
Abstract
The Needleman-Wunsch (NW) is a dynamic programming algorithm used in the pairwise global alignment of two biological sequences. In this paper, three sets of parallel implementations of the NW algorithm are presented using a mixture of specialized software and hardware solutions: POSIX Threads-based, SIMD Extensions-based and a GPU-based implementations. The three implementations aim at improving the performance of the NW algorithm on large scale input without affecting its accuracy. Our experiments show that the GPU-based implementation is the best implementation as it achieves performance 72.5X faster than the sequential implementation, whereas the best performance achieved by the POSIX threads and the SIMD techniques are 2X and 18.2X faster than the sequential implementation, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Bioinformatics is a discipline that applies the principles of computer science, mathematics, and engineering to answer questions related to Biology [9]. This can be fulfilled by applying several techniques to formulate and model the targeted biological problems as computational problems and design algorithms to solve them in an accurate and efficient manner. Sequence alignment is one of the widely studied problems in bioinformatics. It is concerned with finding similarities (patterns) in the same sequence or among different sequences. Sequences can be composed of nucleotides (forming DNA fragments), amino acids (forming proteins), etc. The basic intuition here is that sequences with similar structure are often similar in function as well.
The similarity between two sequences or strings taken from the same alphabet can be measured in several ways. One way is by determining the minimum number of edit operations performed on one sequence to make it identical to the other. Here, the edit operations are insertions, deletions, and substitutions. This is known as the pairwise global alignment problem and it can be generalized to align multiple sequences. If the problem is about aligning subsets of the sequences, then it is called local alignment. Existing algorithms for the alignment problem employ dynamic programming to find the optimal solution in time proportional to the product of the lengths to the sequences to be aligned. This is considered too much for many applications, especially in bioinformatics, where the sequence lengths can be very large (millions of elements). Thus, serious efforts have been invested in improving the performance of alignment algorithms by employing techniques like parallelization or by resorting to heuristics that sacrifice accuracy for better performance [14, 24]. In this paper, the focus is on speeding up the performance of the dynamic programming techniques for the pairwise global alignment problem [8]. More specifically, our goal is to explore improving the performance of the Needleman-Wunsch (NW) sequence alignment algorithm by using different parallelization or victorization techniques [19].
The rest of this paper is organized as follows. Section 2 presents the main concepts that are needed in this paper. Section 3 introduces relevant studies. Section 4 explains the methodology. Section 5 presents our experiments and the analysis of the results. Finally, we conclude in Section 6 and discuss our future directions.
2 Background
In this section, we discuss some background information about the NW algorithm that are necessary to understand this paper. The NW algorithm was introduced by Saul B. Needleman and Christian D. Wunsch in 1970 [19]. Its goal is to align two strings of nucleotides or proteins, considering the probable gaps, and using a representation matrix to represent scores gained by the comparisons of the possible studied pairs [5, 20]. Figure 1 depicts an example of global alignment for two strings.

Global alignment of two strings [14]
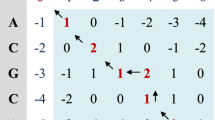
The alignment is performed using dynamic programming. Consider the pairwise alignment of two strings of n and m characters, respectively. A weighted grid network of dimensions (n + 1) × (m + 1) is created where nodes represent intermediate stops (positions) in the alignment and directed edges represent a match, mismatch or indel Footnote 1 in the next position. Note that the edges are directed in a way to allow movement from the upper left corner (position (0,0)) to the lower right corner (position (n,m)) of the grid. The weights are assigned to the edges based on the scoring matrix. In such a grid, each path from position (0,0) to position (n,m) represents an alignment and the objective is to select the longest path [24]. Figure 2 shows an example of the results of this algorithm, where m a t c h = 1, m i s m a t c h = −1, and g a p = −1.

A path representing a global alignment [18]
The NW algorithm relies on filling up a two-dimensional array. Thus, its space complexity is basically the dimensions of the array, i.e., (m + 1) × (n + 1) = O(m n). As for the time complexity, each array entry takes a constant number of operations to be computed (one “minimum of three” operation and three summation operations). Thus, the time complexity is also O(m n). Let the two sequences be represented by two strings x and y over an alphabet Σ. Let f : Σ × Σ → N be a weight function that assigns a score to each possible pair of residues. The algorithm of NW generates a distance matrix H using the following inductive formula [13]:
where
and
At the beginning of the induction, H i,0 = w i ,H 0,j = w j ,E i,0 = w i + v, and F 0,j = w j + v, for 1 ≤ i ≤ n, and 1 ≤ j ≤ m.
