
Abstract
Background
Streptomyces strains degrade many complex organic compounds and produce secondary metabolites. In aerobic organisms such as Streptomyces species, the tricarboxylic acid (TCA) cycle represents an indispensable central carbon metabolic pathway for energy generation and metabolic intermediary replenishment. Although various precursors for antibiotic biosynthesis are derived from this cycle, relatively few studies have focused on determining how a single carbon source can impact this metabolic pathway at different growth phases. In this study, we identified chromosomal genes involved in the TCA cycle in Streptomyces coelicolor and determined their mRNA levels.
Methods and results
We searched the genes involved in the TCA cycle in S. coelicolor through bioinformatic analysis. Growth, glucose concentration quantification and RNA isolation were made from cultures of S. coelicolor grown on minimal medium with glucose along 72 h. mRNA levels of all identified genes were obtained by RT-qPCR. Five enzymes encoded by a single gene each were found, while for the rest at least two genes were found. The results showed that all the genes corresponding to the TCA enzymes were transcribed at very different levels and some of them displayed growth-phase dependent expression.
Conclusion
All TCA cycle-associated genes, including paralog genes, were differentially transcribed in S. coelicolor grown in minimal medium with glucose as carbon source. Some of them, such as succinyl-CoA synthetase and succinate dehydrogenase, have low mRNA levels, which could limit the carbon flux through the TCA cycle. Our findings suggest that the genetic expansion of TCA cycle genes could confer to S. coelicolor the ability to adapt to diverse nutritional conditions and metabolic changes through different paralog genes expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbon metabolism is central to microbial life and supplies all cellular processes with building blocks and energy. Despite having significant differences in lifestyle and occupied niches, fundamental metabolic tasks are highly similar across divergent species [1]. The tricarboxylic acid (TCA) cycle is a major biochemical hub in cells and has two main functions, energy generation and precursor synthesis. In the presence of a terminal electron acceptor, it functions as a proper cycle by oxidising substrate to CO2 and generating NADH for respiration. In addition, intermediates of the TCA cycle serve as precursors for anabolism and secondary metabolism [2].
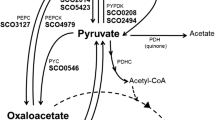
In Streptomyces coelicolor, TCA cycle is essential for maintaining growth and development. Mutations in some genes that encode enzymes associated with this cycle are directly involved in morphological differentiation, indicating that a complete TCA cycle is essential for normal development [3, 4]. This pathway consists of eight consecutive enzyme reactions (Fig. 1) that initiate with the input of acetyl-coenzyme A from glycolysis and its condensation with oxaloacetate to form citrate via citrate synthase (CS) enzyme activity [5]. CSs are classified into two types; type I are found in eukaryotes, archaea, and Gram positive bacteria, and type II are found in algae and Gram negative bacteria [6]. In the next step, aconitase (ACO) catalyses the stereospecific isomerisation of citrate to isocitrate via cis-aconitate [7] while isocitrate dehydrogenase (IDH) catalyses the oxidative decarboxylation of isocitrate to produce 2-oxoglutarate and CO2. Based on their coenzyme specificity and structure, IDHs are classified into two groups i.e., NAD+- or NADP+-dependent enzymes, and in monomeric and homodimeric enzymes, respectively [8].

Genes encoding TCA cycle enzymes in S. coelicolor
The fourth reaction of the cycle is accomplished by 2-oxoglutarate dehydrogenase (2-ODH) complex which catalyses the conversion of 2-oxoglutarate to succinyl-CoA and CO2. This enzyme complex is composed of 3 components, 2-oxoglutarate dehydrogenase (E1 component), dihydrolipoamide succinyltransferase (E2 component), and dihydrolipoamide dehydrogenase (E3 component) [9]. In the next reaction, succinyl-CoA synthetase (SCS) catalyses the breakdown reaction of the coenzyme A ester and the synthesis of GTP-generating succinate. In E. coli, this enzyme is a heterotetramer formed between two alpha and two beta subunits [10] and in C. glutamicum, both SCS subunits are encoded by sucCD operon genes [11]. An oxidation reaction by succinate dehydrogenase (SDH) enzyme converts succinate to fumarate, and in E. coli, SDH is a complex composed of 4 subunits, classified into two domains. The first is the catalytic domain which comprises a flavoprotein subunit and a non-haeme iron subunit and the second is the hydrophobic domain, which consists of two membrane-anchoring subunits [12].
Subsequently, another hydration reaction converts fumarate into malate through fumarase (FH) enzyme catalysis. These enzymes are divided into 2 classes; class I are found mainly in prokaryotes, while class II are distributed in both prokaryotes and eukaryotes [13]. Finally, the last reaction in the TCA cycle is catalysed by malate dehydrogenase (MDH) that oxidises malate into oxaloacetate, using NAD+ or NADP+ as cofactors [14].
Streptomyces coelicolor is the model organism of a genus with the potential to produce a broad diversity of bioactive compounds, hence, most of the studies have focused on the enzymes involved in the production of antibiotics under culture conditions that favours the expression of biosynthetic genes. However, a few studies have focused on the primary metabolism, specifically the TCA cycle, even when is the major supplier of essential precursors, including those for the biosynthesis of antibiotics and other secondary metabolites [15]. Evidence of this is the little proportion of enzymes involved in this pathway that have been characterized, despite the availability of the S. coelicolor genome since 2002 [16]. Besides, a large part of the genes involved in the TCA cycle has not been determined yet and their expression and regulation conditions are poorly understood, even when they could be target to improve carbon metabolism and growth of S. coelicolor and thus the accumulation of metabolic intermediaries to improve secondary metabolite synthesis. Moreover, relatively few studies have compared the transcriptional levels of primary metabolism genes in S. coelicolor over different growth periods using unique carbon sources. In a global transcriptomic analysis of S. coelicolor grown in repressive and non-repressive conditions, only the genes encoding for the putative SDH complex was induced in glucose while the other TCA cycle gene transcripts remained unchanged. However, these comparative analyses were realised using complex media culture in one point time of the exponential growth phase [17]. In another transcriptomic study in S. coelicolor grown in glucose (glycolytic condition) versus tween (gluconeogenic condition), only fumarase Class II encoding gene had a decreased expression on tween [18]. Even when sole carbon sources were used, the cultures were also compared at a single point time during growth. In this study, we identify the chromosomal genes involved in the TCA cycle in the model actinobacteria S. coelicolor grown on minimal medium supplemented with glucose as sole carbon source. Furthermore, in order to ascertain whether genes are expressed and to understand their expression patterns over time, their mRNA expression levels were measured throughout the bacterial growth phases.
