
Abstract
Background
Acute myeloid leukemia (AML) is a type of blood cancer that affects the bone marrow and blood cells. AML is characterized by the rapid growth and accumulation of abnormal white blood cells, known as myeloblasts, which interfere with the production of normal blood cells.
Aims
The main aim was to determine the relationship between these genetic alterations and the clinico-haematological parameters and prognostic factors with therapy for Iraqi patients with AML.
Methods
We used Sanger Sequencing to detect the mutations in 76 AML patients. Clinical data of AML patients were retrospectively analysed to compare the prognosis of each gene mutation group.
Results
Somatic mutations were identified in 47.4% of the enrolled patients in a core set of pathogenic genes, including FLT3 (18 patients, 23.7%), DNMT3A (14, 18.4%), NPM1 (11, 14.5%) and TP53 (5, 6.8%). As multiple mutations frequently coexisted in the same patient, we classified patients into 10 further groups. Two novel mutations were detected in FLT3-ITD, with new accession numbers deposited into NCBI GenBank (OP807465 and OP807466). These two novel mutations were computationally analysed and predicted as disease-causing mutations. We found significant differences between patients with and without the detected mutations in disease progression after induction therapy (remission, failure and death; pv = < 0.001) and statistically significant differences were reported in total leukocyte count (pv = < 0.0001).
Conclusion
These genes are among the most frequently mutated genes in AML patients. Understanding the molecular and clinical significance of these mutations is important for guiding treatment decisions and predicting patient outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a highly aggressive and malignant cancer that affects the blood and bone marrow. It is the most common form of acute leukemia. The World Health Organization (WHO) classifies AML into various subtypes based on genetic and molecular characteristics. AML is characterized by the uncontrolled growth of immature white blood cells called myeloblasts [1]. It is well established that genetic mutations play a critical role in the diagnosis, prognosis and treatment of AML, these mutations are associated with different clinical and biological characteristics of the disease, and they can be used to stratify patients into different risk categories and inform treatment decisions, which is fully reflected in the National Comprehensive Cancer Network (NCCN) guidelines for AML [2]. In de novo acute AML, FLT3, DNMT3A, NPM1, and TP53 mutations are among the most frequent genomic changes. These changes are important for the pathogenesis and progression of the disease, especially when AML-related recurrent cytogenetic abnormalities are absent [3, 4].
The FLT3 receptor, which belongs to the type-III tyrosine kinase family, plays a critical role in cell growth, differentiation, and survival. One of the most frequent mutations in AML is an internal tandem duplication of FLT3’s juxtamembrane domain (FLT3-ITD) which is located on chromosome 13q12 [5]. Approximately 25% of AML cases exhibit this particular feature. It is strongly linked to the disease and is associated with a higher risk of relapse and lower overall survival rates (OS; median 16.2 months) [5, 6]. AML patients with an FLT3-ITD mutation frequently have high bone marrow blast cell counts, total leucocyte counts (WBCs), and elevated rates of relapse, suggesting that FLT3-ITD mutation warn a particularly poor prognosis of AML, with constitutive activation of the kinase within this variation [7].
Additionally, one of the most common chromosomal changes observed in AML is a mutation in the nucleophosmin (NPM1) gene. This gene is situated on chromosome 5q35 and is responsible for encoding a protein that shuttles between the nucleus and the cytoplasm. NPM1 plays a crucial role in regulating pathways related to cell proliferation and the suppression of growth [8]. The mutation in the NPM1 gene has been classified as a Class II mutation, which impacts the transcription and differentiation processes in hematopoiesis. This mutation is found in approximately 20–30% of freshly diagnosed cases of acute myeloid leukemia (AML). Moreover, it is detected in around 50% of cases where the karyotype (chromosomal structure) is considered normal [4, [8]. NPM1 is a chaperone protein that performs a variety of tasks while moving back and forth between the cytoplasm and nucleus. NPM1 mutations have been linked to leukaemia via a number of different mechanisms, but despite the growing understanding of these mechanisms, a focused therapy for NPM1-mutated AML is still lacking [4].
Moreover, Tumor protein 53 (TP53) is a tumor suppressor gene that codes for a protein that regulates cell growth and division; it plays a crucial role in regulating the cell cycle, DNA repair, and apoptosis (programmed cell death), and is located on chromosome 17p13.1. Mutations in the TP53 gene are found in approximately 5–10% of AML cases. The mutations in the TP53 gene locus may be point mutations or deletions and either somatic or germline [9]. TP53 mutations can occur as a primary event or as a secondary event during disease progression or treatment. TP53 mutations can result in the loss of p53 function, leading to the accumulation of DNA damage and genomic instability, which can contribute to leukemogenesis and resistance to chemotherapy [4]. Additionally, mutations or deletions in p53 are associated with a poor prognosis, as they can lead to chemotherapy resistance and disease relapse. Patients with AML and p53 mutations often have a higher risk of treatment failure and shorter overall survival than those without p53 mutations [10, 11].
However, nearly 50% of AML samples have a normal karyotype, and many patients carry no mutation. In contrast, DNA methylation patterns are altered in numerous cancers and often correlate with clinically relevant information such as subtypes, prognosis and drug response. Indeed, aberrant DNA methylation patterns are a hallmark of AML [2, 12]. The de novo methyl transferase 3 A (DNMT3A) gene encodes a 130-kDa protein that is involved in epigenetic regulation. Downstream genes’ expression is decreased as a result of DNMT3A’s ability to catalyse the addition of a methyl group to cytosine residues in CpG islands [12]. R882H is the most common mutation, and it has been shown to act as a dominant-negative on wild-type DNMT3A [12].
