
Abstract
Background
Glioma is the most common type of brain tumors and isocitrate dehydrogenase (IDH1) gene is the most prominent molecular marker about the disease prognosis, response to therapy and patient survival. There are conflicting data about the effect of IDH1 mutation on glial cell proliferation, invasion and migration characteristics. The effect of IDH1 mutation on mTOR signaling pathway, which has key roles in tumorigenesis process, is limited and previous data is controversial. We aimed to explore the effect of wild type and mutant IDH1 overexpression on glioma cells and investigated the correlation with mTOR signaling pathway associated genes.
Methods and Results
U87-MG and A172 cells were transfected with different IDH1 mutant gene overexpressing (R132H, R132L, R132S, R132C) viral vectors. Cell proliferation, cell invasion and migration analysis as well as quantitative PCR analysis with the mutant glioma cell lines were performed. Forty-two patient derived glioma cells were obtained from patients with different glioma subtypes and cancer cells were enriched by culturing cells. Overexpression of both mutant and wild type IDH1 gene promoted the cell proliferation, but only IDH1 mutation increased cell invasion and migration. The expression of IDH1 mutation activated mTOR signaling via upregulation of WNTA, PRKAA2, GSK3B and MTOR genes as well as phosphorylated mTOR protein level.
Conclusions
Our results highlighted IDH1 mutation upregulate mTOR signaling pathway and promote cell proliferation, invasion and migration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioma is the most prevalent primary tumor of the central nervous system (CNS) and various histological forms exist with different molecular, cellular and behavioral characteristics. Recent world health organization (WHO) tumor classification in 2021, has been recently updated with new molecular markers and more detailed classification of brain tumors was aimed for better diagnosis and prognosis [1]. However, isocitrate dehydrogenase 1 (IDH1) gene mutations are still the most usable and reliable molecular marker in glioma classification that is used for accurate diagnosis, disease prognosis and response to treatment [2]. IDH1 protein is a metabolic enzyme and catalyzes the oxidative decarboxylation of isocitrate to α-KG in the tricarboxylic acid cycle (TCA) cycle. α-KG produced by IDH1 has crucial roles in histone modifications, hypoxia-sensing and fatty acid metabolism [3, 4]. Gain of function mutations in IDH1 gene leads to production of oncometabolite 2-hydroxyglutarate (2HG) from α-KG which is the main product of IDH1 enzyme [5]. Although there are different variants of IDH1 mutations (R132H/C/G/S/L), the most prevalent mutation of IDH1 is R132H [6]. Despite the several studies suggesting that IDH1 mutations associated with increased survival in patients with grade II and grade III diffuse glioma including both astrocytic and oligodendroglia subtypes [7]. Other studies propose the notion that IDH1 mutation alters the cellular NADPH/NADP + balance and stimulates the increase of ROS levels and oxidative stress [8] and inhibits the proliferation of glioma cells in vitro [9]. Current standard of care treatment for both IDH mutant and wild type glioma is total or if possible gross total removal of tumor tissue by surgery. Temozolomide is the first choice of chemotherapy with or without anti-angiogenic agents like bevacizumab. For inoperable glioma and partially removed glioma tissues radiotherapy is also used. IDH1 mutation is good prognostic marker for chemo- and radio-sensitivity of tumor as well as longer survival [10].
Mammalian target of rapamycin (mTOR) signaling pathway interconnected with several other pathways in many cell types and it has vital roles in several central processes, like regulation of cell cycle and proliferation, cell growth, intracellular signaling etc. The major effectors of mTOR pathway are receptor tyrosine kinases (RTKs), G-protein coupled receptors, transmembrane integrins, and growth factor receptors [11, 12]. Activation of the mTOR signaling pathway induces cells with malign characteristics, transforms them into a more drug-resistant, proliferative, invasive and malignant phenotype. [13]. Activation of mTOR signaling pathway, especially with the phosphoinositide 3-kinase (PI3K) and Akt axis (PI3K/AKT/mTOR) has been shown in various cancer types [14].
