
Abstract
Oestrogen, the primary female sex hormone, plays a significant role in tumourigenesis. The major pathway for oestrogen is via binding to its receptor [oestrogen receptor (ERα or β)], followed by nuclear translocation and transcriptional regulation of target genes. Almost 70% of breast tumours are ER + , and endocrine therapies with selective ER modulators (tamoxifen) have been successfully applied. As many as 25% of tamoxifen-treated patients experience disease relapse within 5 years upon completion of chemotherapy. In such cases, the ER-independent oestrogen actions provide a plausible explanation for the resistance, as well as expands the existing horizon of available drug targets. ER-independent oestrogen signalling occurs via one of the following pathways: signalling through membrane receptors, oxidative catabolism giving rise to genotoxic metabolites, effects on mitochondria and redox balance, and induction of inflammatory cytokines. The current review focuses on the non-classical oestrogen signalling, its role in cancer, and its clinical significance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
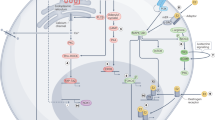
Oestrogens are the primary female sex hormones, mainly synthesised in the ovaries of premenopausal women, and to a lesser extent in the liver, heart, muscle, bone, adipose tissue, and brain. Three physiological oestrogens, namely, oestrone (E1), oestradiol (E2), and oestriol (E3), are formed during cholesterol metabolism. E2 is the predominant and most potent oestrogen found in premenopausal women [1]. The majority of the effects of oestrogen, also known as the genomic effects, are attributed to their binding to the oestrogen receptors (ERα and ERβ) and subsequent translocation to the nucleus. The ligand-bound ER acts as a transcription factor and binds to oestrogen response elements on DNA [2]. ER-dependent oestrogen signalling can also be non-genomic, non-nuclear. Protective effects of E2 on vascular tissues is an example of such signalling, where E2-bound ER interacts with PI3K (phosphoinositide 3-kinase) activating the PI3K/Akt axis resulting in the activation of endothelial nitric oxide synthase [3, 4]. Non-genomic effects of hormones in the extranuclear compartments is rapid and includes important physiological changes in reproductive and nonreproductive cells [5]. ER-independent signalling, which mostly (not exclusively) comprises of non-genomic effects of oestrogen include signalling cascades through membrane receptors and those that directly involve covalent interactions between oestrogens and their metabolites with other biomolecules [6] (Fig. 1). Oestrogens act as endogenous chemical carcinogens through the generation of reactive oxygen species (ROS) and reactive nitrogen species via (a) their direct effect on mitochondria, (b) their oxidative metabolism forming genotoxic metabolites, and (c) their induction of proinflammatory cytokines IL-1β and TNFα [7]. While oestrogens and its receptors also acts as a protective agent in several disease conditions like cardiovascular [8, 9], exercise-induced muscle damage in women [10] and also regulate erythropoiesis under hypoxic condition, thereby can be a target for erythrocytosis [11].
ER-independent oestrogen signalling converges with major cellular signalling pathways. In brain cells derived from mice lacking ERα expression, oestrogen activates Mitogen-activated protein kinases (MAPK) signalling. In a similar model in Breast cancer (BC), exogenous oestrogen treatment increased the levels of phosphorylated protein kinase B (or AKT). Treatment of these cells with ER antagonists did not significantly alter AKT phosphorylation. Moreover, the transient activation of AKT disproved the ER-based nuclear events [12]. Similarly, the interaction of E2 with G-protein-coupled ER 1 (GPER1) culminates in epidermal growth factor receptors (EGFR) [13, 14], PI3/AKT, and MAPK/ERK signalling [15] and NF-κB activation [16]. The current review elaborates on the history and recent reports on ER-independent signalling, the role of oestrogen and its metabolites in cellular physiology, and their clinical significance.

The role of oestrogen in the ER-dependent and ER-independent signalling and the possible therapeutic potential of ER-independent signalling especially in tamoxifen-resistant BC. (ER: oestrogen receptor, PI3K: phosphoinositide 3-kinase, MAPK: Mitogen-activated protein kinases, ROS: reactive oxygen species, RNS: reactive nitrogen species)
Oestrogen metabolism
Oestrogen synthesis in premenopausal women occurs in the ovaries during cholesterol catabolism. The formation of pregnenolone is the first step in oestrogen synthesis via the action of the cytochrome P450 side-chain cleavage enzyme on cholesterol. Pregnenolone is converted into progesterone by 3β-hydroxysteroid dehydrogenase. Testosterone is formed by the sequential action of cytochrome P450 17α-hydroxylase and 17β-hydroxysteroid dehydrogenase (17β-HSD). Androgens are transported from thecal cells to granulosa cells. In the final step, the conversion of testosterone to E2 is catalysed by aromatase, which belongs to the cytochrome P450 superfamily. This enzyme is expressed in the brain, gonads, blood vessels, liver, bones, skin, adipose tissue, and endometrium. The tissue-specific expression of aromatase depends on three factors: alternate splicing, tissue-specific promoters, and different transcription factors. Distinct promoters are differentially regulated by hormones and cytokines. In the brain, aromatase is regulated by post-translational phosphorylation. In non-reproductive women, the main source of E2 is extragonadal organs, where it functions as a paracrine and/or intracrine factor, whereas ovarian E2 is released into the blood [6]. Under physiological conditions, oestrogen metabolism involves conjugation to inactive sulphates and glucuronides [1]. Oestrone sulphate (E1S) is a sulphated oestrogen that serves as the precursor for E2 synthesis. Desulphonation of E1S, a biologically inactive but highly stable derivative, by steroid sulfatase forms E1, which is reduced to E2 by 17β-HSD [17]. E1 and E2 are metabolised via two routes: formation of catechol oestrogen and, to a lesser extent, 16α-hydroxylation [18]. 2-hydroxy oestradiol (2-OHE) and 4-hydroxy oestradiol (4-OHE) are catechol derivatives of E2 that possess tumourigenic potential and are detoxified by catechol-O-methyltransferase (COMT), which has greater catalytic activity toward 2-OHE, thus making it less carcinogenic than 4-OHE [19]. Furthermore, O-methylation of 2-OHE results in the formation of 2-methoxy oestradiol (2-MeOHE) [19]. The inactivation of oestrogen occurs via a conjugation reaction and conversion of E2 (most potent) to E1 and E3 (less active) forms, which help regulate oestrogen activity [1, 20].
In recent decades, advances in research have revealed the involvement of gut microbial flora in the metabolism of oestrogen, especially dietary phytoestrogens. This group of organisms was called astrobleme, broadly classified as Bacteroidetes, Firmicutes, Verrucomicrobia, and Proteobacteria that mainly involve β-glucuronidases and β-glucosidases. The enterohepatic circulation of oestrogen leads to alterations in circulating and excretory oestrogen levels [21]. The use of antibiotics four or more times per year is associated with a moderate increase in BC risk [22, 23]. Antibiotics modulate the gut flora, leading to reduced phytochemical oestrogen metabolism and hence increased circulating oestrogen levels, thereby increasing the cancer risk.
In-vitro and in-vivo studies provide early evidence of ER-independent signalling in cancer
Available data on ER-independent mechanisms of oestrogen are mostly from BC studies, as they are the most implicated in the disease. Direct evidence of oestrogen metabolite genotoxicity was obtained from two independent studies: the big blue rat cell culture mutation assay [20] and Chinese hamster VP-79 cell mutation assay [24]. In-vitro evidence has arisen from studies in which oestrogen metabolites were shown to cause malignant transformation in MCF-10F cells, which are benign breast epithelial cells lacking ER [25, 26]. Furthermore, exposure to physiological concentrations of E2 or 4-OHE in MCF-10F cells resulted in a loss of heterozygosity at common human mammary tumour hot spots. Comparative genomic hybridisation techniques showed that 1 µM 4-OHE induced several damages, including DNA gain (8q24, 9q34) and loss at 13q21 [27]. ER-independent effects of oestrogen were confirmed based on two observations: First, the short time between the stimulus and downstream signalling activation and consistent levels of effector expression, and second, the inability of ER inhibitors to block these actions [14].