2.1 Parallel implementation techniques
This section describes the parallel techniques used in our implementations of the NW algorithm, which are: the POSIX threads technique, the SIMD extension techniques and, finally, the GPU technique.
POSIX threads technique
POSIX threads (sometimes called Pthreads) is a standard for building and manipulating multi-threaded applications. It utilizes the hardware resources effectively. These applications can operate on several operating systems like Microsoft Windows, Linux and Mac OS with reliability and high performance [4].
SIMD techniques
Single Instruction Multiple Data (SIMD) are Intel and AMD x86 instruction set extensions used for designing effective parallel programming applications, where the parallel tasks are performed at the instruction level using a single instruction at a time. It is commonly used in many applications such as speech recognition, graphics and image processing, audio and video processing. SIMD extensions consist of some versions such as: MultiMedia eXtensions (MMX), Streaming SIMD Extensions (SSE, SSE2, SSE3 and SSE4), Visual Instruction Set (VIS) and Advanced Vector Extensions (AVX and AVX2). They support manipulation of short vectors with float and integer variables in addition to supporting 64 bit, 128 bit and 256 bit registers. Following is a discussion of the extensions we use in this work.Footnote 2
-
SSE Extensions: SSE extensions were presented by Intel in 1999 using the Pentium III processor with a support of Hyper-Threading technique, then by AMD in 2001 using the K7 Palomino processor. It uses XMM 128 bit registers to manipulate scalar-packed single precision floating point data, double precision floating point data and packed integer data, and data should be aligned on 16 Byte boundaries when it is saved in the memory.
-
AVX Extensions: AVX instructions were introduced by Intel in 2008 and it got the first support in 2011 also by Intel using the Sandy Bridge processor, and then by AMD using the Bulldozer processor. It promotes the XMM 128 bit integer registers to YMM 256 bit registers, in addition to promoting the mechanism of dealing with variables, where it supports conducting operations between two variables and saving the value in a third variable (such as X = Y + Z). Unlike what happens in the SSE extensions, where the output is overwritten on the source variable (such as X = X + Y) [17].
GPU technique
Graphics Processor Unit (GPU), sometimes called visual processor unit (VPU), is an electronic circuit used for accelerating the manipulation of graphics and image processing, by adopting parallelization techniques to process large amount of data at the same time. For many tasks, it outperforms the general-purpose CPUs because general-purpose CPUs can support only 8, 12, or 16 threads, while GPU supports thousand of threads working concurrently. Physically, a GPU may exist either in a form of an external video card connected to the computer or embedded on the computer main board [6]. The first GPU was produced in 1999 by NVIDIA (who introduced the term GPU) and was called (GeForce 256). Then, in 2002, ATI introduced the term VPU and produced the (Radeon 9700) graphic card. Both GPUs and VPUs are widely used in workstations, embedded systems, personal computers, game consoles and cell phones [22]. Compute Unified Device Architecture (CUDA) is a software platform introduced in 2006 by Intel to operate the GPU (only NVIDIA’s GPUs) resources in order to perform general purpose parallel processing tasks (GPGPU) in a more efficient way than CPUs, using a high level language such as C, C++, OpenACC and FORTRAN [1, 12, 25].
3 Related work
Comparing DNA, RNA and protein strings has been one of the fundamental problems in the field of bioinformatics for decades. Several studies have been conducted by computer scientists, mathematicians and biologists to improve the efficiency and accuracy of the algorithms used to solve these problems. Some of these studies focused on the NW algorithm, while others focused on the Smith-Waterman (SW) algorithm, which is very similar to the NW algorithm, but it is designed for local alignment. Another important issue is the type of alignment. Some papers considered only a pair of sequences while others considered multiple pairwise alignments (e.g., of a “query sequence” against a “database of sequences”) or aligning multiple sequences at the same time.