The Cancer Genome Atlas (TCGA) team recently discovered a subgroup of AML patients in whom DNMT3A, FLT3 and NPM1 mutations coexist at a higher frequency than would be predicted for a chance occurrence as a result of an integrated genomic study of de novo AML [13]. It is common that multiple variations occurs simultaneously in the same patient, indicating the role of concerted interaction of mutations in the pathogenesis of AML [14]. Advances in genetic testing and sequencing technologies have made it possible to identify and analyze multiple mutations simultaneously, leading to a better understanding of the complex genetic landscape of AML and the development of targeted therapies that can improve outcomes for patients [13].
Treatment for AML is a long-term process that depends on several factors, including the patient’s age, overall health, and subtype of AML. The FLT3 status of a patient can be helpful for prognosis and identifying patients who receive treatment with a FLT3 inhibitor, or for alternate consolidations such as stem cell transplants [15]. The most commonly used therapies are chemotherapy and stem cell transplantation; however, the majority of patients die of AML relapse [12].
To enhance outcomes for patients with AML, it is imperative to conduct clinical research investigating new medications and combinations of drugs. Traditionally, drug development efforts have primarily concentrated on conducting dose-finding studies for single-agent therapies in the relapsed setting. This approach has led to the approval of effective targeted treatments, such as FLT3 and DNMT3A inhibitors. By following this approach, researchers aim to identify novel therapies that specifically target the NPM1 mutation, with the goal of improving treatment outcomes for patients with AML who carry this mutation [16, 17]. The fundamentals of intensive chemotherapy continue to be anthracyclines and cytarabine. Alternatives include mitoxantrone-based cytarabine regimens and fludarabine, cytarabine, granulocyte colony-stimulating factor and idarubicin (FLAG-IDA). For patients with FLT3-mutant AML, it has been common practice to include the kinase inhibitor midostaurin (Rydapt) and/or gilteritinib (Xospata) in first-line therapy [16]. Other treatments (i.e. epigenetically targeted therapies) and immunotherapy methods, such as bi-specific T-cell engaging antibodies, checkpoint inhibitors, chimeric antigen receptor T cells, or natural killer cells, are probably most efficient in frontline or early salvage combination approaches when used in the presence of minimal residual disease (MRD) [18]. Hence, the objectives of the present study is to establish the correlation between molecular mutations such as FLT3‑ITD, DNMT3A, NPM1 and TP53 and clinico‑haematological parameters, as well as the prognosis and outcomes of induction therapy in Iraqi AML patients.
Patients, material and methods
Clinical investigations and patients selection
A total of 76 patients with newly diagnosed de novo AML were enrolled at Hiwa Hospital for Cancer in Sulaimania, Iraq, between October 2021 and March 2022. Patients who were on chemotherapy and those who have completed the courses of treatment were excluded. The patients were sourced from various geographic locations within the Iraqi governorates as the hospital serves a wide area. The demographic data captured included age and sex of the patients.
The patients were confirmed to have AML through hematological testing, the diagnosis of AML typically involves the identification and enumeration of blasts based on morphology in the bone marrow (BM) aspirate. This process often requires manual examination. Another laboratory analysis involved a complete blood count (CBC), which included measurements of total WBCs, platelet (Plt) count, and Hemoglobin (Hb) levels. The CBC analysis was performed using a fully automated M-series hematology analyzer called the Medonic CBC Analyzer (manufactured by Boule Medical AB in Stockholm, Sweden). Furthermore, the study included information regarding the outcome of induction therapy for the patients. Induction therapy refers to the initial treatment given to induce remission in AML patients. Reporting the outcome of induction therapy (OIT) provides insights into the effectiveness of the treatment and contributes to understanding the clinical traits of the AML patients in the study. Table 1 provides an overview of the sociodemographic characteristics and clinical traits of the AML patients in the current study.
Genotyping and targeting genes mutations in AML
DNA extraction
In the study, genomic DNA was extracted from EDTA blood samples in a biosafety level 2 laboratory using the Qiagen QIAamp DNA Blood Kit, manufactured in Hilden, Germany. The extraction procedure followed the manufacturer’s recommended protocol.
To assess the concentration and purity of the extracted DNA, the ratio of optical density at 260/280 nm was measured using a NanoDrop spectrophotometer from Thermo Fisher Scientific, located in Waltham, MA, USA. This ratio provides an indication of DNA concentration and purity, with values around 1.8-2.0 considered optimal. The extracted DNA samples were then frozen at -20 °C until they were needed for further analysis or experimentation.
Polymerase chain reaction (PCR)
For PCR amplification of selected genes, primers were designed using the Primer3 + [19] and NCBI Primer-Blast web-based tools [20]. During primer design, criteria such as a higher melting temperature (Tm) were considered to ensure optimal primer annealing and specificity. Hence, we then selected those primers that have a higher melting temperature (Tm) and can specifically and sensitively amplify these regions. The PCR reaction was carried out using a total volume of 20 µl, consisting of 3 µl DNA template, 10 µl nuclease-free water, 1 µl each of 10 µM forward and reverse primers, 2 µl of a 10 mM dNTPs mixture, 2 µl of a 10X reaction buffer, 0.4 µl of 2.5 U/µl Taq DNA polymerase, and 0.6 µl of MgCl2 from AddBio in Daejeon, South Korea. The thermal cycling program included the following steps: initial melting at 94 °C and the other steps for specific PCR condition for each gene were applied as outlined in Table 2. The PCR products were visualized using 2.5% agarose gels stained with SYBR Safe dye from Thermo Fisher Scientific. The gels were examined under ultraviolet light at wavelengths of 329, 660, 348, 408, and 610 nm (Table 2 and supplementary Fig. 1).