Materials and methods
Culture conditions
To prepare fresh S. coelicolor M145 spores, mannitol-soya flour (MS) agar plates were inoculated and cultivated at 30 °C for 3 days. Approximately 108 fresh spores were used to inoculate 200 mL of Luria–Bertani (LB) medium contained in a 1 L Erlenmeyer flask and incubated at 30 °C for 24 h with orbital shaking at 200 rpm. Mycelia from this culture were collected by centrifuging for 5 min at 4200 × g. The mycelium pellet was washed twice with sterile distilled water and resuspended in 5 mL of water. These mycelia suspension were used to inoculate 2 Fernbach siliconised flasks containing 500 mL of minimal medium (5 g of NaCl, 2 g of (NH4)2SO4, 0.3 g of K2HPO4, 0.5 g of MgSO4.7H2O, 0.02 g of FeSO4, 0.05 g of ZnSO4, 0.02 g of CaCl2, 0.001 g of MnCl2, 0.001 g of CoCl2, 20 g of MOPS, 50 g of PEG 8000, and 10 g of glucose as carbon source per litre) with steel coils placed at the bottom. Cultures were incubated at 30 °C with orbital shaking at 200 rpm, and each flask was sampled every 12 h within 72 h to measure growth and extract RNA.
Cell growth and residual glucose measurements
Samples collected in duplicate from the 2 biologically independent batch cultures at different timepoints were used to determine the bacterial growth by measuring the quantity of protein in 3 mL mycelia using the previously described Lowry method [19]. In addition, glucose concentration was evaluated in the supernatant of the samples using the Miller’s method [20].
RNA extraction, DNase I treatment and precipitation
For RNA extraction, mycelia were immediately collected from samples taken from each flask by vacuum filtration and scraping off the filter paper, resuspended in 300 μL of TE buffer (10 mM Tris–HCl; pH 8; 1 mM EDTA) and subsequently sonicated for 10 s on ice by using an ultrasonic homogeniser (Soniprep 150 MSE, London, UK). 100 μL of lysis buffer (TE buffer supplemented with lysozyme, 50 mg/mL) was added to the samples and incubated for 10 min at 37 °C. An additional 450 μL of lysis buffer supplemented with 10 μL of β-mercaptoethanol was added to the samples and 10 s-sonication step was repeated. RNA was extracted from this lysate using the MagJet RNA kit (Thermo Scientific, Waltham, USA) according to the manufacturer’s protocol and treated with a TURBO DNA-free kit (Ambion, Austin, USA) to remove possible traces of DNA. The RNA was precipitated with LiCl following the procedure described by Sambrook et al. [21]. The average RNA concentration was determined using a NanoDrop 1000 spectrophotometer (Thermo Scientific); purity was assessed and controlled at 260/280 and 260/230 ratios within the 1.9–2.1 range.
cDNA synthesis and quantitative PCR
The expression levels of TCA cycle genes (Fig. 1) were determined using a two-step reverse transcription quantitative PCR (RT-qPCR) system. For cDNA synthesis, RNA (0.5 μg) was subjected to reverse transcription using random hexamer primers (Thermo Scientific) and M-MLV reverse transcriptase (Promega, Madison, USA) following the manufacturer’s protocol. The qPCR reaction systems contained 1 μL of each cDNA template, 0.5 μM specific primers designed by Primer-Blast (NCBI) based on the genome sequence GenBank NC_003888 (Table S1), 0.8 μL of DMSO, and an all-in-one qPCR mix (GeneCopoeia, Rockville, USA) at a final volume of 20 μL. All PCR product lengths ranged from 150 to 200 bp. Two biological samples were analysed in duplicate, and control reactions with no template (negative control) and no reverse transcriptase (-RT control) were included to verify the absence of DNA contamination. All RT-qPCR experiments were performed within the tenets of the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines [22]. The standard curve was constructed by plotting the logarithmic concentration of 5 dilution series of a purified PCR product used as standard in triplicate versus threshold cycles, using the same system. Absolute quantification analyses of the samples were processed using Rotor-Gene 6000 series software v1.7 and the experimental data are expressed as the mean value of at least 3 replicates ± the standard deviation of the mean.
Results
Streptomyces coelicolor is the most studied member of the genus due to its high potential to produce numerous antibiotics and bioactive substances. This microorganism grew well on minimal medium containing glucose and ammonium as carbon and nitrogen sources in our culture conditions (Fig. 2, panel A). The biomass increased between 24 and 60 h of culturing, followed by a decrease in growth rate, thereby entering the stationary phase. Approximately 97% of glucose was consumed, and the time at which their consumption ceased coincided with stationary phase.