Siriwardena and Ranasinghe [26] presented a hybrid (CPU-GPU) implementation of NW algorithm, using a parallel CUDA based heterogeneous architecture. They divided the tasks between the CPU and the GPU, by assigning the control task to the CPU to manage the memory size and the number of blocks that will be run on the GPU in each iteration where the NW algorithm will be actually executed. The experiments were conducted on Linux operating system running on an Intel Quad Core processor with a speed of 2.4 MHZ and 3 GB of RAM. The CUDA solution was executed on a single NVIDIA GeForce 8800 GT GPU card with 114 cores, 512 MB of RAM, 16 KB of shared memory/block, and graphics driver version 2.3. Various thread utilization ways, kernel access methods and various memory access pattern levels are compared to pick up the best parallelization mode. The suggested implementation provided a processing speed up of 4.2 times based on the blocking mode, and up to 2 times speed up based on the non-blocking mode compared with the pure CPU implementation.
Liu and Shmidt [16] presented GSWABE, a GPU-accelerated pairwise sequence alignment algorithm, used to retrieve optimal alignments from a collection of short DNA sequences. GSWABE supports all-to-all pairwise global, semi-global, and local alignments using the SW algorithm. GSWABE is implemented on CUDA-enabled GPUs. The algorithm applies dynamic programming for the three types of alignments using the similar computational patterns. GSWABE uses a general tile-based approach in order to achieve fast alignments. The authors evaluated their approach on Kepler-based Tesla K40 GPU and achieved 156 faster execution time than the SSE-based implementation and up to 102.4 faster execution than MSA-CUDA in case of local alignment.
Zhou et al. [29] proposed a hybrid alignment approach based on the SW local alignment algorithm and the NW global alignment algorithm. They tested their approach on a lower-end personal computer with GeForce GTX 750 Ti card and a higher-end personal computer with a GeForce GTX 1070 GPU card. The hybrid parallel implementation provided almost 140 times faster execution when compared to the serial solution.
Rognes and Seeberg [21] implemented the SW based on both MMX and SSE intrinsics. They used Red Hat Linux 6.1 operating system running on an Intel Pentium III single processor with a speed of 500 MHZ and 128 MB of RAM. The authors computed the query profile only once for each existing character in the studied strings. They also used vectors of adjacent cells parallel to the query string, rather than using vectors parallel to the minor matrix diagonal. This gave a processing speed of more than 150 million cell updates per second, which can be considered as 6 times faster than the non-vectorized implementation. This speed was estimated by testing the presented implementation on a collection of 11 various amino acid query strings with a length of 189-567 amino acids for each string.
Rognes [20] introduced a new approach called SWIPE to run the SW algorithm and implemented it using the SSE3 intrinsics, where characters from one query string are aligned with characters from 16 various database strings in a parallel manner, which is 2.5 times faster than the implementation of the alignment with a single thread. This lead to a speed of 106 billion cell updates per second, with a query string of 375 characters length. He used a Linux 64 bit operating system running on 6 cores Intel processor called dual Xeon X5650, and concluded that using the SIMD techniques can increase the speed of local alignment, especially, for the SW database searching.
Wozniak [28] presented a new SW algorithm implementation, using an SUN Ultra SPARC device, based on specialized video instructions (VIS), which are similar to the SIMD instructions in supporting the parallelization manner at the instruction level, in order to accelerate the implementation of the algorithm. The author used vectors of adjacent cells parallel to the minor diagonal of the matrix. The advantage of this approach is the dispense of the conditional part, which is supposed to be used in the inner loop. Thus, the time complexity depends only on both the query and database sequences length. On the other hand, this approach requires computing the query profile individually for each character in the used sequences. However, the presented approach has twice the speed of the conventional implementation.
In [11], Farrar presented an implementation of the SW algorithm for database searching, using a fast approach called Stripped SW based on the SSE2 intrinsics. This author used a Core 2 Duo Xeon processor with a 2.0 GHZ speed to test the presented approach. The stripped SW approach reduced the time needed to perform the sequence comparison by pre-computing the query profile in a stripped reading parallel to the query string for all characters, instead of reading the score of each cell individually, leading to a speed of 3 billion cell updates per second. This approach outperformed the two other SIMD local alignment implementations mentioned previously by 2–8 times.