IDH1 mutations alter the activity of various signaling pathways in glioma. Interaction of IDH1 gene and PI3K/AKT/mTOR signaling pathway has been investigated in various studies. The molecular link between IDH1 and PI3K/AKT/mTOR pathway has previously shown. Generation of 2HG via mutated IDH1 gene inhibits the KDM4A, which is α-KG dependent demethylase enzyme. KDM4A associates with DEPTOR, a negative regulatory protein of mTOR. When KDM4A is inhibited with increased level of 2HG, mTOR pathway is activated [15]. IDH1 mutation shown to be enhanced the migration potential of glioma cells via activation of mTOR signaling pathway and making them vulnerable to chemotherapy [16]. Moreover, inhibition of mTOR signaling pathway in IDH mutant glioma cells reduced oncometabolite 2HG level and increased glioma cell survival [17]. On the other hand, it has also been shown that, overexpression of wild type IDH1 protein, promoted PI3K/AKT/mTOR pathway and ultimately the migration potential of primary glioblastoma cells [18].
Despite the several studies on key molecule IDH1 and central pathway mTOR signaling in glioma, we have limited information on association of IDH1 mutations and individual genes in mTOR signaling pathway. Therefore, hereby we investigated the effect of IDH1 gene on mTOR signaling pathway associated genes, cell proliferation, invasion and migration characteristics of glioma cells. Results were correlated with the tumor samples obtained from different glioma patients with respect to their grades, histopathological subtype and IDH1 status.
Methods
Cell lines and cell culture studies
U87-MG, A172 glioma, HUVEC and HEK 293T cells were purchased from ATCC. Gene expression studies were conducted on U87-MG cells and cell proliferation, invasion and migration assays were performed on A172 and HUVEC cells. Gene expression experiments were conducted on U87-MG cells. Gene expression of both glioma cells were compatible in terms of target genes.
Cells were cultured in Dulbecco’s modified Eagle medium (DMEM)/high glucose (Capricorn, DMEM-HPA) supplemented with 10% fetal bovine serum (Capricorn, FBS-HI-11B) and 1% antibiotic-antimycotic solution, 100X (WISENT, 450-115EL). Cell were maintained at 37 ̊C and 5% CO2 in a humidified incubator (Panasonic, USA).
Lentiviral transfection of cells
Stably IDH1 overexpressing cells were transducted by, lentiviral transfer plasmid vector (pLenti/TO-EYFP-P2A-IDH1(HA) (Addgene, Cat. No: 122,482) containing enhanced yellow fluorescent protein (EYFP), a packaging plasmid psPAX2 (Addgene, Cat. No. 12,260) and VSV-G envelope expressing plasmid pMD2.G (Addgene, Cat. No. 12,259). PFU Ultra II Fusion HS DNA Polymerase (Agilent, AGT-600,670) was used to generate six different point mutations with suitable site directed mutagenesis primers (G395A, G395T, G395C, C394A, C394T and C394G) of IDH1, according to the manufacturer’s instructions.
Wild type (WT) and mutant IDH1 gene expressing viruses were produced in HEK 293T cell line. 2 × 106 cells per well were seeded in each 10 cm culture dish and incubated at 37 ̊C in a 5% CO2 humidified atmosphere. Following 24 h of pre-incubation, cell medium was replaced with fresh DMEM complete (10% FBS and 1% Antibiotic-Antimycotic Solution). Transfection of viral DNA was performed with polyethyleneimine (PEI) and DNA to PEI mixture was as followed: 2 µg of transfer plasmids, 1,5 µg of packaging plasmid, 1,5 µg of envelope plasmid and 22,5 µg PEI (1 mg/ml) in 500µL DMEM medium. The mixture was incubated at room temperature for 15 min and added to culture dish. Twenty-four hour later, medium was replaced with fresh DMEM complete including 2% FBS. Cell medium was collected for three days and centrifuged at 800 g for 10 min at the room temperature and the supernatant was obtained. Viral particles were concentrated by concentrator buffer. The final mixture was incubated with constant racking at 60 rpm for overnight at 4 °C. Supernatant was obtained carefully and viral pellet was resuspended into 1 mL only DMEM by gently pipetting up and down.
U87-MG and A172 glial cells (3 × 105 cells/well) were cultured in 6-well plates as control (non-transfected, NT), or with IDH1 WT and with G395A (R132H), G395T (R132L), C394A (R132S) and C394T (R132C) mutant vectors. After 24 h, viral supernatant was mixed with polybren (5 µg/mL, Santa Cruz, sc-134,220) and infection was performed at 1000 g for 60 min at 30 °C and 6-well plates were placed into incubator at 37 °C with 5% CO2. After 12–16 h, cell mediums were changed with fresh complete DMEM. Blasticidine (Invivogen, ant-bl-05) was added as 3 µg/mL in cell culture medium for antibiotic selection. Cell mediums were replaced with fresh DMEM contained 3 µg/mL blasticidine every 2 or 3 days. Cells were controlled and photographed under a fluorescence microscope (Leica, DM2500).