Growth, residual glucose, and mRNA expression profiles of citrate synthase (CS), aconitase (ACO), isocitrate dehydrogenase (IDH), malate dehydrogenase (MDH) and succinyl-CoA synthetase (SCS) subunits encoding genes, at different growth times in S. coelicolor. Samples obtained from cells grown in MM with 1% glucose were used for protein quantification and RNA extraction. Biomass and transcript levels were determined as described in the “Materials and methods” section. Panel A, biomass (■) and residual glucose concentrations (▲). Panel B, CS sco2736 (■), sco4388 (▲), sco5831 (●) and sco5832 (□). Panel C, MDH sco4827 (■), ACO sco5999 (▲) and IDH sco7000 (●). Panel D, SCS α-subunit sco4809 (■) and sco6586 (▲) and SCS β-subunit sco4808 (□) and sco6585 (●)
Citrate synthase (CS)
Through an in-silico search in the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database, genes of the TCA cycle were identified on the S. coelicolor chromosome (Fig. 1). We found 4 genes associated with citrate synthase (sco2736, sco5831, sco5832, sco4388) and only the product encoded by sco2736 (gltA/citA, 429 amino acids) was classified as a type II citrate synthase in this database. The citA gene encoded a 45 kDa protein insensible to NADH inhibition [3]. We determined the mRNA levels of these 4 genes in S. coelicolor grown on MM with glucose and it was found that all the genes were transcribed, however, there were substantial differences in their levels (Fig. 2, panel B). sco2736 showed the highest transcription levels between 12 and 36 h of culturing, attaining a maximum of 1.7 × 10–2 ng μg−1 of total RNA at 24 h. The RNA level of sco5832 was 80 times lower than those of sco2736 at 24 h, with a decrease in transcription between 12 and 72 h (Fig. 2, panel B). A BLASTp search of the putative sco5832 product showed that it is highly conserved between various Streptomyces species, with identities of more than 95% with citrate synthase/methylcitrate synthases (Fig. S1). Expression levels of sco4388 and sco5831 were 3 and 4 orders of magnitude lower than those of sco2736, respectively (Fig. 2, panel B).
Aconitase (ACO) and Isocitrate dehydrogenase (IDH)
In S. coelicolor, the activity of a single aconitase and its diverse physiological roles in the TCA cycle were described [4]. Consistent with this finding, in the KEGG search, a single gene, sco5999, encoded this enzyme. mRNA levels of sco5999 remained high between 12 and 24 h of culture, subsequently decreasing to almost zero at 72 h (Fig. 2, panel C). Regarding IDH, in a previous study, we showed that only sco7000 codes for monomeric NADP+-IDH in S. coelicolor [23]. The expression profile of this gene showed maximum levels at 12 and 24 h, and a subsequent decline to almost undetectable levels (Fig. 2, panel C).
2-Oxoglutarate dehydrogenase complex (2-ODH)
The 2-ODH complex is formed by 3 subunits and by using the KEGG database, we found that in the S. coelicolor genome, a single gene (sco5281) encoded the E1 component (2-oxoglutarate dehydrogenase), whereas 3 genes (sco1268, sco2181, and sco7123) encoded the putative E2 component (dihydrolipoamide succinyltransferase), and only sco2180 encoded the E3 component (dihydrolipoamide dehydrogenase). The results about mRNA quantification, sco5281 showed an increasing trend between 12 and 24 h, rapidly declining at 36 h, and remaining constant at low levels until 72 h (Fig. 3, panel A). In addition, transcripts for the three E2 subunit encoding genes were detected; sco2181 presented the highest levels (1.76 × 10–3 ng µg−1 of total RNA) between 12 and 24 h, followed by sco1268 with a maximum of 3.32 × 10–5 ng µg−1 of total RNA and sco7123 showed the lowest levels (4.33 × 10–6 ng µg−1 of total RNA). The last two had similar mRNA profiles with levels 2 and 3 orders of magnitude lower than sco2181, respectively (Fig. 3, panel B). sco2180 mRNA levels were high at 12 h (6.84 × 10–3 ng µg−1 of total RNA), and then gradually decreased (Fig. 3, panel C). Even when the intergenic region between sco2180 and sco2181 genes is only 60 nucleotide length implying a possible operonic arrangement, our results showed that these genes had different mRNA levels, suggesting that they were transcribed from different promoters. On the other hand, the pyruvate dehydrogenase (PDH) E1 component was putatively encoded by sco2183; therefore, sco2180, sco2181, and sco2183 could generate a complete PDH or 2-ODH complex since it has been reported that some microorganisms interchange components between these two enzymatic complexes [24, 25]. Through a BLASTp search, sco2182 was found to share identity with GntR-family transcriptional regulators (Fig. S2). Hence, it may be involved in regulating the expression of this cluster.

The mRNA expression profiles of 2-oxoglutarate dehydrogenase (2-ODH) components encoding genes at different growth times in S. coelicolor. RNAs obtained from cells grown in MM with 1% glucose were used for RT-qPCR analysis. Transcript levels were determined as described in the “Materials and methods” section. Panel A, 2-ODH E1 sco5281 (■). Panel B, 2-ODH E2 sco1268 (▲), sco2181 (■), and sco7123 (●). Panel C, 2-ODH E3 sco2180 (■). Panel D, KGOR α-subunit sco4595/6270 (▲) and KGOR β-subunit sco4594/6269 (■)
Besides these enzymatic activities, succinyl-CoA can be synthesised from 2-oxoglutarate in a single step; this reaction is catalysed by 2-oxoglutarate: ferredoxin oxidoreductases (KGOR). In S. coelicolor, 4 genes were found to be involved in this step, i.e., sco4594 and its paralog gene, sco6269, which encodes the KGOR β-subunit, and sco4595 and its paralog gene, sco6270, which encodes the KGOR ⍺-subunit. These two paralog gene pairs showed a high identity (approximately 97% and 99% identity in amino acid and nucleotide sequences, respectively) (Table S2). Because the designed primers could align with both genes, the results obtained correspond to the transcripts of each pair of paralogs. Results showed that sco4594/6269 had a maximum expression level of 1.74 × 10–2 ng per µg−1 of total RNA, while sco4595/6270 had a maximum level of 3.88 × 10–6 ng per µg−1 of total RNA. sco4594/6269 expression levels increased from 12 h, reached a maximum at 36 h, and subsequently declined (Fig. 3, panel D), while the sco4595/6270 expression profile displayed its highest value at 12 h, and then decreased gradually up to 72 h (Fig. 3, panel D).