In addition to the patient group, a control group comprising 25 volunteers was included. The selection of individuals for the control group was based on a questionnaire, ensuring that they were from families with no reported cases or history of cancer. The control group was matched with the patient group based on age and gender to account for potential confounding factors. Hence, we aimed to identify the most common genes that are mutated in AML and determine the regions of high mutation within those genes such as FLT3-ITD [21]. DNMT3A [22], NPM1 [23] gene and TP53 [24] gene.
Sanger sequencing and computational databases
To perform Sanger sequencing, the PCR products obtained for each targeted amplicon were purified using the AddPrep PCR Purification Kit from AddBio. Primers specifically designed for this study were used in the purification process. The purified products were then subjected to nucleotide sequencing using the Sanger sequencing method, which was conducted at a sequencing facility located in South Korea [26].
Following sequencing, the obtained nucleotide sequences were analyzed using the computational FinchTV v.1.4 software (Geospiza, Inc. http://www.geospiza.com/finchtv). This software allowed for manual alignment, editing, and justification of each individual sequence to ensure accuracy. Pairwise alignment of the sequences was further validated using the BLAST software tool, which helps confirm the consistency and reliability of the alignments.
Various computational tools were employed to assess the sequences, identify amino acid variants, and determine the novelty of the mutations reported in the study. These tools included the ExPASy ProtParam online tool [27], the NCBI SNP databases (dbSNP), The 1,000 Genomes Project, HGMD Professional software, and the Ensemble browser. By utilizing these resources, researchers could gather comprehensive information regarding the sequence variations, potential effects on amino acids, and the presence of known mutations or variations reported in existing databases and projects [25]. The relevant accession numbers were obtained from the NCBI GenBank databases.
Statistical analysis
In the analysis of the data, the GraphPad Prism 9 software was used to perform a Two-Way Analysis of Variance (ANOVA). This statistical analysis was employed to assess the differences in the investigated parameters between the two groups: mutated AML cases, unmutated AML cases, and normal cases. The ANOVA helps determine if there are significant variations in the measured parameters among these groups.
To explore the relationships between various factors and the OIT, Pearson correlation and polynomial regression analyses were conducted. These analyses aimed to examine the correlations among gender, age, location, total WBCs, Hb levels, PLT, peripheral blasts, and the OIT. To investigate the relationships between the two groups (AML cases and the control group), as well as between the OIT and patient demographic parameters, Pearson’s chi-square test was used. This statistical test examines the association between categorical variables. The test was employed to determine if there were significant relationships between the groups and the OIT based on the patient demographic parameters. In all statistical analyses, a significance level of p < .05 was used, indicating that results with p-values below this threshold were considered statistically significant.
Results
Study group characteristics
A total of 76 AML cases were included in the study, with 56.2% being male and 43.8% female. The age of the patients ranged from 18 to 89 years, with a mean age of 50 years. The most common age observed among the patients was 59 years, as evidenced by a peak in the age distribution.
Table 1 provides information on the age, gender, WBCs, Hb levels, Plt count, blast percentage, and outcome of remission for all patients.
Importantly, no mutations were detected in any of the 25 control cases. The frequency of germline mutations differed significantly between the tested group (47.4%) and the control group (0%), with a statistically significant p-value of 0.001. This suggests a notable difference in the occurrence of mutations between the AML cases and the control group, emphasizing the relevance of these mutations in AML development.
Molecular landscape and mutation analysis
Frequencies of gene mutations
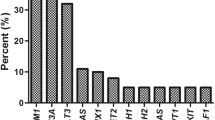
As shown in Fig. 3, the frequencies for all four genes selected in this study were FLT3: 18 patients (23.7%); DNMT3A: 14 patients (18.4%) [25]; NPM1: 11 patients (14.5%) and TP53: 5 patients (6.8%), Fig. 1A. Using combined genetic markers, the AML patients were further classified into 10 groups. The largest group, group 1, accounted for 52.6% of the patients and comprised those without any detected mutations. The other groups were categorized based on specific mutations.
The distribution of patients in the mutation-specific groups is as follows: group 2 consisted of patients with FLT3-ITD mutations only (14.5%); group 3 had DNMT3A mutations only (9.2%); group 4 contained NPM1 mutations only (6.7%); group 5 had TP53 mutations only (5.2%); group 6 comprised patients with FLT3/DNMT3A/NPM1 mutations (4.0%); group 7 consisted of patients with FLT3-ITD/DNMT3A mutations (2.6%); group 8 had FLT3-ITD/NPM1 mutations (2.6%); group 9 contained DNMT3A/NPM1 mutations (1.3%); and group 10 consisted of patients with TP53/DNMT3A mutations (1.3%).
These groups were determined based on the presence or absence of specific mutations, allowing for a more detailed characterization of the genetic profile of AML patients. Figure 1B provides a visual representation of the distribution of patients across these groups.

Frequencies and Distribution of subtypes in AML patients (A) Estimated FLT3, DNMT3A, NPM1 and TP53 variant (allelic) frequencies in a subset of de novo acute myeloid leukaemia samples. (B) Common molecular gene mutation subgroups of de novo acute myeloid leukaemia included in the study
Detection of FLT3-ITD mutation
We found the incidence of FLT3-ITD mutations to be 18 (23.7% of patients). The first FLT3-ITD mutation found in this study was c.1784G > T (R595I) and reported in 8 patients: 44.5%, as previously reported [28] (Fig. 2: A and D, M.1), while the other two mutations are reported here for the first time, and were deposited in NCBI-GenBank as novel mutations with a new accession numbers OP807465 (B:c.1820G > C & c.1831G > C: R607T & E611Q: Fig. 2:B and D, M.2; 4 patients: 22.2%) and OP807466 (C:c.1898 A > G: K634E: D,M.3; 6 patients:33.3%).