Cell proliferation and Migration Assay
xCELLigence RTCA DP system (Agilent, USA) was used to measure cell proliferation continuously. E-plate 16 PET (Agilent, #300,600,890, USA) was used for proliferation assay experiments. For each transfected cell line seeding density experiment was performed initially. Cells were trypsinized, counted, and seeded as 5000, 7500, 10,000, 12,500, 15,000, 20,000 cells per well. Each condition was studied as a triplicate. After deciding the appropriate seeding number, cell proliferation experiments with each condition performed. Briefly, 100 µl medium per well was distributed to each well. Plate was placed in the slot of the system and background measurement was performed. Later cells were seeded accordingly, and plate was let sit in the sterile cell culture hood for 30 min for cells to settle. After 30 min of incubation, plate was placed back in the slot and cell index was measured every 15 min for 96 h.
In order to assess invasion capacity of the cells, RTCA CIM plates (Agilent, #5,665,817,001, USA) were used. Briefly, 160 µl of medium supplemented with FBS was placed in the wells of the lower chamber. Assembled plate was put back to the incubator for an hour. Then, 50 µl of medium without FBS was distributed to the wells and background measurement was performed. Cells were seeded in 100 µl of medium without FBS, and plates were incubated in the cell culture hood for 30 min. After 30 min of incubation, plate was placed back in the slot and cell index was measured every 15 min for 96 h.
In vitro scratch repair assay
Scratch repair assay was performed to assess migration potential of IDH1 transfected and control glioma cells. 1 × 104 cells/well in 12-well plate format was cultured. Scratch was lined on culture dishes using 100 µL pipette tip when cells were confluent on plate surface. Cell media was replaced with fresh DMEM complete including 1%FBS and 1% antibiotic-antimycotic solution. Cells were photographed at 0., 8. and 24. hours. Wound Healing Size Tool in ImageJ Software was utilized to quantify the changes of scratch areas. Images were analyzed and compared with respect to number of cells migrating to scratched area on dish surface using Image J and GraphPad Prism 9.
Patients and tumor cell preparation
Prospective patient tumor sample collection was approved by institutional research ethics committee (2017-14/01). Written consents were obtained from patients to participate in study. Ethics committee reviewed the patient consent forms. The patient selection was only based on diagnosis, and only glioma tumors with a variety of grades were selected. Grade II and III tumors included diffuse or anaplastic astrocytic and oligodendroglia tumors. Glioblastoma samples as grade IV tumors were also included in the patient cohort. The current 2021 World Health Organization (WHO) classification was used for histopathologic diagnosis as IDH wild type astrocytoma (Ast-IDH1 WT, n = 10), R132H mutant IDH1 astrocytoma (Ast R132H, n = 10), oligodendroglioma with IDH1 R132H mutation (Olg R132H, n = 11) and IDH wild type GBM (GBM WT, n = 11) [1].
Patient derived tumor cells were obtained as previously described [19]. Forty-two tumor samples from different patients were obtained by surgical operation. Fresh tumor tissues were sectioned and washed with 1 mL of PBS. Blood vessels were removed off from the tissue. Tissues were crushed with the help of a lancet. Tissues were transferred to 50 mL falcon tubes filled with 10 mL DMEM complete medium including Accutase (BD Biosciences, 561,527), Trypsin (Sigma, 59,429 C) and Collagenase I (Sigma, 1,148,089) enzyme and incubated for 10 min at room temperature. Supernatant was discarded. Following a centrifugation at 200 g for 5 min DMEM complete medium was added to the tube and the pellet was dissolved by pipetting and then dissolved solution was plated into 60 mm cell culture dish and culture dish was placed into the incubator. Cells were incubated for 72 h and passaged maximum of three times to obtain the glial cells at maximum purity.