In-vivo studies in SCID mice showed that oestradiol-exposed, non-malignant MCF-10 cells formed serially transplantable tumours. Direct and concrete evidence in support of this hypothesis was obtained from an experiment with the ERKO/Wnt-1 model, a transgenic Wnt-1 mouse model with ER knockout. The mice were subjected to oophorectomy, and oestradiol was administered at physiological concentrations, which revealed a dose-dependent increase in breast tumour formation. That study was designed such that the confounding effects of progesterone and other ovarian factors were minimised. A limitation of that study was the presence of minimally functional (confirmed by PCR) ERβ. To overcome any leaky expression of ERα, the ER antagonist fulvestrant was used, which did not have any effect on tumour incidence. However, the use of letrozole in combination with a minimally active form of E2 delayed tumour formation, even in animals with intact ovaries [27, 28]. These studies laid the foundation for the possibility of oestrogen-induced tumourigenesis beyond the pretext of ER.
Clinical evidence for the ER-independent effects of oestrogen came from the fact that in patients with BRCA1 mutation, almost 75% of whom were ER − , bilateral oophorectomy reduced the BC risk by 53% [29, 30]. Additionally, post-menopausal hormone therapy increased the risk of both ER + and ER − disease [27, 31]. More recently, growing experimental data suggests ER-independent, immunomodulatory, DNA damaging effect of selective oestrogen receptor modulators (SERMs) like tamoxifen and raloxifene in triple negative BC (TNBC).
Oestrogen metabolites play a vital role in tumourigenesis
17α-Oestradiol (17α-E2) is a weak oestrogen produced in the ovarian follicles that is converted to E1 and further to E2. 17α-E2 was shown to induce VEGFA mRNA expression in a dose- and time-dependent manner via the PI3K/AKT signalling pathway, independent of ER [32]. 4-OHE was found to be tumourigenic in hamster kidney [33]. Repeated exposure to 4-OHE induced ROS production, malignant transformation of MCF10A cells, and growth in nude mice via PI3K/AKT signalling, leading to the overexpression of cell cycle genes cdc2, PRC1, and PCNA and the transcription factor NRF-1 [34]. In addition, 4-OHE induced the invasion of human breast epithelial cells (MCF10F) [35]. The enzyme involved in the formation of 4-OHE, cytochrome P4501B1 (CYP1B1), showed significant activity in the extrahepatic targets of oestrogen, including the breast, where it was upregulated in tumours compared to normal breast. Polymorphism in CYP1B1 at position 432 was observed in BC and endometrial cancer populations [21, 36].
In-vitro studies revealed that both 2-OHE and 2-MeOHE had a negligible affinity to the ER. In MCF-7 cells, 2-OHE significantly enhanced cell growth and protein synthesis in the G2/M phase of the cell cycle compared with E2. However, 2-MeOHE showed cytostatic activity by inhibiting DNA synthesis and mitosis [37]. In cultured endothelial cells, fulvestrant did not alter the dose-dependent inhibition of endothelin-1 synthesis and MAPK activity by 2-MeOHE, indicating an ER-independent mechanism [38]. In-vitro studies using the osteosarcoma (OS) model yielded similar results, and 2-MeOHE was shown to induce time- and dose-dependent apoptosis without considerable changes in normal osteoblasts or decreased resorption in the bone tumour microenvironment. Here, ER-independent action was confirmed in three experiments: (a) only a negligible effect on cell survival was observed with E2, 2-OHE, and oestrone; (b) 2-MeOHE exerted similar effects irrespective of the ER status of the cell; and (c) addition of ICI182780 did not significantly alter the anti-proliferative effect [39,40,41]. This was accompanied by an increase in interferon (IFN) expression in OS cells. 2-MeOHE also increased the phosphorylation of eIF-2α, a downstream effector of IFN-mediated anti-proliferative effects. The analysis of clinical samples also indicated decreased phosphorylation in the tumour compared with that in normal OS tissues, without significant differences in eIF-2α levels [42]. The specificity of 2-MeOHE for OS cells was later demonstrated in-vivo, providing insights into its therapeutic potential [2, 43]. More recently, the mechanism of 2-MeOHE action was reported to be via a decrease in Bcl-2 and VEGF expression and increase in caspase-3 expression, leading to G2/M cell cycle arrest and premature apoptosis [44]. In the bone marrow microenvironment, 2-MeOHE also decreased the expression of cytokines that mediate tumour growth, survival, and angiogenesis [45, 46]. Mechanistically, in-vitro and in-vivo studies have demonstrated macrophage stimulatory protein-1 receptor, a receptor tyrosine kinase, as a target for 2-MeOHE [47]. In summary, unlike other E2 metabolites, 2-MeOHE plays a pronounced tumour-suppressive role by inhibiting cell proliferation and promoting apoptosis (Table 1).
Oestrogens and their genotoxic metabolites impair the cellular redox state
Evidence for the production of genotoxic metabolites via the oxidative metabolism of endogenous and synthetic oestrogens originated in the late 90 s. Oestrogen can act as an endogenous chemical carcinogen, with its oxidative metabolites forming adducts with DNA, which may result in tumour initiation [20]. An imbalance in oestrogen metabolism that leads to oxidative damage can be attributed to various factors. Overexpression of aromatase leads to excessive synthesis of E2, increased production of 4-OHE, loss or decreased function of COMT, low levels of inactivation of the oestrogen quinone pathway and GSH, and/or low levels of quinone oxidoreductase and/or CYP reductase [56]. For instance, BRCA1/BARD1 transcriptionally controls the P450 subsets of genes, namely, CYP1A1 and CYP3A4. Thus, alterations in the expression of either of these proteins may contribute to tumourigenesis [57]. Oestrogen-induced DNA damage can be of three types: generation of free radicals, formation of oestrogen (or its metabolites)—DNA adducts, and endogenous DNA modifications [58].
The oxidation of catechol oestrogens to semiquinones and o-quinones concomitantly produces superoxide anions and hydroxyl radicals, and redox cascades lead to DNA, protein, and lipid damage (Fig. 2). One-electron oxidation of E2 generates reactive phenoxyl radical intermediate, further giving rise to glutathione thiyl, NAD∙ radical, and H2O2. Accumulation of H2O2 leads to DNA base lesions and affects redox-sensitive transcription factors such as NRF2 and NF-κB, which are key players of inflammatory responses [59]. Oestrogen-induced proliferation and activation of macrophages also produce ROS and modify the function of polymorphonuclear leukocytes, resulting in the formation of hypochlorites (OCl–) [7].

Oestrogen metabolites and its association with cancer
Redox cycling of 2-OHE simultaneously accumulates ROS in MCF-10A cells, activating the IκB kinase signalling and thus, anchorage-independent growth [60]. Similarly, this process was found to produce H2O2 and OH. in MCF-7 and MDA-MB-231 cells, leading to oxidative DNA modification expressed by 8–oxo–7,8–dihydroxy–2′–deoxyguanosine [61]. O-Methylation of 2-OHE by COMT to form 2-MeOHE protected BC cells from catechol oestrogen-induced oxidative DNA damage (formation of 8-hydroxy-2′-deoxyguanosine) [19]. Catechol oestrogens and quinones, in the presence of lactoperoxidase, tyrosinase, or prostaglandin H synthase, react with DNA to form stable depurinated adducts, which act as endogenous initiators of breast tumorigenesis [61]. Oestrogen-3,4-quinones react with adenine and guanine in DNA to form depurinated 4-OHE-1-N3 adenine and 4-OHE-1-N7 guanine adducts, respectively [18, 62]. When hamsters were consistently treated with oestrogen, DNA adducts containing endogenous electrophiles were observed in the renal cortex. Furthermore, in that study, when the animals were treated simultaneously with tamoxifen, DNA damage blocking was unsuccessful [58]. Although the authors were unable to conclude that tamoxifen fails to counteract oestrogen-induced damage, this can now be postulated as an ER-independent mechanism.