Serrano et al. [23] conducted a GPU acceleration and power consumption analysis of the SW method, implemented using CUDAlign 4.0 [7], for GPU acceleration on a multi-GPU environment. CUDAlign 4.0 is a parallel technique to obtain optimal alignments of huge DNA sequences on multi-GPU platforms using the SW algorithm. CUDAlign 4.0 is comprised of six execution stages where the first three are accelerated using GPUs. The analysis of performance (execution time) and energy consumption showed a direct correlation between them in most cases. Improving the performance was at the cost of more energy consumption even with cases that did not provide great scalability.
Zhu et al. [30] proposed to use GPUs to speed up the performance of the MAFFT multiple sequence alignment algorithm [15]. In order to overcome the limitations of memory in GPUs. They implemented a memory allocation and reuse strategy that uses an encoding scheme to minimize memory consumption. Moreover, they used high performance shared memory to improve I/O operations. They demonstrated the usefulness of their approach on three different NVIDIA GPUs where the results showed up to 11.28X performance gain when compared to the sequential MAFFT implementation.
In recent papers, the authors of [2, 3] presented their effort to improve the performance of global sequence alignment algorithms. Instead of focusing on the NW algorithms, the authors of [2, 3] focused on the Levenshtein and Damerau Edit Distance Algorithms. The improvement gains achieved were 11X and 12X, respectively. However, when they attempted to use the new unified memory feature provided by NVIDIA, the improvement gains jumped to 61X and 71X, respectively.
4 Methodology
We now describe our parallel implementations of the NW algorithm. However, before we start, we need to discuss the data dependencies in the NW algorithm since such issues are crucial for the development of correct and efficient parallel code.
In the NW algorithm, each cell in the representation matrix has a data dependency with its three upper left adjacent cells (see Fig. 3). Therefore, these cells should be calculated first in order to be able to calculate the value of the targeted cell. This means that, for maximum efficiency, filling the matrix could not be done in an arbitrary manner, but rather, it should be done following a specific order to ensure that there is no violation of the dependencies between the cells.

Data dependencies between cells in the NW algorithm
The most common methods to fill the matrix are: the sequential traversal method and the diagonal traversal method [27]. In sequential traversal, the matrix is filled column by column or row by row. I.e., starting from the cell (0,0), then (1,0) until reaching cell (m,0). Then, the traversal moves to the next column and so on. See Fig. 4a [27]. For traversing a matrix of two strings with the lengths m and n respectively, m × n iterations are needed. For instance, 16 iterations are conducted to fill a 4 × 4 matrix. Note that using parallelization does not help with this situation.

Two different methods to fill the representation matrix [18]
On the other hand, Fig. 4b [27] shows that it is possible to fill all the cells of each minor diagonal at the same time without violating the dependencies between the calculated cells. Thus, using paralellization, seven iterations are needed to fill a 4 × 4 matrix instead of 16 iterations as used in the sequential method. The number of iterations can be simply computed as m + n − 1. Similar to the Wozniak implementation [28], the diagonal traversal method is used in all of our implementations to fill the NW representation matrix, because it is more compatible with the parallelization techniques used during our work.
4.1 Database and query sequences
The performance of the presented programs is analyzed by measuring the overall time required to perform the alignment of the two given DNA sequences, where the first sequence represents the database sequence and the second one represents the query sequence. Both sequences are generated randomly consisting of characters from the alphabet Σ = A, C, G, T with 75% similarity between them, where the global alignment is usually used to align almost two identical sequences. The tests are conducted on sequences with the length of 10,000, 14,992 and 20,000 characters consecutively. Note that all these values are divisible by 16 without remainder to facilitate the treatment of the SIMD vectors.
4.2 Parallel implementations
This section illustrates how the NW algorithm can be parallelized using specialized hardware such as GPUs besides two different CPU approaches: POSIX threads and SIMD.
4.2.1 POSIX threads implementation
Four implementations are conducted, which differ in the number of threads used to determine the best value that can be used. The numbers of threads that we consider are: 2, 4, 8 and 16. During the execution, all cells that belong to the same diagonal are grouped into equal chunks (as much as possible), then each chunk is assigned to a different thread. In order to increase the efficiency of the implementation, diagonals with lengths less than (n/2) are ignored since the time spent in creating new threads is greater than the potential benefit of creating these threads. Figure 5 explains this procedure more clearly.