Sequencing results of the FLT3-ITD mutations and alignment of multiple FLT3-ITD protein sequences. (A) Unmutated: The sequence chromatogram of a wild-type allele with Arginine (AGA) at codon 593: Mutated: the mutant allele with a heterozygous mutation of Arg593Iso (c.1783G > T) in affected members. (B) Unmutated: The sequence chromatogram of a wild-type allele with Arginine (AGA) at codon 607 and Glutamic acid (GAG) at codon 611: Mutated: the mutant allele with a heterozygous mutation of Arg607Thr (c.1820G > C) and Glu611Gln (c.1831G > C) in affected members. (C) Unmutated: The sequence chromatogram of a wild-type allele with Lysine (AAA) at codon 634: Mutated: the mutant allele with a heterozygous mutation of Lys634Glu (c.1898 A > G) in affected members (D) protein sequence alignments illustrating three different FLT3-ITD mutations along with their corresponding frequencies
In this study, computational analysis was performed to assess the potential impact of the two novel mutations on protein stability. The I-mutant tool predicted that the two novel mutations (B: -0.60 and C: -0.70) would decrease protein stability with a Delta Delta G (DDG) value < 0. The VarSite [29] analysis predicted that the mutations (B: 1.15 and C: 1.50) would have a higher probability of being pathogenic than those with values ≤ 1, with a DDG value > 1 [25]. The sequences were aligned using a reliable alignment algorithm to identify conserved and variant regions within the FLT3-ITD mutation Fig. 2D.
Detection of DNMT3A mutation
In our previous study [25], we conducted an examination of a cohort that consisted of both the same patient sample as well as an additional fifteen AML patients with negative DNMT3A detected mutations, as confirmed in the current study. Within the context of our previous research, we made an intriguing discovery concerning three previously novel DNMT3A mutations which have been deposited in the GenBank NCBI for public access (Data availability). These mutations were observed in 14 out of the total 76 patients, accounting for approximately 18.4% of the patient population. These findings highlight the significance of these novel DNMT3A mutations and suggest their potential involvement in the pathogenesis of AML.
Detection of NPM1 mutation
In this study, eleven patients were found to have two different NPM1 mutations (C860-863dupTCTG and C868G > T), accounting for 14.5% of all the currently enrolled patients (Fig. 3A mutation A and B: A.mut). The first mutation found in exon 12 (the most frequently detected mutation in NPM1) [8] was C860-863dupTCTG, accounting for 72.2% of NPM1 mutations, as reported in previous study [30]. The second mutation, c868G > T also in exon 12 accounted for 27.3% of the NPM1 mutations, as reported earlier in Iraq [31] (Fig. 3A mutation x and B: B.mut).

The gene structure of NPM1 along with the mutations associated with AML. (A). changes of tryptophans (288 and 290) and insertion of a new nuclear export signal (NES) motif. These changes cause aberrant cytoplasmic accumulation in NPM1 mutants. (B). The chromatogram shows the representative sequencing results of wild-type and mutant patients
Detection of TP53 mutation
In present study, we detected two types of p53 mutations in 5 of 76 (6.9%) patients with AML (Fig. 4A & B): The first one is I225N substitution caused by the heterozygous c.764T > A, and the next one is A276P substitution that caused by the heterozygous c.826G > C. Importantly, these p53 mutations detected in our study have previously been reported in AML patients, indicating their relevance and significance in the context of AML pathogenesis [32, 33]. Figure 4 provides a visual representation that aids in understanding the specific alterations occurring within the TP53 gene at the protein level. The aligned sequences allow for a comparison between the wild-type TP53 sequence and the mutated variants, highlighting the specific sequence and amino acid changes associated with each mutation in the current study.

Patterns of TP53 mutations: (A) the chromatogram shows the representative sequencing results of wild type and mutant patients (B) Sequences for wild-type and the two detected mutations c.764T > A and c.826G > C (C) Protein sequence alignments illustrating two different TP53 mutations along with their corresponding frequencies
The epidemiological profile
Figure 5 provides a summary of the clinical characteristics of the AML patients (mutated and unmutated) compared to the control group in this study. The analysis examined sociodemographic data such as gender, age, and location, as well as various clinical parameters including total WBCs, PLT, Hb, and peripheral blast count.

Presents the differences between AML patients (both mutated and unmutated cases) and a control group in several key aspects: (a) gender, (b) age, (c) location, (d) total WBCs, (E) platelets (F) Hb and (G) peripheral blasts
Note: Chi-square significant at p ≤ .05. p-Value style: GP: >0.05 (ns), 0.0001 (****) [26]
The results indicated that there were no significant differences between the patients (mutated and unmutated) and the control group in terms of gender, age, and location. However, significant differences were observed between the two groups in terms of other clinical parameters, namely total WBCs, platelets, Hb, and peripheral blast count (all with p-values < 0.0001). Furthermore, the mutation cases showed a significant difference in total WBCs compared to the unmutated cases. A t-test analysis revealed that WBCs were significantly higher in mutated cases compared to unmutated cases (mean difference: -33.36 ± 6.668, p-value < 0.0001).
In contrast, the comparisons of other epidemiological profiles showed only slight differences between the means, without significant data reported for gender (mean difference: 0.02222 ± 0.1160, p-value = 0.8486), age (mean difference: -0.8667 ± 3.403 years, p-value = 0.7997), location (mean difference: 0.06667 ± 0.1120, p-value = 0.5536), platelets (mean difference: -30.42 ± 32.46, p-value = 0.3518), and blasts (mean difference: -4.736 ± 4.724, p-value = 0.3194).
Hence, the analysis revealed significant differences in certain clinical parameters between the mutated and unmutated AML cases, particularly in total WBCs, while no significant differences were observed in gender, age, location, platelets, and blasts.