Quantitative real-time PCR analysis
Total RNA was extracted using NucleoSpin RNA (Macherey-Nagel, #740955.50) total RNA isolation kit by following the manufacturer’s instructions. cDNA synthesis was performed with 2 µg of RNA by using cDNA synthesis kit (A.B.T, #601-005) according to the manufacturer’s instructions. qRT-PCR was done with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, #1,725,271) is 10 µL of enzyme mix added to 1 µL of 10µM forward and reverse primers, 2µL of cDNA and 6 µL nuclease-free H2O. Relative gene expressions were calculated by Livak Method (2- ∆∆Cq) and GAPDH was used as reference gene. Fold change differences of target genes were compared by One-way ANOVA test, Pearson Correlation Test and Simple Linear Regression Analysis was used to compare different groups, by GraphPad Prism 9. Primer sequences were provided in supplementary Table.
Western blotting
Target proteins were evaluated at protein level by using western blotting and quantitative comparison of protein bands. Total protein was isolated by using RIPA solution. Cells expressing WT and mutant IDH1 genes were plated onto in 100 mm culture dish and scraped to obtain total proteins before they become confluent. Cells were collected and centrifugated at 4000 g for 2 min at 4 °C. After centrifugation, supernatant was removed and 200 µL CLB (Cell Lysis Buffer, 10 mM Tris- HCL pH:8, 0,1 M EDTA pH:8 and 0.5% of SDS) was applied to cell pellet. Protein concentrations were determined by Bradford Assay (Bio-Rad, #500-0006). Total of 30 µg protein lysates were loaded into 8–15% of SDS polyacrylamide gel and blotted onto nitrocellulose membrane. Membranes were incubated in %5 BSA blocking for 1 h. Specific monoclonal antibodies for IDH1 (Santa Cruz, sc-515,396), HA-Tag (CST, 3724), p-mTOR (Santa Cruz, sc-293,133), mTOR (Santa Cruz, sc-517,464), and Beta-actin (CST, 3700) were incubated overnight at 4 °C with 1:10000 concentrations and finally appropriate secondary antibodies were used in 1:7000 concentrations. Protein bands were illuminated by using super signal West Femto (Thermofischer) and protein bands were visualized by ChemiDoc MP System (Bio-Rad). Protein band densities were compared by Image J software.
Results
IDH1 mutations promoted cell proliferation and invasion but reduced cell migration in glioma cells
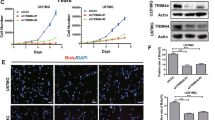
Different IDH1 mutations (R132H, R132L, R132S, R132C) were compared to investigate the effect of different mutations on cell viability, migration and invasion potential of glioma cells. Transfection of glioma cells with either wild type or mutant viral vectors caused 50 to 600-fold overexpression of IDH1 gene compared to non-transfected control cells (Supplementary Fig. 1). Over expression of IDH1 gene promoted proliferation of glioma cells compared to non-transfected controls. All mutations significantly increased cell proliferation compared to wild type overexpressed form of IDH1 gene (p < 0.001). However, R132H and R132L mutations significantly differed from other mutations (p < 0.001), and showed extraordinary proliferation compared to control, wild type (R132) and R132C, R132S overexpressed groups (Fig. 1a).
Invasion potential of cells were assessed by Boyden chamber assay by using xCelligence DP system and RTCA CIM plates. Overexpression of WT IDH1 gene significantly decreased the number of invaded cells compared to controls (p < 0.001). However, all mutant IDH1 forms showed significant increase in invasion compared to both non-transfected controls and overexpressed wild type IDH1 (Fig. 1b).
The effect of IDH1 mutation on cell migration on glioma cell was assessed by scratch repair assay or wound healing assay. First, cell photographs were taken at the beginning of the scratch assay and others were taken at 8th hours and 24th hours. Scratch area was calculated depending on the initial size and number of cells in the defined area for each different cell. Two different photographs were used to calculate the differences in scratch area for each different cell lines (Fig. 1c). Wounded area of non-transfected cells showed similar healing characteristics to that of both wild type and mutant IDH1 proteins, except R132C mutation showed comparably faster healing compared to other groups (Fig. 1d).