POSIX thread implementation with two threads
4.2.2 SIMD extension implementations
Two SIMD implementations are conducted, the first implementation is based on SSE3 extensions while the second one is based on AVX2 extensions. Both implementations follow the same programming model with some differences as shown in Table 1.
During the execution, scores are pre-computed only once before performing the alignment in order to avoid the frequent lookup of δ(x i ,y j ) to be added to H(i − 1,j − 1) values for each cell in the NW representation matrix. Vectors that consist of adjacent cells in parallel to the matrix minor diagonal are used. The number of vectors used in each diagonal may differ from the number of vectors used in the adjacent diagonals, depending on the number of elements that should be processed within each diagonal. For example, when using the SSE3 extensions in the implementation, the size of the vector is 8 elements (according to that, each single instruction can process 8 cells in parallel), while it is 16 in the AVX2 implementation. Thus, the number of required vectors can be computed using the following formula:
where P refers to the desired size of the vector and L refers to the number of elements that should be divided into vectors. The case where the number of elements is not long enough to fit the resulting vectors, the remaining blanks are filled with garbage entries. See Fig. 6.

NW implementation using SIMD extensions
4.2.3 GPU implementation
The GPU implementation is done as follows.
-
1.
The number of blocks and worker threads is optimized (re-sized) separately for each iteration at the CPU side, to avoid wasting time by creating threads more than the volume of the data being processed.
-
2.
The representation matrix is represented as one-dimensional array instead of two-dimensional array (memory optimization) in order to facilitate allocating the processed data in the GPU memory which positively affects the overall performance of the program.
-
3.
The NW algorithm is performed at the GPU side only. To ensure getting the desired improvement of the implementation, all the data are copied from the CPU side in the initialization step. Moreover, arguments that change in each iteration (such as the number of worker threads, number of limited data, etc.) are sent to the GPU at each iteration. This process reduces the time consumed through the transfer of data between CPU and GPU memories.
-
4.
256 threads are used in the implementation, and since the matrix initialization is done at the GPU side, any thread located outside the range set by the CPU is excluded (thus, guaranteeing getting the best running time).
-
5.
Finally, the output (the H matrix) is transferred from the GPU to the RAM in order to display the final result of the alignment.
5 Experiments
In this section, comprehensive tests are conducted to compare the performance of the presented implementations and select the most efficient one. Experiments are repeated 30 times and the average are reported. Moreover, the output of each iteration is carefully checked to verify its correctness. Finally, the performance of the new implementations is computed using the following formula:
where S refers to the running time of the sequential implementation and P refers to the running time of the parallel implementation.
All programs are written in the C language, and the algorithm arguments are assigned to the following values: Match score = 0, Mismatch score = 1, Gap open penalty = 1 and Gap extend penalty = 1. The experimental setup used to test the performance of our implementations can be classified into two environments: the first environment is concerned with the CPU-based implementations, while the second group is concerned with the GPU-based implementation. For the first environment, we an Intel Core i7 processor (3.5 GHz) with 8 GB DDR3 of RAM while, for the second environment, we have an NVIDIA Geforce (GT 740M) graphics card with 2 GB of memory as a GPU. As mentioned early, four POSIX threads implementations are conducted, which differ in the number of threads used to accomplish the work required (2, 4, 8, 16 threads).
The results of this experiment are depicted in Table 2. The table shows that the maximum performance can be gained when using 8 threads during the implementation, where it takes 5.594 seconds, 12.193 seconds and 22.652 seconds, respectively, to perform the global alignment of the strings under consideration. This is considered 49.26% faster that the sequential implementation of the same algorithm.
Although increasing the number of threads has contributed in improving the efficiency of the program, the performance degrades when increasing the number of threads after a certain extent as a result of the communication overhead between these threads. Hence, slowing down the speed of the program.
The second group of experiments uses the SIMD extensions techniques and the results are depicted in Table 3. The table shows that the AVX2 implementation outperforms both the sequential and SSE3 implementations, with running times equal to 0.639 seconds, 1.346 seconds and 2.381 seconds, respectively. This is due to the fact that AVX2 instructions deal with 16 elements at the same time instead of 8 elements as in SSE3 instructions, or one single element as in the sequential implementation.