Succinyl-CoA synthetase (SCS)
SCS is a heterotetramer formed between two alpha and two beta subunits in most bacteria. In the KEGG database, it was found that in the S. coelicolor genome, two pairs of genes encode for hypothetical alpha (sco4809 and sco6586) and beta (sco4808 and sco6585) SCS subunits. The in silico protein translated from sco4808 shared 62.4% identity with the SucC protein from C. glutamicum, while the deduced product of the sco6585 paralog shared less identity (54.9%) with this protein (Table S3). On the other hand, the predicted sco4809 protein showed 70.2% identity with C. glutamicum SucD. In comparison, the product of sco6586 showed less identity (61.9%) with this protein. This evidence indicates that these S. coelicolor genes encode for the SCS alpha and beta subunits (Table S3). Concerning mRNA expression levels, transcripts from the 4 SCS-related genes were detected. sco4808 mRNA profile showed an ascending trend up to a maximum value of 8.31 × 10–6 ng µg−1 of total RNA at 24 h and then decreased to basal levels between 36 and 72 h of culture. sco4809 had a similar profile, but their expression levels differed by one order of magnitude, with a maximum value of 7.17 × 10–5 ng µg−1 of total RNA (Fig. 2, panel D). Furthermore, sco6585 had low but constant mRNA levels between 12 and 48 h of culture (approximately 6.2 × 10–6 ng µg−1 of total RNA on average), which subsequently decreased. In contrast, sco6586 had a maximum value of 6.59 × 10–5 ng µg−1 of total RNA at 12 h of culture, which was 10 times higher than that of sco6585. Our results showed that the 4 SCS encoding genes attained their maximum expression levels in S. coelicolor during the initial growth phase (Fig. 2, panel D).
Succinate dehydrogenase complex (SDH)
The SDH complex consists of four subunits, and ten genes in the S. coelicolor chromosome were found in the KEGG database to be associated with this complex. The SDH iron-sulphur subunit was encoded by 3 genes, sco0922, sco4855, and sco5106; the flavoprotein subunit was encoded by four genes, sco0923, sco4856, sco5107, and sco7109; the cytochrome B subunit was encoded by sco0924 and sco4858, while the membrane anchor subunit was encoded by sco4857. These genes were arranged in two functionally annotated clusters [26], cluster sco4855-sco4858 encoded 4 subunits and cluster sco0922-sco0924 encoded 3 subunits. A third cluster could be sco5106-sco5107 and encoded for probable iron-sulphur and flavoprotein subunits, respectively, while only sco7109 encoded a putative flavoprotein subunit (Fig. S3).
The iron-sulphur subunit protein sequence deduced from sco0922 showed high identity (80%) with SdhB associated with this function in Streptomyces mobaraensis [27]. The products deduced from sco4855 and sco5106 shared 62.5% and 75.7% identity with 1669 and 0417 genes from Mycobacterium smegmatis, respectively (Table S5). In S. coelicolor these 3 genes were transcribed and the mRNA profiles of sco0922 and sco5106 were similar, with their maximum expression levels attained at 24 h, followed by a gradual decrease in their levels (Fig. 4, panel A); these peaks corresponded to 5.1 × 10–5 and 1.03 × 10–5 ng µg−1 of total RNA, respectively. The sco4855 transcript reached its highest expression level of 4.77 × 10–4 ng µg−1 of total RNA at 12 h, and its levels decreased rapidly after this timepoint (Fig. 4, panel A). The identity between iron-sulphur subunit predicted products was low (24–26%) (Table S6), which could indicate divergence after gene duplication events or the acquisition of these copies by horizontal gene transfer. The occurrence of diversification between proteins encoded by redundant copy genes can lead to the acquisition of new functions; in the case of SDH, it has been speculated that SdhA from C. glutamicum can serve as a sensor to maintain cellular redox balance [28].

The mRNA expression profiles of succinate dehydrogenase (SDH) subunits and fumarase (FH) encoding genes at different growth times in S. coelicolor. RNAs obtained from cells grown in MM with 1% glucose were used for RT-qPCR analysis. Transcript levels were determined as described in the “Materials and methods” section. Panel A, SDH Fe-S subunit sco0922 (▲), sco4855 (■), and sco5106 (●). Panel B, SDH flavoprotein subunit sco0923 (■), sco4856 (▲), sco5107 (●) and sco7109 (□). Panel C, SDH cytochrome B subunit sco0924 (■), sco4858 (▲) and SDH membrane anchor subunit sco4857 (●). Panel D, FumB sco5044 (■) and FumC sco5042 (▲)
Multiple genes encoding the SDH flavoprotein subunit were found on the S. coelicolor chromosome. The sequence deduced from sco0923 showed identity between 96 and 100% with SdhA from other Streptomyces species. Among S. coelicolor and M. smegmatis, the product of sco4856 and SdhA showed the highest identity (69.7%), while the amino acid sequence derived from sco5107 showed an identity of 68.3% with 0418 protein (Table S5). In both organisms, there was a gene that was not associated with a cluster; in M. smegmatis, this was gene 1693, whereas in S. coelicolor, it was sco7109. Among the genes that encode this subunit, protein 1693 and sco7109 product showed the highest identity (76%) (Table S5). S. coelicolor flavoprotein subunit encoding genes showed identities ranging from 26 to 37% (Table S6). With respect to the expression levels of their transcripts, sco0923 and sco4856 exhibited similar profiles, with an increase in their expression being observed between 12 and 24 h, followed by a progressively decrease (Fig. 4, panel B). The peak in sco0923 expression corresponded to 4.24 × 10–3 ng µg−1 of total RNA, while that for sco4856 was approximately a third of this value, i.e., 1.61 × 10–3 ng µg−1 of total RNA (Fig. 5, panel B). sco7109 mRNA levels were low throughout S. coelicolor growth phases (Fig. 4, panel B). The sco5107 mRNA expression profile showed an increasing trend at 12 h, then declined until 48 h, and subsequently increased from 60 h, almost attaining twice its initial transcript levels at 72 h. Its maximum expression values at 12 and 72 h were 3 × 10–4 and 5.74 × 10–4 ng µg−1 of total RNA, respectively.