An oestrogen metabolite-induced redox imbalance has also been implicated in tumourigenesis in oestrogen-independent organs. The role of oestrogen in the development of thyroid cancer was evaluated as the prevalence of thyroid cancer was 3–4 times higher in women than in men [20]. Follicular thyroid cells produce thyroid hormones via iodination of thyroglobulin [63]. In-vivo studies have revealed higher exposure to oxidative stress in the thyroid glands of female rats owing to increased ROS production and decreased ROS degradation [64]. Further experimental evidence suggests an imbalance in E2 metabolism, as detected by a higher ratio of depurinated oestrogen-DNA adducts in the urine of women with thyroid cancer [65]. Thus, it can be concluded that an imbalance in oestrogen metabolism impairs the cellular redox balance, contributing to tumourigenesis.
Effects of oestrogen on mitochondria
Mitochondria are the powerhouse of the cell that derive energy for the cells from metabolic fuels. This is achieved through oxidative phosphorylation, in which electrons travel through a series of carrier complexes to the final acceptor, O2, which is reduced to water. Electron transfer is accompanied by the expulsion of protons across the inner mitochondrial membrane, which creates a potential difference across the membrane. The change in free energy upon re-entry of protons into the mitochondrial matrix is coupled with ATP synthesis [66]. Lipid-rich mitochondria have been described to act as a “sink” for the highly lipophilic oestrogen. Moreover, the two main enzymes of oestrogen anabolism, 3-βHSD and aromatase, were found to be localised in the mitochondria in ovarian tumour epithelial cells. [67]. E2, through anchorage and integrin-dependent signalling, was found to induce ROS, especially H2O2 in the perinuclear mitochondria. This ROS induction was accompanied by enhanced cell motility; phosphorylation of c-Jun and CREB; and binding of the oxidant-sensitive transcription factors AP-1, CREB, and NRF1 [67]. Functionally, it also modified G1 to S transition and some of the early G1 genes through an ER-independent, non-genomic signalling pathway [68]. These findings provide a novel perspective on the role of E2-induced changes in mitochondria and their impact on cell cycle regulation.
The treatment of human spermatozoa with endogenous E2 and xenoestrogens genistein and bisphenol A alters the mitochondrial membrane potential and increases O2∙ levels [69]. In isolated rat liver mitochondria, E2 reduces ROS generation, consequently protecting mitochondrial integrity and preventing the release of cytochrome c, thereby inhibiting apoptosis [70]. In ovariectomised mice, E2 directly modulated and protected the mitochondrial functions in the absence of ERα [71]. In-vitro BC study showed that E2 induced the expression of ATG3 and Beclin1 and reduced the expression of p53, leading to mitochondrial damage by autophagy and cellular senescence [72]. Another in-vivo study on cardiovascular disease showed that E2 administration induced E2-mediated autophagy which is related to the Rab9-dependent autophagy pathway [73]. In-vivo study using porcine oocytes found E2 to enhance autophagy, reducing ROS levels and apoptosis activity promoting efficacy of the development of porcine oocytes [74]. Exogenous E2 protected mitochondrial functions in cardiomyocytes of ovariectomized rats having insulin resistance [75]. Menopausal women with reduced E2 levels lead to adiposity and reduced insulin sensitivity and diabetes.
In summary, these studies demonstrate that E2 plays an important role in the regulation of mitochondrial membrane potential, ROS production, autophagy, and apoptosis. The mechanism of E2-mediated regulation of mitochondrial function is yet to be fully elucidated.
Membrane-bound receptor of E2 in cancer
Among the ER family, ERα is expressed abundantly in tissues due to its critical role in cellular processes. ERα dysregulation has been reported in many cancers, including breast [76], ovary [77], and uterus [78]. Apart from ERα and ERβ, the G-protein coupled receptor superfamilies are also known to exert important effects of oestrogen. While ERs are intracellularly localised to the mitochondria, GPER1 or GPR30 are plasma membrane-bound receptors that activate signalling cascades via G-proteins. GPER1 is a 375-amino acid transmembrane receptor that binds to E2 and mediates non-genomic oestrogen responses [79]. E2-bound GPER1 phosphorylates ERK1/2 via EGFR, cAMP, Ca2+, and PKC activation. GPER1 is expressed in normal breast mammary cells (MCF10A) and is considerably downregulated in dedifferentiated BC cells. Additionally, GPER1 was found to reduce cell viability but favoured the migration of metastatic BC cells [80]. This could be due to the different downstream signalling pathways in the two phenotypes and requires further characterisation. Two independent studies confirmed this pattern of expression in clinical tumour samples, with all normal breast epithelia showing positive immunoreactivity for GPER1, whereas in tumours, its expression varied. Crosstalk between GPER1 and CXCR1 was found to be involved in the migration and invasion of BC cells [81]. In tumours, GPER1 expression correlated with histological grade, ER negativity, HER2 positivity, tumour size, and metastasis [82, 83]. Direct in-vivo evidence for the proliferative role of GPER1 was obtained using a mouse model. GPER1 knockout mice were crossed with the transgenic mammary tumourigenesis model, MMTV-PyMT. Interestingly, the initial tumour development process was similar in wild-type and GPER1 deficient mice. After a few weeks, the GPER1 null mice showed smaller tumours, lower proliferation, lower grade, and lesser metastasis [84] GPER1 was reported to have the potential to function as a prognostic marker in various cancers [85]. Plasma membrane-bound GPER1 expression is a negative prognostic factor in ER + , tamoxifen-treated, and high-risk BC [86, 87].
Anti-oestrogens tamoxifen and fulvestrant, natural compounds hydroxytyrosol and oleuropein, phytoestrogen coumestrol, and endocrine disruptor bisphenol A bind to and act as non-selective agonists of GPER1 [88]. The selectivity of receptors and cross-reactivity of ligands depend on the effective ligand concentration [89]. E2 and genistein were reported to protect fibroblasts and keratinocytes from ROS-induced damage by activating GPER1 [90]. Genistein promoted the proliferation of thyroid squamous cell carcinoma SW579 cells via GPER1 activation [91]. Chlorobisphenol A, an oestrogenic compound, was found to induce cell proliferation in neuronal cells in-vitro via both ERα and GPER1 pathways [92]. C–C motif chemokine ligand 18 (CCL18), a cytokine overexpressed in tumours, was also shown to bind to GPER1 [93]. GPER1 antagonists such as G-15 [94], G-36 [95], and C4PY (meso-(p-acetamidophenyl)-calix[4]pyrrole) and GPER1 agonists such as 131I-fulvestrant [96] and 5408–0877 [97] are among the GPER1 modulating compounds identified previously (Table 2).
In osteoblasts, E2 inhibits autophagy and apoptosis via the GPER1/AKT axis [103]. Evidence shows lower mortality rates in women with colorectal cancer (CRC), which may be due to the role of oestrogen. While ERβ is downregulated in CRC, public datasets on CRC suggest that the overexpression of GPER1 can selectively rescue tumourigenic effects via the Wnt/β-catenin pathway [104]. Increased expression of GPER1 was observed in gastric cancer and reported to be associated with EMT and poor prognosis [105, 106]. In NSCLC A549 cells, GPER1 agonist decreased the proliferation and enhanced the apoptotic activity by modulating the redox enzymes of the cell [107]. Histological analysis of NSCLC tissues revealed that higher nuclear localisation of GPER1 correlated with poor recurrence-free and overall survival [108]. In papillary thyroid cancer, GPER1 has been implicated in tumourigenesis and is a potential therapeutic target [109]. GPER1 is overexpressed in the cell membrane and cytoplasm of uterine cervical adenocarcinoma in situ and adenocarcinoma, unlike the strong nuclear expression of GPER1 in normal tissue. GPER1-positive patients have poor overall survival [110]. In leiomyosarcoma cells, insulin was found to increase the levels of GPER1 mRNA and protein via the PRKCD/MAPK1/c-Fos/AP axis [87]. GPER1 was reported to be overexpressed in prostate cancer-associated fibroblasts and to regulate its interaction with tumour-associated macrophages, which have the potential to regulate the tumour microenvironment [111]. In glioblastoma, premenopausal women with high GPER1 levels showed better prognosis [112]; however, there is a need to understand the complex signalling pathways that occur parallelly inducing the genomic and non-genomic effects of GPER1 signalling.