Mutant IDH1 overexpression enhanced cell proliferation, invasion but not migration in U87-MG cells. (a) Cell proliferation, (b) cell invasion, (c) cell migration assay microscope photographs (d) cell migration assay analysis of IDH1 wild type and mutation transfected U87-MG cells. Data were obtained for cell proliferation and invasion assays for each 15 min via electrical impedance based measurement. Each line represents different mutation transfected glial cells. R132H (green), R132L (dark blue), R132S (pink), R132G (damson color), G394T (light purple), C394A (light blue), wild type (red). IDH1 mutations induce cell proliferation in U87-MG cell line. Cells were photographed at the beginning, 8. hour and 24. hour for cell migration assay. (*indicate statistical significance: *p < 0.05, **p < 0.001, ***p < 0.0001) Cells were photographed under 40X magnification. The size of scale bar is 100 μm
Data suggested that overexpression of mutant IDH1 increased cell proliferation, invasion but not significantly affect cell migration potential of glioma cells. R132H and R132L mutations showed more malign cancer characteristics compared to other mutations and wild type overexpression of IDH1 gene. On the other hand, overexpression of IDH1 mutations in non-cancerous HUVEC cells did not show the same effect on cell phenotype. WT IDH1 overexpressing HUVEC cells showed less cell proliferation compared to non-transfected control cells, nonetheless overexpression of R132H, R132L, R132C and R132S mutations suppressed the cell proliferation (Supplementary Fig. 2).
IDH1 mutation hyperactivated mTOR signaling pathway
The effect of IDH1 on mTOR signaling pathway, the relative mRNA levels of target genes enrolled in pathway, were analyzed via quantitative real time PCR (qRT-PCR). WNT5A gene express Wnt5a protein which activates canonical WNT signaling pathway through binding to different receptors and exerts oncogenic properties in several cancers [20]. Wild type IDH1 overexpression led to increased level of WNT5A and R132H mutation led to even more increased level in glioma cells (Fig. 2a). AMP-activated protein kinase is an enzyme expressed by PRKAA2 gene and activated when there is cellular stress. Several isoforms exist and have controversial roles in cancer, some as tumor suppressor and some as oncogene [21]. The level of PRKAA2 gene expression was significantly increased in both wild type and mutant IDH1 overexpressed glioma cells, however the R132H mutant IDH1 overexpression was significantly more compared wild type IDH1 (Fig. 2b). TSC1 and TSC2 genes express Hamartin and Tuberin proteins which forms a tumor suppressive complex and controls cell cycle progression and proliferation by signaling the mTOR pathway [22, 23]. The level of TSC1 and TSC2 gene expression was down regulated in both wild type and mutant IDH1 overexpressed glioma cells, moreover TSC1 level was significantly downregulated in mutant IDH1 cells, whereas TSC2 did not significantly differ in between mutant and wild type IDH1 expressing U87-MG cells (Fig. 2c and d). Glycogen synthase kinase 3 beta protein is coded by GSK3B gene and involved in energy metabolism of cells and positively interacts with TSC complex by phosphorylating it [24]. IDH1 mutation led to significant upregulation of GSK3B gene compared to wild type IDH1 over expressing and control cells (Fig. 2e). mTOR is kinase enzyme that is coded by MTOR gene forms two distinct complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) and regulates several central processes in the cell like promoting cell proliferation, cell motility, cell survival and transcription regulation [25]. mRNA level of MTOR gene decreased in wild type IDH1 overexpressing cells whereas mutant IDH1 overexpressing cells significantly upregulated mTOR gene expression (Fig. 2f). The level of mTOR and phospo-mTOR (p-mTOR) protein were also significantly upregulated in mutant IDH1 overexpressing cells whereas untreated and wild type IDH1 cells did not (Fig. 2 g, 2 h). The ratio of p-mTOR to mTOR protein level was upregulated in mutant IDH1 overexpressing U87-MG cells (Fig. 2i).

Overexpression of IDH1 mutation upregulated mTOR signaling pathway genes in U87-MG cells (a) WNT5A, (b) PRKAA2, (c) TSC1, (d) TSC2, (e) GSK3B, (f) MTOR quantitative gene expression results. (g) Western blot images of mTOR, phosphorylated mTOR (p-mTOR) and Actin (Housekeeping protein) proteins. (h) quantitative analysis of mTOR protein, (i) quantitative analysis of p-mTOR protein to total mTOR protein. Control refers to non-transfected, IDH1 WT refers to IDH1 wild type overexpressing, IDH1 R132H refers to R132H mutant IDH1 overexpressing U87-MG cells. (* indicate statistical significance: *p < 0.05, **p < 0.001, ***p < 0.0001, ns: non-significant)
Overall, when mutant IDH1 gene is expressed, WNT5A, PRKAA2 and GSK3B mRNA levels increased where as TSC1 and TSC2 mRNA levels decreased. Taken together, upregulation of these gene indicated the activation of mTOR signaling pathway.