Panoramic view of mRNA expression profiles of the TCA cycle genes in S. coelicolor grown on minimal medium supplemented with 1% of glucose as carbon source. Growth times at 24 and 48 h are indicated with dotted lines
The cytochrome B subunit was found to be encoded by sco0924 and sco4858; because there is no homology between them, they may not be paralogs. In addition, sco0924 did not show homology with any M. smegmatis or E. coli genes. A BLASTp search of the predicted protein encoded by this gene showed that it was conserved across Streptomyces species, sharing identity with the cytochrome B subunit of SDH. In contrast, the protein sequence of SCO4858 shared 55.7% identity with M. smegmatis SdhC (Table S5). Quantification of sco0924 mRNA showed that its levels in the samples were high at 12 and 24 h, with the highest expression levels of all TCA cycle-associated genes being observed during the initial growth phase (Figs. 2, panel A and 4, panel C). These expression levels subsequently decreased to 0 at 72 h. Furthermore, sco4858 levels were approximately 10 times lower than those of sco0924, with its maximum levels observed at 24 h, and this was followed by a decrease in its levels (Fig. 4, panel C).
Finally, the membrane anchor subunit of SDH was encoded solely by sco4857. The predicted protein product of this gene shared a 47.9% identity with M. smegmatis 1671 gene (Table S5). sco4857 showed mRNA levels of 1.51 × 10–3 ng µg−1 of total RNA at 24 h, and these levels subsequently decreased until 72 h of culture (Fig. 4, panel C). All genes involved in the formation of the SDH complex in S. coelicolor grown on a 1% glucose-containing medium were transcribed in the first hours of growth under this condition. This finding is different from that reported in E. coli and C. glutamicum, in which the expression levels of SDH-associated genes are repressed by glucose [12, 28].
Fumarase (FH) and Malate dehydrogenase (MDH)
In S. coelicolor, the enzymes involved in the last reactions of the TCA cycle have already been characterised due to their essential roles in synthesizing intermediates of industrial interest. Fumarases are encoded by sco5042 and sco5044, which encode FumC and FumB fumarases, and are assigned to the Class II and Class I in the KEGG database, respectively. The predicted protein encoded by sco5042 shares 47.6% identity with FumC, while that of sco5044 shares approximately 63% identity with E. coli FumA and FumB (Table S7). The mRNA profiles of sco5042 and sco5044 were highly similar, showing high expression levels during the first hours of growth and a subsequent decrease (Fig. 4, panel D). However, there was an approximately ninefold difference in expression level between them, with sco5044 and sco5042 mRNAs having maximum expression values of 2.26 × 10–3 and 2.55 × 10–4 ng µg−1 of total RNA, respectively (Fig. 4, panel D). On the other hand, MDH is encoded by sco4827, which has already been cloned and the MDH enzyme has been purified [14]. sco4827 mRNA levels peaked at 24 h and subsequently declined (Fig. 2, panel C), and it was one of the genes that showed the highest expression levels between 12 and 24 h of growth.
Discussion
Acetyl-CoA oxidation, induced by glucose breakdown through the TCA cycle, is essential in microorganisms because it aids them to generate energy and reduce levels of the coenzymes, NADH and FADH2. In Streptomyces species, this pathway plays an indispensable role as the supplier of precursors and cofactors necessary for the secondary metabolism. The type of secondary metabolite that will be synthesised determines the source of the precursors, which could be central metabolites, derivates of anabolic reactions as building blocks, or specifically synthesised precursors. The TCA cycle intermediates such as α-ketoglutarate, fumarate, and oxaloacetate are precursors for the synthesis of glutamate and aspartate, functioning as metabolite building blocks. Furthermore, the regenerated NADPH via IDH catalysis reaction is used as reducing power in polyketides and β-lactam synthesis [29]. In Streptomyces clavuligerus the accumulation of succinate, oxaloacetate, malate and acetate is associated with the production of clavulanic acid [30], while in Streptomyces toxytricini, supplementation of cultures with TCA cycle intermediates in stationary phase enhanced lipstatin production [31]. Besides, due to the close relationship between TCA cycle, development and antibiotic production, alterations in one of them can have a deleterious effect on the others, as occurred with ACO or CS mutants of S. coelicolor, which are unable to sporulate and synthesize antibiotics in glucose media [3, 4].
Despite these essential metabolic functions, the expression of genes involved in the eight reactions of the TCA cycle in Streptomyces has not been studied in detail. Using S. coelicolor as a model and the KEGG database, it was possible to identify the genes that encode each of the enzymes that participate in glucose oxidation in this organism. Aconitase, isocitrate dehydrogenase, malate dehydrogenase enzymes and some subunits of 2-oxoglutarate dehydrogenase and succinate dehydrogenase, were each encoded by a single gene. However, gene multiplicity was found in some genes in this microorganism, e.g., four gene copies encoded the citrate synthase and the flavoprotein subunit of succinate dehydrogenase, three genes encoding for the E2 component of 2-oxoglutarate dehydrogenase, three for the iron-sulphur subunit of succinate dehydrogenase, and two gene copies were found encoding for the other enzymes and subunits of the complexes that participate in the TCA cycle. This genetic redundancy or genetic expansion is a common feature of the central metabolic enzymes in Streptomyces [32], and endows organisms with cellular flexibility and the ability to adapt to dynamic environments. The competence of Streptomyces species to synthesise a wide variety of metabolites as an adaptive response to challenging conditions stems from genetic expansion, thereby providing adaptive robustness [33, 34].