Oestrogen signaling via GPER1 has been studied in several non-cancer models [113, 114]. Mechanistically, these studies showed the involvement of necroptosis, ERK signalling, which are crucial in tumorigenesis as well. In-vitro studies also showed the protective role of Genistein/E2 on mitochondria via GPER1 pathway in non-alcoholic fatty liver disease in postmenopausal women and mice muscle cells [115]. GPER1 also partially mediates genistein stimulation and improves glucose tolerance in-vivo [116]. In-vivo studies showed that E2 protects the mitochondrial functions of muscle cells. Estrogen-supplemented postmenopausal women and ovariectomized mice restored complex I function in muscle and liver cells [117]. In both cancer and other diseases, estrogen signaling via GPER1 confirmed to have protective effects over mitochondria functions. However, more research on the mechanism of GPER1 pathway on mitochondria will help to identify novel therapeutic approaches. Further studies are required to shed light on the relationship and balance between ER and GPER1 activities. The role of GPER1 in TNBC might be of clinical significance, that requires experimental evidence.
Therapeutic significance of ER-independent oestrogen signalling
ER-independent oestrogen signalling provides a plausible explanation for resistance to ER antagonist therapies, such as RET signalling mediated Raloxifene resistance demonstrated in-vitro [118]. A recent study demonstrated novel analogues of Tamoxifen, showing potential toxicity against TNBC cell lines MDA-MB-231 and MDA-MB-468 [119]. With the emerging success of drug repurposing, off-target effects of these ER modulators are being explored for their roles in other diseases [120]. 2-MeOHE sensitises BC cells to taxane treatment by centrosome de-clustering [121]. Apart from cell growth arrest and apoptosis, 2-MeOHE also demonstrated the potential to overcome drug resistance in multiple myeloma (MM). Similarly, in OS cells, it inhibited mitochondrial biogenesis via PPARγ, coactivator 1α, and cytochrome c oxidase I. It was also proved to be a potent inhibitor of succinate dehydrogenase complex subunit A and sirtuin 3 [122]. In-vitro, MM models of melphalan (LR-5), doxorubicin (Dox-40 and Dox-6), and dexamethasone (MM.1R) resistance were shown to undergo apoptosis upon treatment with 2-MeOHE. Its therapeutic potential was evaluated in clinical trials involving patients with hormone-refractory prostate cancer and reported to be a well-tolerated drug, with non-linear pharmacokinetics and suboptimal plasma levels. Despite its poor availability for oral administration, it showed some anti-cancer activity at 1200 mg/day [123]. In prostate cancer, the administration of 2-MeOHE in combination with eugenol showed better efficacy, thus reducing the therapeutic concentration and adverse effects (1). 2-MeOHE was effective against melanoma in-vitro and in-vivo and acted synergistically with PD-1 blockade immunotherapy [124]. Synergistic treatment with 2-MeOHE and erlotinib induced apoptosis in hepatocellular cancer (HCC) cells and inhibited the stemness of erlotinib-resistant hypoxic cells [125]. To improve the efficacy of 2-MeOHE, polymeric micelles were used for delivery, which increased the cellular uptake and enhanced the anti-tumour activity in-vitro [126]. Similarly, the micellar administration of 2-MeOHE showed a beneficial effect in lung cancer cells by modulating hypoxia, cell cycle, and apoptosis [126, 127]. Liposomal delivery of 2-MeOHE nanoparticles was effective in the treatment of uterine leiomyoma in-vivo using patient-derived xenografts. There was an almost two-fold difference in the tumour size, proliferation, and apoptosis [128]. There is a need to conduct rigorous clinical trials to establish 2-MeOHE as a therapeutic agent in conjunction with existing therapies.
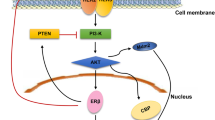
An in-vitro study showed that overexpression of ERα36 and GPER1 can affect responsiveness to tamoxifen and promote tumour progression, thereby not supporting this treatment in ER + BC [129]. E2-mediated GPR30 activation was reported to induce the mTOR signalling pathway, promoting p62 phosphorylation which, in turn, increased ESR1 expression. Targeting this signalling pathway to decrease ESR1 expression holds therapeutic potential in oestrogen-sensitive cells [130]. The antidepressant drug fluoxetine was found to exert oestrogen-like actions through GPER1 activation, leading to the activation of PI3/AKT and ERK signalling cascades [131]. The n-butanol extract of Huaier was found to inhibit gastric cancer proliferation, invasion, and metastasis in-vitro by inhibiting GPER1-mediated PI3/AKT signalling [105]. siRNA-mediated knockdown of GPER1 suppressed the proliferation, invasion, and migration of gastric cancer cells by attenuating PI3/AKT-mediated EMT [106]. The GPER agonist G1 altered the mitochondrial membrane potential, DNA damage, and apoptosis in mantle cell lymphoma (MCL), thereby confirming the potential of G1 in combination with ibrutinib as a candidate therapy for MCL [132]. Targeting GPER1 is a promising approach for modulating cisplatin resistance in gastric cancer. In-vitro GPER1 knockdown increased sensitivity to cisplatin and induced changes in EMT [133]. Cepharanthine hydrochloride was reported to induce mitophagy by binding to GPER1 in HCC primary cells, cell lines, and mouse xenograft models [134]. The GPER1 agonist G1 showed a potential anti-tumour effect in glioblastoma in-vivo [112]. However, there are few data from mechanistic studies on targeting GPER1 and sufficient clinical evidence to prove its specificity to GPER1.
In summary, the expression of ER and GPER1 was reported to be independent in BC, and both receptors showed distinct binding affinities to various oestrogen metabolites [135]. In addition, most ER antagonists serve as GPER1 agonists; hence, treatment modalities require a deep understanding of these pathways, especially in BC [135]. GPER1/EGFR signalling plays an important role in increasing tamoxifen resistance in ER-positive BC [136]. Increased expression of GPER1 and GPER1-EGFR mediated signalling play a major role in TNBC progression [137, 138] modulating the GPER1 signalling pathway could be an effective approach in the treatment of both hormone-positive cancer and TNBC. Recent advances in research have provided other options such as proteolysis-targeting chimeras (PROTACs) to target specific proteins of interest. Hence, GPER1 holds potential as a therapeutic target in cancer, and the modulation of its expression can complement existing anti-oestrogen therapies and help manage drug resistance in oestrogen-driven cancers.