Although AVX2 deals with double the number of elements processed by the SSE3, the amount of superiority is not too large due to the absence of some built-in instructions (like shift instructions), which require the use of some built-in instructions combined with each other to perform the desired task. This results in consuming more time and affecting the performance of the whole program. The AVX2 implementation is considered as 94.52% faster than the sequential implementation, whereas the SSE3 is 92.82% faster than the sequential implementation.
The final group of experiments (which were already reported in our earlier paper [10]) is done to compare between the CPU and GPU based implementations. From the results of this experiment, which are depicted in Table 4, it is clearly shown that the GPU-based implementation significantly outperforms the CPU-based implementation. The running times that have been spent from the GPU-based implementation to perform the alignment are 0.065 seconds, 0.222 seconds and 0.599 seconds consecutively, which is considered as 98.62% faster than the CPU-based implementation.
The summary of results shown in Fig. 7 clearly shows that the GPU-based implementation is the best implementation as it achieves performance 72.5X faster than the sequential implementation, whereas the best performance achieved by the POSIX threads and the SIMD techniques are 2X and 18.2X faster than the sequential implementation, respectively. The GPU implementation outperforms the other implementations due to the effective architecture that it uses. In GPUs, threads are ordered into blocks that are all organized into a grid, where execution and management of possibly thousands of threads is conducted by the GPU itself, preventing any thread management overhead that could negatively affect the performance of the implementation. On the other hand, the POSIX threads technique uses a number of separate threads to perform the parallelism task, which require more communication and synchronization operations, thus, limiting the possibility to use larger number of threads during execution.

The performance of three parallel implementations presented
6 Conclusion
Existing approaches for genetic sequence alignment (such as the NW SW algorithms) suffer from poor performance with large scale input. For this reason, we have presented three different sets of parallel implementations: POSIX threads based, SIMD extensions based and GPU based implementations, to speed up the global alignment with the improved NW algorithm. Our experiments show that the best performance is gained when using the GPU-based architecture, due to the high number of threads it can utilize. However, the performance of the GPU implementation is correlated with the memory optimization and the amount of mutual transformation of data between the CPU and GPU memories. The presented GPU-based implementation significantly outperforms the other implementations where it is considered as 4.1% faster than the SIMD implementation represented by the AVX2 intrinsics, 49.36% faster than the POSIX threads implementation and as 98.62% faster than the sequential implementation. We also confirm that it outperforms other previous GPU implementations because of the better design and optimization methods that are used in our approach.
As a future work, we plan to design a new parallel implementation of the NW algorithm using Open Computing Language (OpenCL) environment, which is supported by AMD, NVIDIA and other brands. This should open the door to experiment with our approach on several hardware. Furthermore, we plan to present another new implementation, which could be done by combining both the SIMD approach and the GPU approach (based on CUDA or OpenCL framework) to exploit the parallelism capability provided by each type of them.
Notes
Indel is a term used for both the insertion and deletion operations in biological sequences.
References
Alsmirat MA, Jararweh Y, Al-Ayyoub M, Shehab MA, Gupta BB (2017) Accelerating compute intensive medical imaging segmentation algorithms using hybrid cpu-gpu implementations. Multimed Tools Appl 76(3):3537–3555
Balhaf K, Alsmirat MA, Al-Ayyoub M, Jararweh Y, Shehab MA (2017) Accelerating levenshtein and damerau edit distance algorithms using gpu with unified memory. In: 2017 8th international conference on information and communication systems (ICICS). IEEE, pp 7–11
Balhaf K, Shehab MA, Wala’a T, Al-Ayyoub M, Al-Saleh M, Jararweh Y (2016) Using gpus to speed-up levenshtein edit distance computation. In: 2016 7th international conference on information and communication systems (ICICS). IEEE, pp 80–84