The expression of redundant genes is required to accomplish their function as an adaptive reservoir. In order to demonstrate this, we performed the absolute quantification of the levels of TCA cycle-associated genes throughout the S. coelicolor growth phases in minimal medium with glucose and ammonium as sole carbon and nitrogen source. One of the most significant observations was the differential gene expression over time. To enable the dynamic elucidation of these metabolic changes, we elaborated a mini chart table depicting the expression profiles of all TCA cycle-associated genes at different growth periods (Fig. 5). Among the CS genes, sco2736 showed high transcript levels in the active growth phase. This higher sco2736 expression was expected because previous data showed that this gene encoded the main CS enzyme in S. coelicolor during the exponential growth phase [3]. Besides, in a global S. coelicolor transcriptomic analysis in R5 complete medium, sco2736 was the most expressed CS encoding gene, and this corroborates our observations [35]. sco5832 also expressed maximum mRNA levels in the first hours of growth. This gene showed identity with citrate synthase (CS)/2-methylcitrate synthases (2MCS), and to the best of our knowledge, none of these enzymes have been reported in Streptomyces species. In C. glutamicum, two paralogous genes, prpC1 and prpC2, encode enzymes with both CS and 2MCS activities [36]. Studies on E. coli have demonstrated the presence of a dimeric protein with these activities; the authors speculated that both activities were elicited by the same enzyme, and in some organisms, the genes were duplicated and diverged to give separate enzymes [37]. Both fumarase encoding genes, sco5044 and sco5042, were expressed in these conditions. In E. coli, fumA exhibited low transcription levels in the presence of glucose as carbon source, while fumC had constant but lower expression levels than those of fumA. The authors proposed that FumC could replace FumA under conditions of iron limitation and oxidative stress; therefore, differential transcription of the 2 fumarases permits E. coli to be able to adapt to these environmental changes [38]. Similarly, in S. coelicolor, the transcript levels of sco5044 were higher than those of sco5042, which showed lower but constant mRNA levels in the presence of glucose.
Among the enzymes that did not have gene multiplicity, malate dehydrogenase had the highest mRNA levels in the first hours of growth. This is contrary to that observed in several organisms, including E. coli, in which mdh gene expression is regulated through glucose catabolic repression [39]. Aconitase and isocitrate dehydrogenase genes showed very similar transcript levels and profiles (Fig. 5). Viollier et al. [4] evaluated aconitase promoter gene activity in S. coelicolor grown on YEME medium and described a peak in activity during the early exponential phase, followed by a gradual decrease. These results are consistent with the mRNA profiles obtained in the present study.
In enzymes constituted of many subunits, low transcription levels of some of its components can reduce the availability of the active complex, thereby creating a bottleneck in the carbon flux through the TCA cycle. This could be the case for SCS and SDH. The expression levels of the SCS β-subunit encoding genes, were low but detectable. This finding differs from that reported for E. coli and C. glutamicum, in which the transcription of the sucCD operon is repressed by glucose [40, 41]. Gene products from sco4808 and sco4809 shared high identity with those of their respective paralogs i.e., sco6585 and sco6586 (60–70% and 70–80% similarity, respectively) (Table S4). Therefore, it would be possible that an active complex be formed from alpha subunits originating from either sco4809 or sco6586; similarly, beta subunits may originate from either sco4808 or sco6585. Hence, the catalytic reaction would constantly be active and generate GTP energy. However, the expression levels of the α-subunits encoding genes (sco4809 or sco6586) were approximately 10 times higher than those of the β-subunits encoding genes. Therefore, the availability of the β-subunit could limit the formation of active SCS and the flow of carbon through the TCA cycle.
In the case of succinate dehydrogenase complex, the transcript levels of the genes that encode for its iron-sulphur subunit were one or two orders of magnitude lower than those genes that encode for its other subunits, while the flavoprotein, membrane anchor and cytochrome B subunits encoding genes showed similar profiles and mRNA levels, which could suggest that they were arranged in an operon. For 2-ODH complex, E2 component encoding genes showed the lowest levels; based on these results, it can be speculated that most of the E2 component came from sco2181, compensating the negligible expression of sco7123. In the case of KGOR activity, the maximum levels of sco4594/6269 mRNA obtained was approximately 4500 times higher than those of sco4595/6270. These results suggest that the alpha subunit could be the limiting factor for the formation of the KGOR complex, showing a minimal contribution towards the decarboxylation of 2-oxoglutarate.
Although it has been previously reported that there is a correlation between the transcript and translation levels of the genes that participate in the primary metabolism in S. coelicolor [35], it is important to consider that the mRNA levels is just one of the multiple factors influencing carbon flux in the TCA cycle, which is the result of different regulation levels, both at the post-transcriptional level as mRNA stability and translation efficiency, and at the post-translational level because the activity of enzymes of primary metabolism can be regulated through post-translational modifications, such as acetylation in response to a decrease in the demand to reduce power and energy load on the cell [15, 42]. Hence, our study contributes with the identification of transcripts that can restrict the carbon flux during different stages of growth on a single carbon source and represents a prelude to a possible modification of the expression levels of the identified target genes to make carbon flux through the TCA cycle more efficient in S. coelicolor grown in the presence of glucose, thereby enhancing the synthesis of secondary metabolites of biotechnological interest.
Data availability
All data generated or analysed in this study are included in this published article [and its supplementary information files].
Code availability
Not applicable.
References
Chubukov V, Gerosa L, Kochanowski K, Sauer U (2014) Coordination of microbial metabolism. Nat Rev Microbiol 12(5):327–340
Vuoristo KS, Mars AE, Sanders JPM, Eggink G, Weusthuis RA (2016) Metabolic Engineering of TCA Cycle for Production of Chemicals. Trends Biotechnol 34(3):191–197. https://doi.org/10.1016/j.tibtech.2015.11.002
P. H. Viollier, W. Minas, G. E. Dale, M. Folcher, and C. J. Thompson, "Role of acid metabolism in Streptomyces coelicolor morphological differentiation and antibiotic biosynthesis," (in eng), J Bacteriol, vol. 183, no. 10, pp. 3184–92, doi: https://doi.org/10.1128/JB.183.10.3184-3192.2001.