Conclusion
Almost 70% of breast tumours are ER + , and endocrine therapies with selective ER modulators (tamoxifen) or downregulators (fulvestrant) are a common treatment modality for such subtypes. Approximately 25% of tamoxifen-treated patients present with disease recurrence 5 years post-therapy. Several mechanisms, including MAPK- and AKT-induced downregulation of ER and epigenetic modifications, have been implicated in tamoxifen resistance. Furthermore, 20–30% of ER + patients develop either de-novo or acquired resistance to tamoxifen. As described previously, ER-independent actions may represent a mode of resistance to anti-ER therapy. Furthermore, modulating the oestrogen metabolism imbalance leading to free radical-mediated DNA damage, addressing the therapeutic potential of oestrogen-mitochondrial interactions, and targeting GPER1 offer novel treatment strategies, especially for TNBC.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Okoh V, Deoraj A, Roy D (2011) Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim Biophys Acta - Rev Cancer 1815:115–133
Maran A, Zhang M, Kennedy AM, Turner RT (2003) ER-independent actions of estrogen and estrogen metabolitesin bone cells. J Musculoskel Neuron Interact 3:367–369
Simoncini T, Hafezi-Moghadam A, Brazil DP et al (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407:538–541. https://doi.org/10.1038/35035131
Haynes MP, Sinha D, Russell KS et al (2000) Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 87:677–682. https://doi.org/10.1161/01.res.87.8.677
Levin ER (2005) Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol 19:1951–1959
Cui J, Shen Y, Li R (2013) Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med 19:197–209. https://doi.org/10.1016/J.MOLMED.2012.12.007
Roy D, Cai Q, Felty Q, Narayan S (2007) Estrogen-induced generation of reactive oxygen and nitrogen species, gene damage, and estrogen-dependent cancers. J. Toxicol. Environ Heal - Part B Crit Rev 10:235–257
Iorga A, Cunningham CM, Moazeni S et al (2017) The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ 8:33. https://doi.org/10.1186/s13293-017-0152-8
Aryan L, Younessi D, Zargari M, et al (2020) The Role of Estrogen Receptors in Cardiovascular Disease. Int. J. Mol. Sci. 21
Kendall B, Eston R (2002) Exercise-induced muscle damage and the potential protective role of estrogen. Sports Med 32:103–123. https://doi.org/10.2165/00007256-200232020-00003
Azad P, Villafuerte FC, Bermudez D et al (2021) Protective role of estrogen against excessive erythrocytosis in Monge’s disease. Exp Mol Med 53:125–135. https://doi.org/10.1038/s12276-020-00550-2
Tsai E-M, Wang S-C, Lee J-N, Hung M-C (2001) Akt Activation by Estrogen in Estrogen Receptor-negative Breast Cancer Cells
Luo H, Yang G, Yu T et al (2014) GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr Relat Cancer 21:355–369. https://doi.org/10.1530/ERC-13-0237
Filardo EJ, Quinn JA, Bland KI, Frackelton ARJ (2000) Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649–1660. https://doi.org/10.1210/mend.14.10.0532
De Marco P, Cirillo F, Vivacqua A, et al (2015) Novel Aspects Concerning the Functional Cross-Talk between the Insulin/IGF-I System and Estrogen Signaling in Cancer Cells . Front. Endocrinol. 6
Zhu P, Liao L-Y, Zhao T-T et al (2017) GPER/ERK&AKT/NF-κB pathway is involved in cadmium-induced proliferation, invasion and migration of GPER-positive thyroid cancer cells. Mol Cell Endocrinol 442:68–80. https://doi.org/10.1016/j.mce.2016.12.007
Purohit A, Woo LWL, Potter BVL (2011) Steroid sulfatase: a pivotal player in estrogen synthesis and metabolism. Mol Cell Endocrinol 340:154–160. https://doi.org/10.1016/j.mce.2011.06.012
Li KM, Todorovic R, Devanesan P et al (2004) Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis 25:289–297. https://doi.org/10.1093/carcin/bgg191
Lavigne JA, Goodman JE, Fonong T, et al (2001) The Effects of Catechol-O-Methyltransferase Inhibition on Estrogen Metabolite and Oxidative DNA Damage Levels in Estradiol-treated MCF-7 Cells 1
Cavalieri E, Chakravarti D, Guttenplan J et al (2006) Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta - Rev Cancer 1766:63–78
Parida S, Sharma D (2019) The microbiome-estrogen connection and breast cancer risk. Cells 8:1642. https://doi.org/10.3390/CELLS8121642
Xuan C, Shamonki JM, Chung A et al (2014) Microbial dysbiosis is associated with human breast cancer. PLoS One 9:e83744. https://doi.org/10.1371/journal.pone.0083744
Wu AH, Tseng C, Vigen C et al (2020) Gut microbiome associations with breast cancer risk factors and tumor characteristics: a pilot study. Breast Cancer Res Treat 182:451–463. https://doi.org/10.1007/s10549-020-05702-6
Kong LY, Szaniszlo P, Albrecht T, Liehr JG (2000) Frequency and molecular analysis of hprt mutations induced by estradiol in Chinese hamster V79 cells. Int J Oncol 17:1141–1149. https://doi.org/10.3892/ijo.17.6.1141
Fernandez SV, Russo IH, Russo J (2006) Estradiol and its metabolites 4-hydroxyestradiol and 2-hydroxyestradiol induce mutations in human breast epithelial cells. Int J cancer 118:1862–1868. https://doi.org/10.1002/IJC.21590
Russo J, Fernandez SV, Russo PA et al (2006) 17-Beta-estradiol induces transformation and tumorigenesis in human breast epithelial cells. FASEB J 20:1622–1634. https://doi.org/10.1096/FJ.05-5399COM
Yue W, Yager JD, Wang JP et al (2013) Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids 78:161–170
Yue W, Wang JP, Li Y et al (2010) Effects of estrogen on breast cancer development: Role of estrogen receptor independent mechanisms. Int J Cancer 127:1748–1757. https://doi.org/10.1002/ijc.25207
Rebbeck TR, Kauff ND, Domchek SM (2009) Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J Natl Cancer Inst 101:80–87. https://doi.org/10.1093/jnci/djn442
Eisen A, Lubinski J, Klijn J et al (2005) Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. J Clin Oncol 23:7491–7496. https://doi.org/10.1200/JCO.2004.00.7138
Lakhani SR, Van De Vijver MJ, Jacquemier J et al (2002) The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol 20:2310–2318. https://doi.org/10.1200/JCO.2002.09.023
Banerjee S, Saxena N, Sengupta K, Banerjee SK (2003) 17alpha-estradiol-induced VEGF-A expression in rat pituitary tumor cells is mediated through ER independent but PI3K-Akt dependent signaling pathway. Biochem Biophys Res Commun 300:209–215. https://doi.org/10.1016/s0006-291x(02)02830-9
Sasaki M, Tanaka Y, Kaneuchi M, et al (2003) CYP1B1 Gene Polymorphisms Have Higher Risk for Endometrial Cancer, and Positive Correlations with Estrogen Receptor and Estrogen Receptor Expressions
Okoh VO, Felty Q, Parkash J et al (2013) Reactive Oxygen Species via Redox Signaling to PI3K/AKT Pathway Contribute to the Malignant Growth of 4-Hydroxy Estradiol-Transformed Mammary Epithelial Cells. PLoS One. https://doi.org/10.1371/journal.pone.0054206
Russo J, Lareef MH, Balogh G et al (2003) Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J Steroid Biochem Mol Biol 87:1–25
Zheng W, Xie D-W, Jin F, et al (2000) Genetic Polymorphism of Cytochrome P450–1B1 and Risk of Breast Cancer 1
Lottering ML, Haag M, Seegers JC (1992) Effects of 17 beta-estradiol metabolites on cell cycle events in MCF-7 cells. Cancer Res 52:5926–5932
Dubey RK, Jackson EK, Keller PJ et al (2001) Estradiol metabolites inhibit endothelin synthesis by an estrogen receptor-independent mechanism. Hypertension 37(2):640–644