Performed Pearson correlation and polynomial regression analyses (A). Correlations between mutated and unmutated AML patients (B). Correlations between AML patients with control group regarding clinical parameters (gender, age, location, total WBCs, Hgb platelets, peripheral blasts and OIT)
The analysis conducted on the epidemiological profile using Pearson correlation and polynomial regression revealed several correlations among the variables. In Fig. 6, the red boxes represent variables with a negative relationship, while the blue boxes represent variables with a positive relationship.
In AML mutated and unmutated patients, the total WBC count exhibited a highly positive correlation with FLT3-ITD and DNMT3A mutations, with correlation coefficients of 0.48 and 0.35, respectively. This indicates that as the total WBC count increases, the likelihood of having FLT3-ITD and DNMT3A mutations also increases. The outcome of induction therapy showed significant correlations as well, particularly in patients with DNMT3A mutations, with a correlation coefficient of 0.48 (Fig. 6A). This suggests that patients with DNMT3A mutations had similar outcomes in induction therapy.
Furthermore, in AML patients compared to the control group, there were high positive correlations observed, particularly between WBCs and variables such as overall induction therapy (OIT) (correlation coefficient of 0.60), FLT3 mutation (correlation coefficient of 0.53), and blast count (correlation coefficient of 0.56). Additionally, correlations were observed between DNMT3A mutation and OIT (correlation coefficient of 0.50) and between blast count and OIT (correlation coefficient of 0.64) (Fig. 6B). These findings indicate that there is a strong positive relationship between these variables in AML patients compared to the control group.
Overall, these results highlight the associations between various variables in AML patients, including the total WBC count, specific gene mutations (FLT3-ITD and DNMT3A), and the outcome of induction therapy. The correlations found in the study provide insights into the relationships between these factors, which can contribute to a better understanding of AML and potentially aid in its management and treatment.

Clinical Outcome of studied cases after 60 days of complete treatment and induction therapy
Treatment response analysis and follow up
In the treatment of AML, the therapeutic process can be lengthy, typically taking 6 to 12 months to complete, which includes chemotherapy and other treatment modalities. In this study, we considered two different outcomes based on the presence or absence of the investigated mutations.
Among the 76 AML patients with detected mutations, the outcomes of 60 days after complete treatment with induction therapy were as follows: 26 (72.2%) achieved remission, 7 (19.5%) experienced treatment failure and 3 (8.3%) unfortunately passed away. On the other hand, for the patients without the detected mutations, the outcomes were 34 (85.0%) remissions, 4 (10.0%) failures, and 2 (5.0%) deaths. Significant differences were observed in the outcomes between patients with and without detected mutations (p-value < 0.001) Fig. 7. This suggests that the presence of these specific mutations may have an impact on the response to treatment and overall prognosis in AML patients.
Discussion
In this retrospective cohort study, our aim was to investigate the frequency and prognostic impact of FLT3-ITD, DNMT3A, NPM1, and TP53 mutations in patients with newly diagnosed AML. These genetic mutations play a significant role in disease pathogenesis and are considered important predictors of treatment response and outcomes. To the best of our knowledge, there has been limited evaluation of these mutations in this specific region. Therefore, our study aimed to fill this knowledge gap and provide insights into the prevalence and prognostic implications of these mutations in AML patients. The mutation frequencies observed in our study were as follows: FLT3-ITD: 23.7%, DNMT3A: 18.4%, NPM1: 14.5%, and TP53: 6.7%. These mutations were associated with worse outcomes independent of other clinical markers such as age, total WBCs counts, blasts, and response rates during induction therapy.
These findings highlight the importance of genetic investigation in the diagnosis, prognosis, and treatment of AML. Identifying these mutations can provide valuable information for tailoring treatment strategies and predicting patient outcomes [34]. However, the varied results between studies may be due to possible differences in sample size as well as different methods used to evaluate these mutations [35].
The most common genetic abnormalities found in patients with AML are FLT3 mutations. In fact, FLT3-ITD is one of the current molecular genetic markers used for the diagnosis and risk assessment of AML [35]. We found the incidence of FLT3-ITD mutations to be 23.7%, which was marginally similar to that found in a Japanese cohort (23%) [36], higher than that found in an Indian cohort (10.8%) [37] and lower than that in a study from the United Kingdom (27%) [38]. In this study, the first mutation detected (c.1751delc) was previously reported [28], while the other two mutations are reported for the first time, and were deposited in NCBI-GenBank as novel mutations with the accession numbers OP807465 and OP807466. These novel mutations were predicted to be ‘disease-causing’ according to the mutant tester tools. Both mutations were computationally predicted by I-Mutant software to alter the protein function and decrease protein stability, with predicted DDG values < 0 [25, 39]. Moreover, the VarSite tool showed that amino acid residues for both mutations with a disease propensity value of 1.15 and 1.50, were predicted a disease-associated variants [25]. These computational predictions provide insights into the potential functional consequences of the identified mutations and contribute to our understanding of their impact on the protein’s structure and stability in the context of the disease.
The FLT3 was found to be overexpressed in leukemia and to be a prognostic factor for poor prognosis in AML. Patients with high FLT3 expression also have high levels of leukemic oncogenes. As a result, high FLT3 expression is a risk factor for leukaemia [40]. Cases with FLT3-ITD mutations are associated with a higher level of WBCs and high blasts percentage, with lower hemoglobin and platelets [28, 41].
