni0.13Mn0.54O2 in the voltage range of 1.5−4.8 V. The first charge-discharge curve indicates the successful preparation of Co-based lithium-rich ternary materials.

a Cycling performance and b charge-discharge curves of the prepared Li1.2Co0.13Ni0.13Mn0.54O2 material in the voltage range of 1.5−4.8 V. c Cycling performance and d rate capability of the prepared Li1.2Ni0.2Mn0.6O2 material in the voltage range of 2−4.8 V
The cycling stability of Li1.2Ni0.2Mn0.6O2 material at 0.1 C in the voltage range of 2−4.8 V is clearly displayed in Fig. 7c. The corresponding discharge curves at the first cycle and the 40th cycle are also illustrated in the inserted image. The first discharge specific capacity reaches to 216.4 mAh g−1, while the discharge specific capacity at the 40th cycle is 193.8 mAh g−1. Thus, the prepared Li1.2Ni0.2Mn0.6O2 material displays a high-capacity retention of 90%. Moreover, the rate capability of the prepared Li1.2Ni0.2Mn0.6O2 material in the voltage range of 2−4.8 V is also compared in Fig. 7d. The half cell is charged and discharged at 0.1 C, 0.2 C, 0.5 C, 1.0 C, 2.0 C, 5.0 C, and 10 C for five cycles, respectively, and then at 0.1 C for 5 additional cycles. Mainly due to the increase in polarization and electrode overpotential, the discharge specific capacity decreases with increasing current density. Even at 10 C, the discharge specific capacity of the material is still 20 mAh g−1, which indicates that the prepared Li1.2Ni0.2Mn0.6O2 material has the possibility of high-rate discharge. When the rate returns to 0.1 C, the material still shows high discharge capacity of 174 mAh g−1, indicating its good structural stability and high current withstand ability.
In order to compare and analyze the electrochemical kinetics of Li+ in P-Li2CO3 and P-LiNO3, EIS spectra of the two electrodes at open circuit potential before cycling have been illustrated in Fig. 8. According to EIS curves, Li+ diffusion coefficient has also been calculated. As in standard Nyquist plots, EIS spectrum includes a semicircle at high-frequency region and a slope at low-frequency region. The intercept of horizontal axis (Z′) with the semicircle at high frequency is related to uncompensated ohmic resistance of the electrolyte (RΩ). Charge transfer resistance (Rct) can be revealed by the diameter of the semicircle. The rising line in the low-frequency section represents Warburg impedance (ZW), which is associated with the diffusion of Li+ into the bulk material.

a EIS of P-Li2CO3 and P-LiNO3 at open circuit potential. b The relationship between Z′ and ω−1/2 for the Li/ P-Li2CO3 and Li/ P-LiNO3 cells
Clearly, Rct value of P-LiNO3 is about 15 Ω, which is lower than that of P-Li2CO3 (68 Ω), indicating its fast reaction kinetics. Generally, Li+ diffusion coefficient (DLi+) can be calculated according to the following equation:
where R is the gas constant, T is the absolute temperature, n is the number of transferred electrons, F is the Faraday constant, A is the surface area of the electrode, C is the concentration of Li+ in the electrode, and σ is the Warburg factor obeying the relationship: Zre = σω−1/2. DLi+ of P-LiNO3 is calculated to be 6.19 × 10−15 cm2 s−1, which is higher than that of P-Li2CO3 (3.09 × 10−16 cm2 s−1), demonstrating better lithium intercalation kinetics in the P-LiNO3 electrode.
In conclusion, through the above analysis, it can be seen that the type of lithium source and the electrochemical window range can directly affect the electrochemical performances of lithium-rich manganese-based cathode materials. Firstly, the type of lithium source can affect the crystal structure parameters of Li-rich materials. The wider layer spacing and better layered structure are more conducive to the Li+ insertion/extraction. Secondly, the type of lithium source can affect the morphological characteristics of Li-rich materials. The material with hierarchical structure can increase the contact area between electrode and electrolyte and reduce the interfacial side reaction. Thirdly, the materials prepared by different lithium sources have different reaction kinetics. Fast electrochemical kinetics is conducive to charge transfer, leading to the improvement of rate capability. In addition, the test results in different electrochemical window ranges show that wide electrochemical window is beneficial to stimulate new electrochemical reaction to promote the specific capacity of Li-rich material, while narrow electrochemical window is beneficial to maintain the structural stability of Li-rich material so as to improve the cyclic stability of the electrode.