Maran A, Zhang M, Kennedy AM et al (2002) 2-methoxyestradiol induces interferon gene expression and apoptosis in osteosarcoma cells. Bone 30:393–398. https://doi.org/10.1016/S8756-3282(01)00681-0
Benedikt MB, Mahlum EW, Shogren KL et al (2010) 2-Methoxyestradiol-mediated anti-tumor effect increases osteoprotegrin expression in osteosarcoma cells. J Cell Biochem 109:950–956. https://doi.org/10.1002/JCB.22473
Maran A, Shogren KL, Benedikt M et al (2008) 2-methoxyestradiol-induced cell death in osteosarcoma cells is preceded by cell cycle arrest. J Cell Biochem 104:1937–1945. https://doi.org/10.1002/JCB.21758
Wimbauer F, Yang C, Shogren KL et al (2012) Regulation of interferon pathway in 2-methoxyestradiol-treated osteosarcoma cells. BMC Cancer 12:1–10. https://doi.org/10.1186/1471-2407-12-93/TABLES/1
Przychodzen P, Wyszkowska R, Gorzynik-Debicka M et al (2019) Anticancer potential of oleuropein, the polyphenol of olive oil, with 2-methoxyestradiol, separately or in combination, in human osteosarcoma cells. Anticancer Res 39:1243–1251. https://doi.org/10.21873/ANTICANRES.13234
Tang X, Tao F, Xiang W et al (2020) Anticancer effects and the mechanism underlying 2-methoxyestradiol in human osteosarcoma in vitro and in vivo. Oncol Lett 20:1. https://doi.org/10.3892/OL.2020.11925/HTML
Chauhan D, Catley L, Hideshima T et al (2002) 2-Methoxyestradiol overcomes drug resistance in multiple myeloma cells. Blood 100:2187–2194. https://doi.org/10.1182/BLOOD-2002-02-0376
Dubey RK, Imthurn B, Jackson EK (2007) 2-Methoxyestradiol: a potential treatment for multiple proliferative disorders. Endocrinology 148:4125–4127. https://doi.org/10.1210/EN.2007-0514
Batth IS, Huang SB, Villarreal M et al (2021) Evidence for 2-methoxyestradiol-mediated inhibition of receptor tyrosine kinase RON in the management of prostate cancer. Int J Mol Sci. https://doi.org/10.3390/IJMS22041852
Zhang X, Huang H, Xu Z, Zhan R (2010) 2-Methoxyestradiol blocks cell-cycle progression at the G2/M phase and induces apoptosis in human acute T lymphoblastic leukemia CEM cells. Acta Biochim Biophys Sin (Shanghai) 42:615–622. https://doi.org/10.1093/abbs/gmq065
Pal P, Hales K, Hales DB (2020) The pro-apoptotic actions of 2-methoxyestradiol against ovarian cancer involve catalytic activation of PKCδ signaling. Oncotarget 11:3646. https://doi.org/10.18632/ONCOTARGET.27760
Sawicka E, Saczko J, Roik J et al (2020) Effect of Interaction between 17β-Estradiol, 2-Methoxyestradiol and 16α-Hydroxyestrone with Chromium (VI) on Ovary Cancer Line SKOV-3: Preliminary Study. Mol 25:5214. https://doi.org/10.3390/MOLECULES25215214
Rincón-Rodriguez R, Mena D, Mena J et al (2019) F-Spondin Is the Signal by Which 2-Methoxyestradiol Induces Apoptosis in the Endometrial Cancer Cell Line Ishikawa. Int J Mol Sci 20:3850. https://doi.org/10.3390/IJMS20163850
Ghosh R, Ganapathy M, Alworth WL et al (2009) Combination of 2-methoxyestradiol (2-ME2) and eugenol for apoptosis induction synergistically in androgen independent prostate cancer cells. J Steroid Biochem Mol Biol 113:25–35. https://doi.org/10.1016/J.JSBMB.2008.11.002
Sharma N, Raut PW, Baruah MM, Sharma A (2021) Combination of quercetin and 2-methoxyestradiol inhibits epithelial–mesenchymal transition in PC-3 cell line via Wnt signaling pathway. Futur Sci OA. https://doi.org/10.2144/fsoa-2021-0028
Zhang S, Yu H, Li J et al (2022) 2-Methoxyestradiol combined with ascorbic acid facilitates the apoptosis of chronic myeloid leukemia cells via the microRNA-223/Fms-like tyrosine kinase 3/phosphatidylinositol-3 kinase/protein kinase B axis. Bioengineered 13:3470–3485. https://doi.org/10.1080/21655979.2021.2024327
Musial C, Knap N, Zaucha R et al (2022) Induction of 2-hydroxycatecholestrogens O-methylation: a missing puzzle piece in diagnostics and treatment of lung cancer. Redox Biol 55:102395. https://doi.org/10.1016/J.REDOX.2022.102395
Cavalieri E, Rogan E, Chakravarti D (2004) The Role of Endogenous Catechol Quinones in the Initiation of Cancer and Neurodegenerative Diseases. https://doi.org/10.1016/S0076-6879(04)82017-2
Stewart MD, Zelin E, Dhall A et al (2018) BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proc Natl Acad Sci U S A 115:1316–1321. https://doi.org/10.1073/PNAS.1715467115/SUPPL_FILE/PNAS.1715467115.SAPP.PDF
Roy D, Liehr JG (1999) Estrogen DNA damage and mutations. Mutation Res/Fund Mol Mech Mutagen. https://doi.org/10.1016/S0027-5107(99)00012-3
Maiti S, Nazmeen A (2019) Impaired redox regulation of estrogen metabolizing proteins is important determinant of human breast cancers. Cancer Cell Int 19:1–13. https://doi.org/10.1186/S12935-019-0826-X/FIGURES/5
Na HK, Park SA, Kim EH et al (2009) 4-hydroxyestradiol induces anchorage-independent growth of human mammary epithelial cells via activation of IkappaB kinase: potential role of reactive oxygen species. Cancer Res 69:2416–2424. https://doi.org/10.1158/0008-5472.CAN-08-2177
Starek-Świechowicz B, Budziszewska B, Starek A (2021) Endogenous estrogens—breast cancer and chemoprevention. Pharmacol Reports 73:1497–1512. https://doi.org/10.1007/S43440-021-00317-0/FIGURES/3
Cavalieri EL, Stack DE, Devanesan PD et al (1997) Molecular origin of cancer: Catechol estrogen-3 4-quinones as endogenous tumor initiators. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.94.20.10937
Faria CC, Peixoto MS, Carvalho DP, Fortunato RS (2019) The emerging role of estrogens in thyroid redox homeostasis and carcinogenesis. Oxid Med Cell Longev 2019:2514312. https://doi.org/10.1155/2019/2514312
Fortunato RS, Braga WMO, Ortenzi VH et al (2013) Sexual dimorphism of thyroid reactive oxygen species production due to higher NADPH Oxidase 4 expression in female thyroid glands. Thyroid 23:111–119. https://doi.org/10.1089/THY.2012.0142
Zahid M, Goldner W, Beseler CL et al (2013) Unbalanced estrogen metabolism in thyroid cancer. Int J cancer 133:2642–2649. https://doi.org/10.1002/ijc.28275
Sastre-Serra J, Valle A, Company MM et al (2010) Estrogen down-regulates uncoupling proteins and increases oxidative stress in breast cancer. Free Radic Biol Med 48:506–512. https://doi.org/10.1016/j.freeradbiomed.2009.11.025
Felty Q, Roy D (2005) Mitochondrial signals to nucleus regulate estrogen-induced cell growth. Med Hypotheses 64:133–141. https://doi.org/10.1016/j.mehy.2003.12.056
Felty Q, Singh KP, Roy D (2005) Estrogen-induced G1/S transition of G0-arrested estrogen-dependent breast cancer cells is regulated by mitochondrial oxidant signaling. Oncogene 24:4883–4893. https://doi.org/10.1038/sj.onc.1208667
Skibińska I, Jendraszak M, Borysiak K et al (2016) 17β-estradiol and xenoestrogens reveal synergistic effect on mitochondria of human sperm. Ginekol Pol 87:360–366. https://doi.org/10.5603/GP.2016.0005
Borrás C, Gambini J, López-Grueso R et al (2010) Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta 1802:205–211. https://doi.org/10.1016/j.bbadis.2009.09.007
Torres MJ, Kew KA, Ryan TE et al (2018) 17β-Estradiol directly lowers mitochondrial membrane microviscosity and improves bioenergetic function in skeletal muscle. Cell Metab 27:167-179.e7. https://doi.org/10.1016/j.cmet.2017.10.003
Bajbouj K, Shafarin J, Taneera J, Hamad M (2020) Estrogen signaling induces mitochondrial dysfunction-associated autophagy and senescence in breast cancer cells. Biology (Basel). https://doi.org/10.3390/biology9040068