Additionally, the frequency of NPM1 mutations was 14.5%, as reported previously in India (14.4%) [37]. However, this frequency was lower than that reported in the United Kingdome (21%) [15] and in a separate study in India (27.9%) [8], and higher than that previously reported in Iraq (10%) [31]. A duplication of TCTG at positions 956 to 959 in the reference sequence (c860-863dupTCTG), known as NPM1 mutation A, accounts for 75–80% of instances [30]. In our study, it accounted for 72.2%. Additionally, another mutation in exon 12 (c868G > T) was found, accounting for 27.3% of NMP1 mutations, as reported earlier in Iraq [31]. Exon 12 was the location of the majority of examined NPM1 mutations. Regardless of the mutation, all variations result in changes at the C-terminus of the NPM leukemic mutants that cause the protein to dislocate into the cytoplasm. These changes include tryptophan(s) 288 and 290 and the creation of an additional nuclear export signal (NES), and this protein has a tendency to be stable and accumulate in the cytoplasm [42].
Furthermore, we observed two missense TP53 mutations in five patients, c764T > A and c826G > C. Both mutations were reported previously in other studies [32, 33]. They are associated with distinct clinico-biological features and were a poor prognostic factor in this study’s AML patients, independent of age, WBC count, karyotype and other genetic markers. Most TP53 mutations were found in exons 5–8 [11]. TP53 is noted in 6–14% of AML cases, and in general, TP53 mutations confer an adverse prognosis with documented chemo-resistance. TP53 mutations may be mainly responsible for the very poor prognosis of complex karyotype AML [43, 44].
In our previous study, which included the same patient sample along with an additional fifteen AML patients who did not have a DNMT3A mutation, we identified three novel DNMT3A mutations. These mutations were found in 14 out of the total number of patients, accounting for approximately 18.4% of the patient population [25]. This result was lower than that obtained in a US cohort (21.9%) [45] and higher than that in a study in China (14.0%) [46]. The mutations that were detected in the DNMT3A gene included heterozygous V877I and L888Q, which were caused by c2629G > A and c2663T > A substitutions, respectively. Additionally, a frameshift mutation c2635delA was identified, resulting in protein truncation with a stop codon at position N879T*. Furthermore, promoter methylation analysis of the DNMT3A gene was performed using the methylation-specific PCR (MSP) technique. The results revealed that promoter methylation was present in 9 out of the total number of patients, accounting for approximately 64.28% of the sample. Importantly, a significant correlation was observed between DNMT3A mutations and promoter methylation (pv = 8.52 × 10^5) [25].
Multiple mutations frequently coexist in the same patient, indicating that coordinated interactions between mutations may play a role in the development of AML [47]. The difference in the frequency of these mutations might be due to the geographical distribution of the population studied [37]. In the present study, AML were patients divided into 10 groups based on genetic markers. Interactions of genetic alterations in AML frequently were reported, except the NPM1 mutation, which was not observed in patients with TP53 mutations. In a previous study, AML patients with three FLT3/NPM1/DNMT3A gene mutations (AML FLT3/NPM1/DNMT3A) presented with a high disease burden and had an average elevated WBC (> 100.000 × 10+ 9/L) compared to other groups [48].
The clinico-haematological characteristics observed in this study are consistent with findings from other studies conducted globally. The study examined the relationships and correlations between gene mutation status and parameters such as Hb, total WBCs, PLT, location, and blasts percentage. It is worth noting that age and gender did not exhibit statistically significant differences between the mutation and non-mutation groups. This finding is in line with a previous study that also reported no difference in mutation frequency between males and females [49]. However, mutated and unmutated patients had elevated blast cells and low platelets and haemoglobin counts, with statistically significant differences and correlations were reported in total WBC count in patients with gene mutations compared with unmutated patients [41]. Indeed, elevated WBCs can be caused by a variety of conditions, including infections and autoimmune disorders. Furthermore, the presence of blasts in the blood is the best indicator of acute leukaemia. Blasts are cancerous cells that are still immature but replicate rapidly, causing tiredness from anemia and a risk of bleeding from a low platelet count. They slow down the production of red blood cells and platelets [50]. However, current study reported a significant differentiation and correlations in AML cases compared to a control group enrolled in the genetic and hemato-pathological investigations (Fig. 5A and B).
Importantly, we found significant correlations between patients with detected mutations and patients without mutations in disease progression after clinical induction therapy (remission, failure and death; p = < 0.001) [51]. These correlations indicate that low responses to treatment and higher mortality rates after 60 days of induction therapy (Fig. 6). The general approach for therapy has not substantially changed in recent years [44]. Intensive clinical induction therapy is conducted with 3 days of an anthracycline and 7 days of cytarabine (commonly referred to as ‘713’ regimens) that aims at killing off all leukemic cells and forcing the patient into remission. Sometimes several rounds of induction with increasing potency must be undertaken in order to obtain the desired result. Complete remission (CR) is achieved in 60–80% of younger adults and in 40–60% of older adults (60 years or above) [1]. In addition to their function in the pathogenesis of the disease, molecular mutations in AML are considered important predictors of patients’ response to therapy and outcomes [37]. Efforts to develop protein kinase inhibitors, that target mutated forms of the FLT3 receptor, have led to successive generations of FLT3 inhibitors [44]. FLT3 inhibitors have received FDA approval and have demonstrated increased clinical response in AML patients when used alone or in combination with chemotherapy [52]. Therefore, the European Leukaemia Network (ELN) recomended that all AML patients have access to FLT3 mutational test data within 72 h to guide treatment decisions based on the patient’s mutation status [44]. Every AML subtype described under the heading ‘AML with recurrent genetic abnormalities’ has a specific genetic mutation that must be identified to determine prognosis and guide treatment. For example when there is no concurrent FLT3-ITD mutation, NPM1 mutations consistently indicate a favorable prognosis in AML patients under 60 years of age; however, in AML patients 70 years of age or older, NPM1 mutations seem to be the sole factor influencing prognosis in multivariate analysis [16]. Patients with NPM1-mutated AML who have high FLT3-ITD (ratio ≥ 0.5) should receive conventional chemotherapy as well as a FLT3 inhibitor (which was not approved at the time our patients were treated). These patients may benefit from the incorporation of GO into frontline therapy [53].