P. H. Viollier, K. T. Nguyen, W. Minas, M. Folcher, G. E. Dale, and C. J. Thompson, "Roles of aconitase in growth, metabolism, and morphological differentiation of Streptomyces coelicolor," (in eng), J Bacteriol, vol. 183, no. 10, pp. 3193–203, , doi: https://doi.org/10.1128/JB.183.10.3193-3203.2001.
Remington SJ (1992) “Structure and mechanism of citrate synthase,” (in eng). Curr Top Cell Regul 33:209–229. https://doi.org/10.1016/b978-0-12-152833-1.50017-4
Ge Y, Cao Z, Song P, Zhu G (2015) “Identification and characterization of a novel citrate synthase from Streptomyces diastaticus No. 7 strain M1033,” (in eng). Biotechnol Appl Biochem 62(3):300–308. https://doi.org/10.1002/bab.1372
Gruer MJ, Artymiuk PJ, Guest JR (1997) “The aconitase family: three structural variations on a common theme,” (in eng). Trends Biochem Sci 22(1):3–6. https://doi.org/10.1016/s0968-0004(96)10069-4
Imabayashi F, Aich S, Prasad L, Delbaere LT (2006) “Substrate-free structure of a monomeric NADP isocitrate dehydrogenase: an open conformation phylogenetic relationship of isocitrate dehydrogenase,” (in eng). Proteins 63(1):100–112. https://doi.org/10.1002/prot.20867
Frank RA, Price AJ, Northrop FD, Perham RN, Luisi BF (2007) “Crystal structure of the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex,” (in eng). J Mol Biol 368(3):639–651. https://doi.org/10.1016/j.jmb.2007.01.080
Fraser ME, James MN, Bridger WA, Wolodko WT (1999) “A detailed structural description of Escherichia coli succinyl-CoA synthetase,” (in eng). J Mol Biol 285(4):1633–1653. https://doi.org/10.1006/jmbi.1998.2324
Kind S, Becker J, Wittmann C (2013) “Increased lysine production by flux coupling of the tricarboxylic acid cycle and the lysine biosynthetic pathway–metabolic engineering of the availability of succinyl-CoA in Corynebacterium glutamicum,” (in eng). Metab Eng 15:184–195. https://doi.org/10.1016/j.ymben.2012.07.005
Park SJ, Tseng CP, Gunsalus RP (1995) “Regulation of succinate dehydrogenase (sdhCDAB) operon expression in Escherichia coli in response to carbon supply and anaerobiosis: role of ArcA and Fnr,” (in eng). Mol Microbiol 15(3):473–482. https://doi.org/10.1111/j.1365-2958.1995.tb02261.x
Yang ST, Zhang K, Zhang B, Huang H (2019) 3.16 - Fumaric Acid. In: Moo-Young M (ed) Comprehensive Biotechnology (Third Edition). Pergamon, Oxford, pp 188–207
Ge YD, Cao ZY, Wang ZD, Chen LL, Zhu YM, Zhu GP (2010) “Identification and biochemical characterization of a thermostable malate dehydrogenase from the mesophile Streptomyces coelicolor A3(2),” (in eng). Biosci Biotechnol Biochem 74(11):2194–2201. https://doi.org/10.1271/bbb.100357
Liao G, Xie L, Li X, Cheng Z, Xie J (2014) “Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling,” (in eng). J Proteomics 106:260–269. https://doi.org/10.1016/j.jprot.2014.04.017
Bentley SD et al (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417(6885):141–147. https://doi.org/10.1038/417141a
Romero-Rodríguez A et al (2016) “Transcriptomic analysis of a classical model of carbon catabolite regulation in Streptomyces coelicolor,” (in eng). BMC Microbiol 16:77. https://doi.org/10.1186/s12866-016-0690-y
Schniete JK et al (2020) Differential transcription of expanded gene families in central carbon metabolism of Streptomyces coelicolor A3(2), (in eng). Access Microbiol. https://doi.org/10.1099/acmi.0.000122
Flores ME, Sánchez S (1985) Nitrogen regulation of erythromycin formation in Streptomyces erythreus. FEMS Microbiol Lett 26(2):191–194
Miller GL (1959) Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal Chem 31(3):426–428. https://doi.org/10.1021/ac60147a030
J. Sambrook, Molecular cloning : a laboratory manual. Third edition. Cold Spring Harbor, N.Y. : Cold Spring Harbor Laboratory Press, [2001] ©2001, 2001.