Conclusion
In this paper, Fe-containing Li-rich Mn-based cathode materials (Li1.2Fe0.16Ni0.24Mn0.4O2) have been successfully synthesized by a facile oxalate co-precipitation method using two different lithium resources. The electrochemical performances of the materials are systematically investigated in different voltage window ranges. The prepared materials show micro-nano hierarchical morphology and typical crystal structure characteristics of lithium-rich materials. The type of lithium source has a certain influence on the specific capacity and cycle stability of the material. A wide voltage range is conducive to the specific capacity of the material, while a narrow voltage range is conducive to maintaining the cycling stability of the material. In addition, the oxalate co-precipitation method employed in this study is universal and can be applied to the synthesis of other lithium-rich manganese-based materials. This study further expands the preparation route of lithium-rich materials, and provides useful help for the development of high-capacity cathode materials for lithium-ion batteries.
References
Armstrong AR, Holzapfel M, Novák P, Johnson CS, Kang S-H, Thackeray MM, Bruce PG (2006) Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li[Ni0.2Li0.2Mn0.6]O2. J Am Chem Soc 128:8694–8698. https://doi.org/10.1021/ja062027+
Ates MN, Mukerjee S, Abraham KM (2014) A Li-rich layered cathode material with enhanced structural stability and rate capability for Li-ion batteries. J Electrochem Soc 161:A355–A363. https://doi.org/10.1149/2.070403jes
Cao K, Shen T, Wang K, Chen D, Wang W (2017) Influence of different lithium sources on the morphology, structure and electrochemical performances of lithium-rich layered oxides. Ceram Int 43:8694–8702. https://doi.org/10.1016/j.ceramint.2017.03.203
Cui Y, Liu K, Man J, Cui J, Zhang H, Zhao W, Sun J (2019) Preparation of ultra-stable Li[Ni0.6Co0.2Mn0.2]O2 cathode material with a continuous hydroxide co-precipitation method. J Alloys Compd 793:77–85. https://doi.org/10.1016/j.jallcom.2019.04.123
Fu C, Li G, Luo D, Zheng J, Li L (2014) Gel-combustion synthesis of Li1.2Mn0.4Co0.4O2 composites with a high capacity and superior rate capability for lithium-ion batteries. J Mater Chem A 2:1471–1483. https://doi.org/10.1039/c3ta13920d
Huang W, Wang G, Luo C, Xu Y, Xu Y, Eckstein BJ, Chen Y, Wang B, Huang J, Kang Y, Wu J, Dravid VP, Facchetti A, Marks TJ (2019) Controllable growth of LiMn2O4 by carbohydrate-assisted combustion synthesis for high performance Li-ion batteries. Nano Energy 64:103936. https://doi.org/10.1016/j.nanoen.2019.103936
Kim J-S, Johnson CS, Vaughey JT, Thackeray MM, Hackney SA, Yoon W, Grey CP (2004) Electrochemical and structural properties of xLi2M‘O3·(1 − x)LiMn0.5Ni0.5O2 electrodes for lithium batteries (M‘=Ti, Mn, Zr; 0 ≤ x ≤ 0.3). Chem Mater 16:1996–2006. https://doi.org/10.1021/cm0306461
Kim D, Kang S-H, Balasubramanian M, Johnson CS (2010) High-energy and high-power Li-rich nickel manganese oxide electrode materials. Electrochem Commun 12:1618–1621. https://doi.org/10.1016/j.elecom.2010.09.009
Lee S-H, Kim H-S, Jin B-S (2019) Recycling of Ni-rich Li(Ni0.8Co0.1Mn0.1)O2 cathode materials by a thermomechanical method. J Alloys Compd 803:1032–1036. https://doi.org/10.1016/j.jallcom.2019.06.229