In patients with AML, the treatment is usually divided into two chemotherapy phases: remission induction and consolidation. Remission induction aims to achieve a complete remission by reducing the number of leukemic cells. Consolidation therapy is then given to eliminate any remaining leukemic cells and lower the risk of relapse. Currently, it was observed that the presence of mutations had an impact on the remission rate in AML patients. Certain genetic alterations were found to influence the response to treatment and the achievement of remission, death often occurs due to complications of marrow failure, such as sepsis or significant hemorrhage. Furthermore, undiagnosed, rapidly evolving cases may present with fatal multi-organ failure [54, 55].
Conclusion
The study highlights that FLT3-ITD, DNMT3A, and NPM1 are frequently observed genetic alterations in Iraqi adult patients with AML. Molecular testing at various stages of the disease, including diagnosis, remission, and relapse, can provide valuable data to guide personalized clinical therapy. These genetic panels generate extensive data beyond the current diagnostic and prognostic criteria for AML patients. As our understanding of the impact of somatic mutations and their complex interactions continues to improve, this information may be utilized in the future to optimize and individualize treatment strategies, increasing the likelihood of achieving complete remission. It is therefore crucial to preserve AML patient samples in biobanks for future genomics and proteomics analyses, enabling the exploration of mutational patterns and the development of new targeted therapies.
Data Availability
The datasets regarding FLT3-ITD gene generated during the study were deposited into the NCBI GenBank Banklt database and can be accessed with the accession numbers OP807465 and OP807466 (https://www.ncbi.nlm.nih.gov/nuccore/OP807465/https://www.ncbi.nlm.nih.gov/nuccore/OP807466). Furthermore, the data related to DNMT3A gene in our study released as: (https://www.ncbi.nlm.nih.gov/nuccore/ON881282.1) (https://www.ncbi.nlm.nih.gov/nuccore/ON881281.1) and (https://www.ncbi.nlm.nih.gov/nuccore/ON881280.1).
Code Availability
Not Applicable.
References
Arber DA et al (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405
Tang S et al (2017) FLT3-ITD with DNMT3A R882 double mutation is a poor prognostic factor in chinese patients with acute myeloid leukemia after chemotherapy or allogeneic hematopoietic stem cell transplantation. Int J Hematol 106:552–561
Loghavi S et al (2014) Clinical features of de Novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J Hematol Oncol 7:1–10
Papaemmanuil E et al (2016) Genomic classification and prognosis in Acute myeloid leukemia. N Engl J Med 374:2209–2221
Volpe G et al (2015) Regulation of the flt3 gene in haematopoietic stem and early progenitor cells. PLoS ONE 10:1–18
Schlenk RF et al (2014) Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 124:3441–3449
Verson CA (2020) Profiling FLT3 mutations in mexican Acute Myeloid Leukemia Pediatric Patients: impact on overall survival. 8,
Bhattacharyya J et al (2018) Prevalence and clinical significance of FLT3 and NPM1 mutations in Acute myeloid leukaemia patients of Assam, India. Indian J Hematol Blood Transfus 34:32–42
George B, Kantarjian H, Baran N, Krocker JD, Rios A (2021) Tp53 in acute myeloid leukemia: molecular aspects and patterns of mutation. Int J Mol Sci 22,
Huang X et al (2013) A novel PTEN/Mutant p53/ c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia (United States) 15:952–965
Hou H et al (2015) TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. https://doi.org/10.1038/bcj.2015.59
DiNardo C, Lachowiez C (2019) Acute myeloid leukemia: from mutation profiling to treatment decisions. Curr Hematol Malig Rep 14:386–394
Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia (2013) N Engl J Med 368:2059–2074
Bullinger L, Konstanze D (2022) Genomics of Acute Myeloid Leukemia Diagnosis and Pathways 35,
Lazenby M et al (2014) The prognostic relevance of flt3 and npm1 mutations on older patients treated intensively or non-intensively: a study of 1312 patients in the UK NCRI AML16 trial. Leukemia 28:1953–1959
Döhner H et al (2022) Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 140:1345–1377
Stein EM et al (2021) Safety and Efficacy of Menin Inhibition in patients (pts) with MLL-Rearranged and NPM1 mutant Acute Leukemia: a phase (ph) 1, first-in-human study of SNDX-5613 (AUGMENT 101). Blood 138:699–699
Uy GL et al (2021) Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 137:751–762
Untergasser A et al (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:71–74
Kalendar R, Khassenov B, Ramankulov Y, Samuilova O, Ivanov K (2017) I. FastPCR: an in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 109:312–319
Manachai N et al (2022) Activating Mutation in the Receptor Tyrosine Kinase FLT3 with Clinicopathological Relevance in Canine Mast Cell Tumors. Vet. Med. Int (2022)
Park DJ et al (2020) Characteristics of DNMT3A mutations in acute myeloid leukemia. 55,
Gamaleldin MA, Imbaby SA, E. (2021) T. The role of tumor necrosis factor receptor superfamily member 4 (TNFRSF4) gene expression in diagnosis and prognosis of acute myeloid leukemia. Mol Biol Rep 48:6831–6843
Kadia TM et al (2016) TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer 122:3484–3491
Ali AM, Salih GF (2022) Identification of three novel DNMT3A mutations with compromising methylation capacity in human acute myeloid leukaemia. Mol Biol Rep. https://doi.org/10.1007/s11033-022-07977-y
Ali KM et al Clinical outcomes and phylogenetic analysis in reflection with three predominant clades of SARS-CoV-2 variants. 0–2 https://doi.org/10.1111/eci.14004