Sasaki Y, Ikeda Y, Uchikado Y et al (2021) Estrogen plays a crucial role in rab9-dependent mitochondrial autophagy delaying arterial senescence. J Am Heart Assoc 10:e019310. https://doi.org/10.1161/JAHA.120.019310
Duan J, Chen H, Xu D et al (2021) 17β-estradiol improves the developmental ability, inhibits reactive oxygen species levels and apoptosis of porcine oocytes by regulating autophagy events. J Steroid Biochem Mol Biol 209:105826. https://doi.org/10.1016/j.jsbmb.2021.105826
Gorbenko NI, Borikov AY, Ivanova OV et al (2014) Effect of 17β-estradiol on bioenergetic processes in the heart mitochondria of ovariectomized rats with insulin resistance. Biomed Khim 60:576–580. https://doi.org/10.18097/pbmc20146005576
Kim S, Lee J-Y, Shin SG et al (2021) ESRRA (estrogen related receptor alpha) is a critical regulator of intestinal homeostasis through activation of autophagic flux via gut microbiota. Autophagy 17:2856–2875. https://doi.org/10.1080/15548627.2020.1847460
Laws MJ, Kannan A, Pawar S et al (2014) Dysregulated estrogen receptor signaling in the hypothalamic-pituitary-ovarian axis leads to ovarian epithelial tumorigenesis in mice. PLoS Genet 10:e1004230. https://doi.org/10.1371/journal.pgen.1004230
Kasoha M, Dernektsi C, Seibold A et al (2020) Crosstalk of estrogen receptors and Wnt/β-catenin signaling in endometrial cancer. J Cancer Res Clin Oncol 146:315–327. https://doi.org/10.1007/s00432-019-03114-8
Tutzauer J, Gonzalez de Valdivia E, Swärd K et al (2021) Ligand-independent g protein-coupled estrogen receptor/g protein-coupled receptor 30 activity: lack of receptor-dependent effects of G-1 and 17β-estradiol. Mol Pharmacol 100:271–282. https://doi.org/10.1124/molpharm.121.000259
Segura-Bautista D, Olivares A, Casas-González P et al (2020) GPR30 expression and function in breast cancer cells are induced through a cis-acting element targeted by ETS factors. Oncol Rep 43:1669–1682. https://doi.org/10.3892/or.2020.7540
Jiang Q-F, Wu T-T, Yang J-Y et al (2013) 17β-Estradiol promotes the invasion and migration of nuclear estrogen receptor-negative breast cancer cells through cross-talk between GPER1 and CXCR1. J Steroid Biochem Mol Biol 138:314–324. https://doi.org/10.1016/j.jsbmb.2013.07.011
Filardo EJ, Graeber CT, Quinn JA et al (2006) Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin cancer Res an Off J Am Assoc Cancer Res 12:6359–6366. https://doi.org/10.1158/1078-0432.CCR-06-0860
Ignatov T, Weißenborn C, Poehlmann A et al (2013) GPER-1 expression decreases during breast cancer tumorigenesis. Cancer Invest 31:309–315. https://doi.org/10.3109/07357907.2013.789901
Marjon NA, Hu C, Hathaway HJ, Prossnitz ER (2014) G Protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol Cancer Res 12:1644–1654. https://doi.org/10.1158/1541-7786.MCR-14-0128-T
Ulhaq ZS, Soraya GV, Milliana A, Tse WKF (2021) Association between GPER gene polymorphisms and GPER expression levels with cancer predisposition and progression. Heliyon 7:e06428. https://doi.org/10.1016/j.heliyon.2021.e06428
Tutzauer J, Sjöström M, Bendahl P-O et al (2020) Plasma membrane expression of G protein-coupled estrogen receptor (GPER)/G protein-coupled receptor 30 (GPR30) is associated with worse outcome in metachronous contralateral breast cancer. PLoS One 15:e0231786. https://doi.org/10.1371/journal.pone.0231786
De Marco P, Romeo E, Vivacqua A et al (2014) GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer 21:739–753. https://doi.org/10.1530/ERC-14-0245
DeLeon C, Wang DQ-H, Arnatt CK (2020) G Protein-Coupled Estrogen Receptor, GPER1, Offers a Novel Target for the Treatment of Digestive Diseases . Front. Endocrinol. 11
Prossnitz ER, Arterburn JB (2015) International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev 67:505–540. https://doi.org/10.1124/pr.114.009712
Savoia P, Raina G, Camillo L et al (2018) Anti-oxidative effects of 17 β-estradiol and genistein in human skin fibroblasts and keratinocytes. J Dermatol Sci 92:62–77. https://doi.org/10.1016/j.jdermsci.2018.07.007
Chen Z, Xuan Q, Zhao D et al (2020) Roles of G protein-coupled receptor 30 in the effects of genistein on apoptosis and cell cycle in human thyroid squamous cells SW579. Wei Sheng Yan Jiu 49:780–784. https://doi.org/10.19813/j.cnki.weishengyanjiu.2020.05.015
Adeyemi SA, Choonara YE, Kumar P et al (2019) Folate-decorated, endostatin-loaded, nanoparticles for anti-proliferative chemotherapy in esophaegeal squamous cell carcinoma. Biomed Pharmacother 119:109450. https://doi.org/10.1016/j.biopha.2019.109450
Schmidt-Wolf R, Zissel G (2020) Interaction Between CCL18 and GPR30 Differs from the Interaction Between Estradiol and GPR30. Anticancer Res 40:3097–3108. https://doi.org/10.21873/anticanres.14291
Bai L-Y, Weng J-R, Hu J-L et al (2013) G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem Biol Interact 206:375–384. https://doi.org/10.1016/j.cbi.2013.10.014
Rudelius M, Rauert-Wunderlich H, Hartmann E et al (2015) The G protein-coupled estrogen receptor 1 (GPER-1) contributes to the proliferation and survival of mantle cell lymphoma cells. Haematologica 100:e458–e461
Yin G, Zeng B, Peng Z et al (2018) Synthesis and application of 131I-fulvestrant as a targeted radiation drug for endocrine therapy in human breast cancer. Oncol Rep 39:1215–1226. https://doi.org/10.3892/or.2018.6212
Broughton BRS, Miller AA, Sobey CG (2010) Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. Am J Physiol Heart Circ Physiol 298:H1055–H1061. https://doi.org/10.1152/ajpheart.00878.2009
Pratiwi RIA, Widyarti S, Sumitro SB (2023) Potential of Kesambi Active Compound (Schleichera oleosa) as Antagonist G-Protein Estrogen Receptor 1 (GPER1) by In Silico. J Exp Life Sci 13:43–51. https://doi.org/10.21776/ub.jels.2023.013.01.07
Masuhara M, Tsukahara T, Tomita K et al (2016) A relation between osteoclastogenesis inhibition and membrane-type estrogen receptor GPR30. Biochem Biophys Reports 8:389–394. https://doi.org/10.1016/j.bbrep.2016.10.013
Xu KJ, Loganathan N, Belsham DD (2022) Bisphenol S induces Agrp expression through GPER1 activation and alters transcription factor expression in immortalized hypothalamic neurons: A mechanism distinct from BPA-induced upregulation. Mol Cell Endocrinol 552:111630. https://doi.org/10.1016/j.mce.2022.111630
Segovia-Mendoza M, Mirzaei E, Prado-Garcia H, et al (2022) The Interplay of GPER1 with 17-Aminoestrogens in the Regulation of the Proliferation of Cervical and Breast Cancer Cells: A Pharmacological Approach. Int. J. Environ. Res. Public Health 19
Khan SU, Ahemad N, Chuah L-H et al (2019) Sequential ligand- and structure-based virtual screening approach for the identification of potential G protein-coupled estrogen receptor-1 (GPER-1) modulators. RSC Adv 9:2525–2538. https://doi.org/10.1039/C8RA09318K
Zhang Y, Jiang T, Ni S et al (2022) Effects of Estrogen on Proliferation and Apoptosis of Osteoblasts through Regulating GPER/AKT Pathway. Cell Mol Biol 68:124–129. https://doi.org/10.14715/cmb/2022.68.6.20
Abancens M, Harvey BJ, McBryan J (2022) GPER Agonist G1 Prevents Wnt-Induced JUN Upregulation in HT29 Colorectal Cancer Cells. Int J Mol Sci. https://doi.org/10.3390/ijms232012581
Wang X-F, Hu C, Mo S-W et al (2022) GPR30 Activation promotes the progression of gastric cancer and plays a significant role in the anti-GC effect of Huaier. J Oncol 2022:2410530. https://doi.org/10.1155/2022/2410530