Butenhof DR (1997) Programming with POSIX threads. Addison-Wesley Professional, Boston
Chan S, Wong A, Chiu D (1992) A survey of multiple sequence comparison methods. Bull Math Biol:563–598
Cook S (2012) CUDA programming: a developer’s guide to parallel computing with GPUs. Newnes, p 16–25
De Oliveira Sandes E, Miranda G, Martorell X, Ayguade E, Teodoro G, Magalhaes Alves de Melo AC (2016) Cudalign 4.0: incremental speculative traceback for exact chromosome-wide alignment in GPU clusters. IEEE Trans Parallel Distrib Syst 27(10):2838–2850
Durbin R, Eddy S, Krogh A, Mitchison G (1998) Biological sequence analysis. Cambridge University Press, Cambridge
El-Metwally S, Ouda O, Helmy M (2014) Next generation sequencing technologies and challenges in sequence assembly. Springer Sci Bus 7:16–25
Fakirah M, Shehab MA, Jararweh Y, Al-Ayyoub M (2015) Accelerating needleman-wunsch global alignment algorithm with gpus. In: 2015 IEEE/ACS 12th international conference of computer systems and applications (AICCSA). IEEE, pp 1–5
Farrar M (2007) Striped smith-waterman speeds database searches six times over other simd implementations. Bioinformatics 23(2):156–161
Gebali F (2011) Algorithms and parallel computing, vol 84. Wiley, New York
Gotoh O (1982) An improved algorithm for matching biological sequences. J Mol Biol 162(3):705–708
Jones C, Pevzner P (2004) An introduction to bioinformatics algorithms. MIT press, Cambridge
Katoh K, Toh H (2008) Recent developments in the mafft multiple sequence alignment program. Brief Bioinform 92:86–98
Liu Y, Schmidt B (2015) Gswabe: faster gpu-accelerated sequence alignment with optimal alignment retrieval for short dna sequences. Concurr Comput: Pract Exper 27 (4):958–972
Lomont C (2011) Introduction to intel advanced vector extensions. White paper, Intel
Needleman-wunsch algorithm. https://en.wikipedia.org/wiki/Needleman-Wunsch_algorithm [Online; accessed July-2015]
Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48(3):443–453
Rognes T (2011) Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation. BMC Bioinform 12:221. https://doi.org/10.1186/1471-2105-12-221
Rognes T, Seeberg E (2000) Six-fold speed-up of smith-waterman sequence database searches using parallel processing on common microprocessors. Bioinformatics 16(8):699–706
Sanders J, Kandrot E (2010) CUDA by example: an introduction to general-purpose GPU programming. Addison-Wesley Professional, Boston
Serrano JP, De Oliveira Sandes E, Magalhaes Alves de Melo A, Ujaldon M (2017) Smith-waterman acceleration in multi-gpus: a performance per watt analysis. In: Bioinformatics and biomedical engineering - 5th international work-conference, IWBBIO 2017, Granada, Spain, April 26–28, 2017, Proceedings, Part II, pp 512–523
Setubal J, Meidanis J (1997) Introduction to computational molecular biology. PWS Pub, Boston
Shehab MA, Al-Ayyoub M, Jararweh Y (2015) Improving fcm and t2fcm algorithms performance using gpus for medical images segmentation. In: 2015 6th international conference on information and communication systems (ICICS), pp 130–135
Siriwardena P, Ranasinghe N (2010) Accelerating global sequence alignment using cuda compatible multi-core gpu. In: 5th international conference in information and automation for sustainability (ICIAFS), pp 201–206
Vermij P (2011) Genetic sequence alignment on a supercomputing platform. Doctoral dissertation, TU Delft, Delft University of Technology
Wozniak A (1997) Using video-oriented instructions to speed up sequence comparison. Comput Appl Biosci: CABIOS 13(2):145–150
Zhou W, Zhanxiu C, Lian B, Wang J, Jianping M (2017) Protein database search of hybrid alignment algorithm based on gpu parallel acceleration. J Supercomput. https://doi.org/10.1007/s11227-017-2030-x
Zhu X, Li K, Salah A, Shi L, Li K (2015) Parallel implementation of MAFFT on cuda-enabled graphics hardware. IEEE/ACM Trans Comput Biol Bioinform 12(1):205–218. https://doi.org/10.1109/TCBB.2014.2351801
Acknowledgments
The authors would like to thank the Deanship of Research at the Jordan University of Science and Technology for funding this work, grant number 20150050. Also, they would like to thank Alexandros Stamatakis and Tomas Flouri from HITS, Germany, for their support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jararweh, Y., Al-Ayyoub, M., Fakirah, M. et al. Improving the performance of the needleman-wunsch algorithm using parallelization and vectorization techniques. Multimed Tools Appl 78, 3961–3977 (2019). https://doi.org/10.1007/s11042-017-5092-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11042-017-5092-0