ProtParam tool. https://web.expasy.org/protparam/
Yunus NM et al (2015) Characterisation and clinical significance of FLT3-ITD and non-ITD in Acute myeloid leukaemia patients in Kelantan. Northeast Peninsular Malaysia. https://doi.org/10.7314/APJCP.2015.16.12.4869
Laskowski RA, Thornton JM, VarSite (2020) Disease variants and protein structure. 111–119. https://doi.org/10.1002/pro.3746
Falini B et al (2006) Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood 108:1999–2005
Othman GO, Mohammad NS, Saeed CH (2021) Molecular study of nucleophosmin 1 (NPM1) gene in acute myeloid leukemia in kurdish population. 21:687–692
Yanada M et al (2016) TP53 mutations in older adults with acute myeloid leukemia. Int J Hematol 103:429–435
Dutta S et al (2020) Functional classification of tp53 mutations in acute myeloid leukemia. Cancers (Basel) 12:1–16
Article O (2020) Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the Outcomes of patients with newly diagnosed Acute myeloid leukemia. 765–774. https://doi.org/10.1002/cncr.32566
Hamed HR, Al-jumaily RMK, Kadhom AE (2021) Characterization of NPM1 and FLT3-ITD mutations in iraqi patients with AML. 21:133–139
Kiyoi BH et al (1999) Prognostic implication of FLT3 and N- RAS gene mutations in Acute myeloid leukemia. 93:3074–3080
Sazawal S et al (2017) NPM1 and FLT3 mutations in acute myeloid leukemia with normal karyotype. Indian perspective. https://doi.org/10.4103/IJPM.IJPM
Kottaridis PD et al (2001) The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United King. Blood 98:1752–1759
Rashid PMA, Salih GF (2022) Molecular and computational analysis of spike protein of newly emerged omicron variant in comparison to the delta variant of SARS – CoV – 2 in Iraq. Mol Biol Rep. https://doi.org/10.1007/s11033-022-07545-4
Cheng J, Qu L, Wang J, Cheng L, Wang Y (2018) High expression of FLT3 is a risk factor in leukemia. Mol Med Rep 17:2885–2892
Naseem S et al (2021) NPM1 and FLT3-ITD / TKD gene mutations in. 15:15–26
Hindley A, Catherwood MA, McMullin MF, Mills K (2021) I. significance of npm1 gene mutations in aml. Int J Mol Sci 22,
Sasaki K et al (2020) Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer 126:765–774
Döhner H et al (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129:424–447
Gary Gilliland D, Griffin JD (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100:1532–1542
Hou HA et al (2012) DNMT3A mutations in acute myeloid leukemia: Stability during disease evolution and clinical implications. Blood 119:559–568
Naoe T, Kiyoi H (2013) Gene mutations of acute myeloid leukemia in the genome era. 165–174. https://doi.org/10.1007/s12185-013-1257-4
Elrhman HAEA, El-Meligui YM, Elalawi SM (2021) Prognostic impact of concurrent DNMT3A, FLT3 and NPM1 gene mutations in Acute myeloid leukemia patients. Clin Lymphoma Myeloma Leuk 21:e960–e969
Easwar A, Siddon AJ (2021) Genetic landscape of myeloproliferative neoplasms with an emphasis on molecular diagnostic laboratory testing. Life 11:1–22
Bumbea H et al (2022) Platelet Defects in Acute Myeloid Leukemia — Potential for Hemorrhagic Events.
Bond J et al (2019) T-cell acute lymphoblastic leukemia 104:1617–1625
Stone RM et al (2017) Midostaurin plus chemotherapy for Acute myeloid leukemia with a FLT3 mutation. N Engl J Med 377:454–464
Thiede C et al (2020) Impact of NPM1 / FLT3 -ITD genotypes de fi ned by the 2017 european LeukemiaNet in patients with acute myeloid leukemia. 135:371–380
Barranco R, Fossati F, Candosin S, Orcioni GF, Ventura F (2016) Unexpected sudden death due to acute myeloid leukemia subtype M5: a case report and review of the literature. 207–211. https://doi.org/10.4323/rjlm.2016.207
Kiani BH et al (2020) Artemisinin and its derivatives: a promising cancer therapy. Mol Biol Rep 47:6321–6336
Acknowledgements
Not applicable.
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
AMA and GFA were involved in the conception of the study idea and the experimental design. AMA conducted the laboratory work, performed data analysis, and prepared the manuscript. GFA provided supervision and guidance throughout the process. Both authors participated in discussions regarding the results and contributed to the finalization of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethical aspects of this study were given utmost importance and followed the established guidelines and regulations. Prior to enrollment, all patients provided verbal consent for the collection of blood samples. The study protocol was reviewed and approved by the Ministry of Health’s health directorate, with the approval obtained on August 23, 2021 (protocol NO. 9811). The research investigations were conducted at the University of Sulaimani, College of Science, in accordance with the applicable rules and regulations. The university granted approval for the study (No. 1917/259 on 01/08/2021), ensuring that ethical standards were upheld and necessary protocols were followed throughout the study. The study was conducted in compliance with the research ethical guidelines set by the Ministry of Health. The ethical standards outlined in the Declaration of Helsinki, which governs medical research involving human subjects, were strictly followed. Participants, including the parents of patients under the age of 18, provided both verbal and written consent after being provided with the necessary information and having the opportunity to review the Declaration of Helsinki.
Consent to participant
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ali, A.M., Salih, G.F. Molecular and clinical significance of FLT3, NPM1, DNMT3A and TP53 mutations in acute myeloid leukemia patients. Mol Biol Rep 50, 8035–8048 (2023). https://doi.org/10.1007/s11033-023-08680-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08680-2