Mutant IDH1 gene expression positively correlated with mTOR signaling pathway genes and disease severity
Expression of IDH1 and mTOR pathway associated genes were analyzed on tumor samples that were obtained from patients and tumor cells were enriched by culturing cells and eliminating the nontumoral cells (blood cells, endothelial cells etc.). mRNA was obtained from primary cell culture of patient tumors. IDH1 gene was significantly more expressed in mutant histopathological subtypes of glioma Ast(R132H) and Olg(R132H) compared to wild type glioma subtypes Ast(WT) and GBM(WT) (Fig. 3a). Grade III glioma showed maximum level of IDH1 gene mRNA (Fig. 3b). The level of WNT5A gene expression correlated with IDH1 gene and showed maximum expression in mutant glioma as well as grade III subtypes (Fig. 3c and d). The level of TSC1 mRNA was maximum in GBM(WT) group and grade IV tumors (Fig. 3e and f). However, TSC2 gene showed maximum gene expression in Olg(132 H) subtype and grade III tumors (Fig. 3 g and 3 h). Interestingly, Ast(R132H) subtype showed significantly increased TSC2 gene expression than Ast(WT) subtype (Fig. 3 g). The level of PRKAA2 gene expression correlated with IDH1 gene and showed maximum expression in mutant glioma as well as grade III subtypes (Fig. 3i and j). GSK3B gene expression was only significantly increased in Ast(R132H) subtype (Fig. 3k) and grade II and grade III tumors (Fig. 3 L). Finally, the level of MTOR gene expression was significantly increased in Ast(R132H), Olg(132 H) and GBM(WT) subtypes compared to Ast(WT) subtype (Fig. 3 m). Higher the grade of tumors, the more MTOR gene expression was observed (Fig. 3n).

mTOR signaling pathway associated genes upregulated in patient derived glioma cells and correlated with tumor histopathology and grades. Expression of IDH1, WNT5A, TSC1, TSC2, PRKAA2, GSK3B, MTOR genes between histological subtypes (a, c, e, g, i, k, m) and tumor grades (b, d, f, h, j, l, n). o) Gene expression correlation matrix. Colors indicate degree of correlation. Ast(IDH1 WT): IDH1 wild type astrocytoma, Ast(R132H): R132H IDH1 mutant astrocytoma, Olg(R132H): IDH mutant oligodendroglioma, GBM (IDH WT): IDH1 wild type glioblastoma
Correlation matrix of expressed genes showed that IDH1 gene expression is positively correlated with the genes analyzed, except for TSC2 gene. The maximum correlation between IDH1 and MTOR (R2 = 0.94) and WNT5A (R2 = 0.92) (Fig. 3o). Taken together, it can be stated that IDH1 expression was highly correlated with mTOR pathway genes in patient derived glioma cells.
Discussion
The mTOR and associated proteins are currently hot spots for anti-cancer therapeutics. Since mTOR pathway enrolled in several central mechanisms in both normal and cancer cells. There are several reports indicating the hyperactivation of mTOR pathway in several cancer forms. One of the primary members of the pathway is Wnt protein that indirectly inactivates GSK3β protein through Dishevelled (Dvl) protein [26]. GSK3β and AMP-activated protein kinase (AMPK protein coded by PRKAA2 gene) upregulates and phosphorylates TSC1/TSC2 complex [27]. Phosphorylated TSC1/TSC2 complex suppresses the activity of mTORC1 complex via Rheb inhibition [28]. Finally, mTOR activity promotes several intracellular signaling cascades that are enrolled in microtubule organization, cell death, survival and differentiation. Interaction between these key players of mTOR pathway can be bypassed by aberrant activity of other molecules having role in the pathway. Here it is shown that when R132H mutant IDH1 is overexpressed in glioma cells WNT5A gene was downregulated PRKAA2 was upregulated, TSC1 and TSC2 were downregulated and GSK3β was upregulated. Finally, mTOR and its phosphorylated form was significantly upregulated. Gene expression data obtained from cell line studies and patient tumors, altogether showed that mTOR signaling pathway was activated in IDH1 mutant glioma (Fig. 4). Furthermore, the data is confirmed in patient derived glioma cells and activation of mTOR pathway was relatively more in IDH1 mutant glioma patients compared to IDH wild type glioma subtypes.