Li T, Chen ZX, Ai XP, Cao YL, Yang HX (2012) LiF/Fe nanocomposite as a lithium-rich and high capacity conversion cathode material for Li-ion batteries. J Power Sources 217:54–58. https://doi.org/10.1016/j.jpowsour.2012.05.111
Liu J, Hou M, Yi J, Guo S, Wang C, Xia Y (2014) Improving the electrochemical performance of layered lithium-rich transition-metal oxides by controlling the structural defects. Energy Environ Sci 7:705–714. https://doi.org/10.1039/c3ee41664j
Peng Z, Mu K, Cao Y, Xu L, Du K, Hu G (2019) Enhanced electrochemical performance of layered Li-rich cathode materials for lithium ion batteries via aluminum and boron dual-doping. Ceram Int 45:4184–4192. https://doi.org/10.1016/j.ceramint.2018.11.087
Sankar Devi V, Athika M, Duraisamy E, Prasath A, Selva Sharma A, Elumalai P (2019) Facile sol-gel derived nanostructured spinel Co3O4 as electrode material for high-performance supercapattery and lithium-ion storage. J Energy Storage 25:100815. https://doi.org/10.1016/j.est.2019.100815
Sun Z, Xu L, Dong C, Zhang H, Zhang M, Ma Y, Liu Y, Li Z, Zhou Y, Han Y, Chen Y (2019) A facile gaseous sulfur treatment strategy for Li-rich and Ni-rich cathode materials with high cycling and rate performance. Nano Energy 63:103887. https://doi.org/10.1016/j.nanoen.2019.103887
Surace Y, Simões M, Karvonen L, Yoon S, Pokrant S, Weidenkaff A (2015) Freeze drying synthesis of Li3MnO4 cathode material for Li-ion batteries: A physico-electrochemical study. J Alloys Compd 644:297–303. https://doi.org/10.1016/j.jallcom.2015.04.221
Tran N, Croguennec L, Labrugère C, Jordy C, Biensan P, Delmas C (2006) Layered Li1 + x(Ni0.425Mn0.425Co0.15 )1-xO2 positive electrode materials for lithium-ion batteries. J Electrochem Soc 153:A261–A269. https://doi.org/10.1149/1.2138573
West WC, Soler J, Ratnakumar BV (2012) Preparation of high quality layered-layered composite Li2MnO3–LiMO2 (M = Ni, Mn, Co) Li-ion cathodes by a ball milling–annealing process. J Power Sources 204:200–204. https://doi.org/10.1016/j.jpowsour.2012.01.011
Yamada A, Chung SC, Hinokuma K (2001) Optimized LiFePO4 for lithium battery cathodes. J Electrochem Soc 148:A224–A229. https://doi.org/10.1149/1.1348257
Yamashita T, Hayes P (2008) Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl Surf Sci 254:2441-2449. https://doi.org/10.1016/j.apsusc.2007.09.063
Yang Z, Lu J, Bian D, Zhang W, Yang X, Xia J, Chen G, Gu H, Ma G (2014) Stepwise co-precipitation to synthesize LiNi1/3Co1/3Mn1/3O2 one-dimensional hierarchical structure for lithium ion batteries. J Power Sources 272:144–151. https://doi.org/10.1016/j.jpowsour.2014.08.052
Yao X, Xu Z, Yao Z, Cheng W, Gao H, Zhao Q, Li J, Zhou A (2019) Oxalate co-precipitation synthesis of LiNi0.6Co0.2Mn0.2O2 for low-cost and high-energy lithium-ion batteries. Mater Today Commun 19:262-270. doi:https://doi.org/10.1016/j.mtcomm.2019.02.001
Zhang L, Wu B, Li N, Mu D, Zhang C, Wu F (2013) Rod-like hierarchical nano/micro Li1.2Ni0.2Mn0.6O2 as high performance cathode materials for lithium-ion batteries. J Power Sources 240:644–652. https://doi.org/10.1016/j.jpowsour.2013.05.019
Zhang B, Ou X, Zheng J-c, Shen C, Ming L, Han Y-d, Wang J-l, Qin S-e (2014) Electrochemical properties of Li2FeP2O7 cathode material synthesized by using different lithium sources. Electrochim Acta 133:1–7. https://doi.org/10.1016/j.electacta.2014.03.188
Zhao T, Chen S, Li L, Zhang X, Chen R, Belharouak I, Wu F, Amine K (2013) Synthesis, characterization, and electrochemistry of cathode material Li[Li0.2Co0.13Ni0.13Mn0.54]O2 using organic chelating agents for lithium-ion batteries. J Power Sources 228:206–213. https://doi.org/10.1016/j.jpowsour.2012.11.099