Bustin SA et al (2009) “The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments,” (in eng). Clin Chem 55(4):611–622. https://doi.org/10.1373/clinchem.2008.112797
Takahashi-Iñiguez T, Cruz-Rabadán S, Burciaga-Cifuentes LM, Flores ME (2014) “Molecular cloning, purification, and biochemical characterization of recombinant isocitrate dehydrogenase from Streptomyces coelicolor M-145,” (in eng). Biosci Biotechnol Biochem 78(9):1490–1494. https://doi.org/10.1080/09168451.2014.923290
Spencer ME, Guest JR (1985) Transcription analysis of the sucAB, aceEF and lpd genes of Escherichia coli (in eng). Mol Gen Genet 200(1):145–154. https://doi.org/10.1007/BF00383328
Kinugawa H, Kondo N, Komine-Abe A, Tomita T, Nishiyama M, Kosono S (2020) In vitro reconstitution and characterization of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase hybrid complex from Corynebacterium glutamicum (in eng). Microbiologyopen. https://doi.org/10.1002/mbo3.1113
Strakova E, Bobek J, Zikova A, Vohradsky J (2013) Global features of gene expression on the proteome and transcriptome levels in S. coelicolor during germination (in eng). PLoS ONE. https://doi.org/10.1371/journal.pone.0072842
Zhang L et al (2019) Comparative proteome analysis of Streptomyces mobaraensis under MgCl (in eng). Food Res Int 121:622–632. https://doi.org/10.1016/j.foodres.2018.12.027
Lee DS, Park JS, Kim Y, Lee HS (2014) Corynebacterium glutamicum sdhA encoding succinate dehydrogenase subunit A plays a role in cysR-mediated sulfur metabolism (in eng). Appl Microbiol Biotechnol 98(15):6751–6759. https://doi.org/10.1007/s00253-014-5823-3
Gunnarsson N, Eliasson A, Nielsen J (2004) Control of fluxes towards antibiotics and the role of primary metabolism in production of antibiotics (in eng). Adv Biochem Eng Biotechnol 88:137–178. https://doi.org/10.1007/b99260
H. Ramirez-Malule, S. Junne, M. Nicolás Cruz-Bournazou, P. Neubauer, and R. Ríos-Estepa, "Streptomyces clavuligerus shows a strong association between TCA cycle intermediate accumulation and clavulanic acid biosynthesis," (in eng), Appl Microbiol Biotechnol, vol. 102, no. 9, pp. 4009–4023, 2018
Kumar P, Dubey KK (2016) Modulation of fatty acid metabolism and tricarboxylic acid cycle to enhance the lipstatin production through medium engineering in Streptomyces toxytricini (in eng). Bioresour Technol 213:64–68. https://doi.org/10.1016/j.biortech.2016.01.133
G. van Keulen and P. J. Dyson, "Chapter Six - Production of Specialized Metabolites by Streptomyces coelicolor A3(2)," in Advances in Applied Microbiology, vol. 89, S. Sariaslani and G. M. Gadd Eds.: Academic Press, 2014, pp. 217–266.
Hiltner JK, Hunter IS, Hoskisson PA (2015) “Tailoring specialized metabolite production in streptomyces,” (in eng). Adv Appl Microbiol 91:237–255. https://doi.org/10.1016/bs.aambs.2015.02.002
Schniete JK et al (2018) Expanding primary metabolism helps generate the metabolic robustness to facilitate antibiotic biosynthesis in Streptomyces (in eng). MBio. https://doi.org/10.1128/mBio.02283-17
Jeong Y et al (2016) The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2) (in eng). Nat Commun 7:11605. https://doi.org/10.1038/ncomms11605
Claes WA, Pühler A, Kalinowski J (2002) “Identification of two prpDBC gene clusters in Corynebacterium glutamicum and their involvement in propionate degradation via the 2-methylcitrate cycle,” (in eng). J Bacteriol 184(10):2728–2739. https://doi.org/10.1128/jb.184.10.2728-2739.2002
Gerike U, Hough DW, Russell NJ, Dyall-Smith ML, Danson MJ (1998) “Citrate synthase and 2-methylcitrate synthase: structural, functional and evolutionary relationships,” (in eng). Microbiology (Reading) 144(Pt 4):929–935. https://doi.org/10.1099/00221287-144-4-929
Park SJ, Gunsalus RP (1995) “Oxygen, iron, carbon, and superoxide control of the fumarase fumA and fumC genes of Escherichia coli: role of the arcA, fnr, and soxR gene products,” (in eng). J Bacteriol 177(21):6255–6262. https://doi.org/10.1128/jb.177.21.6255-6262.1995
Park SJ, Cotter PA, Gunsalus RP (1995) “Regulation of malate dehydrogenase (mdh) gene expression in Escherichia coli in response to oxygen, carbon, and heme availability,” (in eng). J Bacteriol 177(22):6652–6656. https://doi.org/10.1128/jb.177.22.6652-6656.1995
Park SJ, Chao G, Gunsalus RP (1997) “Aerobic regulation of the sucABCD genes of Escherichia coli, which encode alpha-ketoglutarate dehydrogenase and succinyl coenzyme A synthetase: roles of ArcA, Fnr, and the upstream sdhCDAB promoter,” (in eng). J Bacteriol 179(13):4138–4142. https://doi.org/10.1128/jb.179.13.4138-4142.1997
Han SO, Inui M, Yukawa H (2008) “Effect of carbon source availability and growth phase on expression of Corynebacterium glutamicum genes involved in the tricarboxylic acid cycle and glyoxylate bypass,” (in eng). Microbiology (Reading) 154(Pt 10):3073–3083. https://doi.org/10.1099/mic.0.2008/019828-0
Ishigaki Y, Akanuma G, Yoshida M, Horinouchi S, Kosono S, Ohnishi Y (2017) “Protein acetylation involved in streptomycin biosynthesis in Streptomyces griseus,” (in eng). J Proteomics 155:63–72. https://doi.org/10.1016/j.jprot.2016.12.006
Acknowledgements
We thank Dr. María E. Gonsebatt for kindly providing us with the qPCR analysis equipment and Dr. Renato L. Rodríguez for technical assistance. We thank Lic. Omar Rangel, Biol. Sandra L. Hernández and Martha Cariño for their technical support.
Funding
This study was partially supported by the grants, PAPIIT IN210019 and IN214116 (DGAPA-UNAM).
Author information
Authors and Affiliations
Contributions
MEF conceived and designed the experiments, TTI performed the experiments, and MEF and TTI analysed the data, and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
Toshiko Takahashi-Iñiguez and María Elena Flores declare that they have no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (DOCX 960 kb)
Additional Supporting Information may be found in the online version of this article: Fig. S1-S3 and Tables S1-S7.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Takahashi-Iñiguez, T., Flores, M.E. mRNA levels of tricarboxylic acid cycle genes in Streptomyces coelicolor M145 cultured on glucose. Mol Biol Rep 50, 719–730 (2023). https://doi.org/10.1007/s11033-022-08068-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-08068-8