Xu E, Xia X, Jiang C, et al (2020) GPER1 Silencing Suppresses the Proliferation, Migration, and Invasion of Gastric Cancer Cells by Inhibiting PI3K/AKT–Mediated EMT . Front Cell Dev Biol. 8
Kurt Hakan A, Çelik A, Kelleci Mehmet B (2015) Oxidative/antioxidative enzyme-mediated antiproliferative and proapoptotic effects of the GPER1 agonist G-1 on lung cancer cells. Oncol Lett 10:3177–3182. https://doi.org/10.3892/ol.2015.3711
Li Z-H, Liu C, Liu Q-H et al (2022) Cytoplasmic expression of G protein-coupled estrogen receptor 1 correlates with poor postoperative prognosis in non-small cell lung cancer. J Thorac Dis 14:1466–1477. https://doi.org/10.21037/jtd-22-29
Bertoni APS, de Manfroi P, A Tomedi J et al (2021) The gene expression of GPER1 is low in fresh samples of papillary thyroid carcinoma (PTC), and in silico analysis. Mol Cell Endocrinol 535:111397. https://doi.org/10.1016/j.mce.2021.111397
Ino Y, Akimoto T, Takasawa A et al (2020) Elevated expression of G protein-coupled receptor 30 (GPR30) is associated with poor prognosis in patients with uterine cervical adenocarcinoma. Histol Histopathol 35:351–359. https://doi.org/10.14670/HH-18-157
Zhang R, Zong J, Peng Y et al (2021) GPR30 knockdown weakens the capacity of CAF in promoting prostate cancer cell invasion via reducing macrophage infiltration and M2 polarization. J Cell Biochem. https://doi.org/10.1002/jcb.29938
Hirtz A, Bailly Y, Rech F, et al (2022) Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma. Int. J. Mol. Sci. 23
Li Z, Chen L, Chu H et al (2022) Estrogen alleviates hepatocyte necroptosis depending on GPER in hepatic ischemia reperfusion injury. J Physiol Biochem 78:125–137. https://doi.org/10.1007/s13105-021-00846-5
Bopassa JC, Eghbali M, Toro L, Stefani E (2010) A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 298:H16-23. https://doi.org/10.1152/ajpheart.00588.2009
Sbert-Roig M, Bauzá-Thorbrügge M, Galmés-Pascual BM et al (2016) GPER mediates the effects of 17β-estradiol in cardiac mitochondrial biogenesis and function. Mol Cell Endocrinol 420:116–124. https://doi.org/10.1016/j.mce.2015.11.027
Vásquez-Reyes S, Vargas-Castillo A, Noriega LG et al (2022) Genistein stimulation of white adipose tissue thermogenesis is partially dependent on GPR30 in mice. Mol Nutr Food Res 66:e2100838. https://doi.org/10.1002/mnfr.202100838
Torres MJ, Ryan TE, Lin C-T et al (2018) Impact of 17β-estradiol on complex I kinetics and H(2)O(2) production in liver and skeletal muscle mitochondria. J Biol Chem 293:16889–16898. https://doi.org/10.1074/jbc.RA118.005148
Pecar G, Liu S, Hooda J et al (2023) RET signaling in breast cancer therapeutic resistance and metastasis. Breast Cancer Res 25:26. https://doi.org/10.1186/s13058-023-01622-7
Elbagoury RM, Shenouda MA, Elnakib HE et al (2023) Design, synthesis, and metabolite identification of Tamoxifen esterase-activatable prodrugs. Bioorg Chem 131:106303. https://doi.org/10.1016/j.bioorg.2022.106303
Ralf B, T. HM, Gabriel F-C, et al (2022) Repurposing tamoxifen as potential host-directed therapeutic for tuberculosis. MBio 14:e03024-e3122. https://doi.org/10.1128/mbio.03024-22
El-Zein R, Thaiparambil J, Abdel-Rahman SZ (2020) 2-methoxyestradiol sensitizes breast cancer cells to taxanes by targeting centrosomes. Oncotarget 11:4479. https://doi.org/10.18632/ONCOTARGET.27810
Gorska-Ponikowska M, Kuban-Jankowska A, Eisler SA et al (2018) 2-Methoxyestradiol affects mitochondrial biogenesis pathway and succinate dehydrogenase complex flavoprotein subunit a in osteosarcoma cancer cells. Cancer Genomics Proteomics 15:73–89. https://doi.org/10.21873/cgp.20067
Sweeney C, Liu G, Yiannoutsos C et al (2005) A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin Cancer Res 11:6625–6633. https://doi.org/10.1158/1078-0432.CCR-05-0440
Hua W, Huang X, Li J et al (2022) 2-methoxyestradiol inhibits melanoma cell growth by activating adaptive immunity. Immunopharmacol Immunotoxicol. https://doi.org/10.1080/0892397320222062380
Zheng S, Ni J, Li Y et al (2021) 2-Methoxyestradiol synergizes with Erlotinib to suppress hepatocellular carcinoma by disrupting the PLAGL2-EGFR-HIF-1/2α signaling loop. Pharmacol Res 169:105685. https://doi.org/10.1016/J.PHRS.2021.105685
Alhakamy NA, Ahmed OAA, Fahmy UA, Md S (2021) Development and In Vitro Evaluation of 2-Methoxyestradiol Loaded Polymeric Micelles for Enhancing Anticancer Activities in Prostate Cancer. Polym 13:884. https://doi.org/10.3390/POLYM13060884
Awan ZA, AlGhamdi SA, Alhakamy NA et al (2022) Optimized 2-methoxyestradiol invasomes fortified with apamin: a promising approach for suppression of A549 lung cancer cells. Drug Delivery 29:1536–1548. https://doi.org/10.1080/10717544.2022.2072412
Borahay MA, Vincent KL, Motamedi M et al (2020) (2020) Liposomal 2-methoxyestradiol nanoparticles for treatment of uterine leiomyoma in a patient-derived xenograft mouse model. Reprod Sci 281(28):271–277. https://doi.org/10.1007/S43032-020-00248-W
Xu Z, Zhao D, Zheng X et al (2022) Low concentrations of 17β-estradiol exacerbate tamoxifen resistance in breast cancer treatment through membrane estrogen receptor-mediated signaling pathways. Environ Toxicol 37:514–526. https://doi.org/10.1002/tox.23417
Tsai C-L, Lin C-Y, Chao A et al (2021) GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor α Protein Expression by Inducing Its Release from a Complex Formed with KEAP1. J Pers Med. https://doi.org/10.3390/jpm11090906
Lei B, Xu L, Zhang X et al (2021) The proliferation effects of fluoxetine and amitriptyline on human breast cancer cells and the underlying molecular mechanisms. Environ Toxicol Pharmacol 83:103586. https://doi.org/10.1016/j.etap.2021.103586
Zhou L, Yu T, Yang F et al (2021) G Protein-Coupled Estrogen Receptor Agonist G-1 Inhibits Mantle Cell Lymphoma Growth in Preclinical Models. Front Oncol 11:668617. https://doi.org/10.3389/fonc.2021.668617
Wang X, Xu Z, Sun J et al (2020) Cisplatin resistance in gastric cancer cells is involved with GPR30-mediated epithelial-mesenchymal transition. J Cell Mol Med 24:3625–3633. https://doi.org/10.1111/jcmm.15055
Wang Y, Su G-F, Huang Z-X et al (2020) Cepharanthine hydrochloride induces mitophagy targeting GPR30 in hepatocellular carcinoma (HCC). Expert Opin Ther Targets 24:389–402. https://doi.org/10.1080/14728222.2020.1737013
Filardo EJ, Thomas P (2012) Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 153:2953–2962. https://doi.org/10.1210/en.2012-1061
Ignatov A, Ignatov T, Roessner A et al (2010) Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat 123:87–96. https://doi.org/10.1007/s10549-009-0624-6
Xu S, Yu S, Dong D, Lee LTO (2019) G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front Endocrinol (Lausanne) 10:725. https://doi.org/10.3389/fendo.2019.00725
Girgert R, Emons G, Gründker C (2012) Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast Cancer Res Treat 134:199–205. https://doi.org/10.1007/s10549-012-1968-x
Funding
Cancer Institute (WIA)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gopinath, P., Oviya, R.P. & Gopisetty, G. Oestrogen receptor-independent actions of oestrogen in cancer. Mol Biol Rep 50, 9497–9509 (2023). https://doi.org/10.1007/s11033-023-08793-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08793-8