Zhao T, Li L, Chen R, Wu H, Zhang X, Chen S, Xie M, Wu F, Lu J, Amine K (2015) Design of surface protective layer of LiF/FeF3 nanoparticles in Li-rich cathode for high-capacity Li-ion batteries. Nano Energy 15:164–176. https://doi.org/10.1016/j.nanoen.2015.04.013
Zhao T, Zhou N, Zhang X, Xue Q, Wang Y, Yang M, Li L, Chen R (2017) Structure evolution from layered to spinel during synthetic control and cycling process of Fe-containing Li-rich cathode materials for lithium-ion batteries. ACS Omega 2:5601–5610. https://doi.org/10.1021/acsomega.7b00689
Zhao T, Gao X, Wei Z, Guo K, Wu F, Li L, Chen R (2018a) Three-dimensional Li1.2Ni0.2Mn0.6O2 cathode materials synthesized by a novel hydrothermal method for lithium-ion batteries. J Alloys Compd 757:16–23. https://doi.org/10.1016/j.jallcom.2018.05.058
Zhao T, Zhang Y, Li Y, Sun X, Li Y, Ji R (2018b) Electrochemical activation of novel Fe-based Li-rich cathode material for lithium-ion batteries. J Alloys Compd 741:597–603. https://doi.org/10.1016/j.jallcom.2018.01.161
Zhao T, Meng Y, Ji R, Wu F, Li L, Chen R (2019a) Maintaining structure and voltage stability of Li-rich cathode materials by green water-soluble binders containing Na+ ions. J Alloys Compd 811:152060. https://doi.org/10.1016/j.jallcom.2019.152060
Zhao T, Ji R, Meng Y, Zhang G, Si H, Wang Y, Yang M, Wu F, Li L, Chen R (2019b) In situ generated spinel-phase skin on layered Li-rich short nanorods as cathode materials for lithium-ion batteries. J Mater Sci 54:9098–9110. https://doi.org/10.1007/s10853-019-03425-8
Zhao T, Ji R, Yang H, Zhang Y, Sun X, Li Y, Li L, Chen R (2019c) Distinctive electrochemical performance of novel Fe-based Li-rich cathode material prepared by molten salt method for lithium-ion batteries. J Energy Chem 33:37–45. https://doi.org/10.1016/j.jechem.2018.08.005
Zhao T, Ji R, Meng Y (2019d) The role of precipitant in the preparation of lithium-rich manganese-based cathode materials. Chem Phys Lett 730:354–360. https://doi.org/10.1016/j.cplett.2019.06.034
Zhong X-B, Yang Z-Z, Wang H-Y, Lu L, Jin B, Zha M, Jiang Q-C (2016) A novel approach to facilely synthesize mesoporous ZnFe2O4 nanorods for lithium ion batteries. J Power Sources 306:718–723. https://doi.org/10.1016/j.jpowsour.2015.12.102
Zhou H, Yang Z, Yin C, Yang S, Li J (2018) Fabrication of nanoplate Li-rich cathode material via surfactant-assisted hydrothermal method for lithium-ion batteries. Ceram Int 44:20514–20523. https://doi.org/10.1016/j.ceramint.2018.08.048
Funding
This study was funded by the National Natural Science Foundation of China (51902213), the Natural Science Foundation of Hebei Education Department (BJ2020046, ZC2016045), the Nature Science Foundations of Hebei Province (B2016210071, B2019210358), and the National College Students’ Innovative Entrepreneurial Training Project of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PDF 362 kb).
Rights and permissions
About this article

Cite this article
Zhao, T., Shen, J., Si, H. et al. Effects of lithium source and electrochemical window on the properties of Li1.2Fe0.16Ni0.24Mn0.4O2 cathode material prepared by oxalate co-precipitation method. J Nanopart Res 23, 43 (2021). https://doi.org/10.1007/s11051-021-05159-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-021-05159-8