Interaction of IDH1 mutation and mTOR signaling pathway. Blue color indicates increased gene expression, red color indicates decreased gene expression
Previous studies have also reported that IDH mutant glioma activates PI3K/mTOR pathway for transformation and eventually worse prognosis of the disease by modulating cell proliferation, cell survival and migration [16, 29]. Further, inhibition of PI3K/mTOR signaling reduced 2HG production and increased the survival [17]. On the other hand, another study with 354 glioma patients reported that mutant IDH1 inhibited PI3K/Akt signaling pathway [30]. Study by Birner P. et al., indicated that IDH1 mutation and active PI3K/Akt signaling reversely correlated and mutant IDH1 inhibited the Akt phosphorylation [30]. Despite the apparently opposite findings, previous data confirmed that mTOR signaling is regulated through several central proteins and obviously Wnt, Akt and PI3K have different roles in glioma for mTOR activation. In this study, activation of mTOR signaling was shown through Wnt, AMPK, GSK3β and mTOR and it supports the idea of IDH1 mutation activated the mTOR signaling pathway.
Cell proliferation, invasion and migration are important phenotypic characteristics of glioma cells as like the most cancer cells. However, previous studies reported conflicting results about the effect of IDH1 on glial tumor characteristics. While some studies reported that IDH1 mutation reduced cell proliferation, invasion and migration potential via downregulating Wnt/β catenin pathway [31] or via HIF1α [32, 33], many other studies indicated that IDH1 mutation promoted all these characteristics via epithelial mesenchymal transition (EMT) [34], or Akt/mTOR pathway [16, 35] or methylating various target gene expressions [36]. Additionally, it was shown that overexpression of wild type IDH1 caused G1 stage cell cycle arrest and transformed cells into chemo resistant cell types whereas, overexpression of R132H mutant IDH1 transformed glioma cells into chemo sensitive cell type [37, 38]. Considering the previous controversial results about the IDH1 mutation effect on glioma, it can be speculated that, overexpression of IDH1 gene either in wild type or mutant form has an impact on tumor characteristics. Here, we showed that IDH1 overexpression promoted cell proliferation compared to untreated control (non-transfected) cells, moreover mutant IDH1 expressing cells showed increased cell proliferation compared to both wild type overexpressing and control groups. However, cell invasion was significantly reduced in wild type group and increased in mutant IDH1 overexpressing groups compared to controls. Cell migration was significantly reduced in IDH1 mutant overexpressing cells compared to controls and wild type IDH1 overexpressing cells (Fig. 1). The increased proliferation and cell invasion behavior of mutant glioma cells may be induced by activated mTOR signaling pathway. On the other hand, migration characteristics of cells may have reduced due to other cellular processes apart from the mTOR pathway.
Frequency of IDH1 mutations in glioma subtypes varied in different studies in ranging between 70 and 100%. Many studies reported that the most frequent mutation is R132H, subsequently R132C, R132G, R132L and R132S mutations [6, 39]. Previous studies proved that production of 2HG was inversely correlated with the frequency of the mutations among all IDH1 mutations with respect to neoplastic activity as well as malign properties of glioma cells. R132G mutation induced the maximum level of 2HG production, R132L and R132C promoted least level of cell survival in glioma cells [40]. Here, we showed that R132L mutation showed the maximum cell proliferation, R132H, R132S and R132C showed somewhat similar cell proliferation potential (Fig. 1a). Unlike cell proliferation, cell invasion did not significantly differ in between different IDH1 mutations (Fig. 1b), but cell migration was minimum in R132S compared to other mutations. As Pusch et al., stated the difference on cellular behavior of different IDH1 mutations may be due to an increased or decreased enzyme kinetics [40] and that leads to possible natural selection of R132H that favor increased survival and sensitivity to chemo- and radiotherapy in glioma patients.
Presented study has also several limitations. Major limitation is the lack of western blot or protein level evidences for each of tested genes. However, we aimed to observe the effect of IDH1 mutations in gene expression level. mTOR signaling pathway is a central cellular pathway and lots of proteins involved in it for various cellular interactions. We could only analyze the key genes and proteins. Another limitation is the absence of IDH1 suppressed cells to compare with overexpressed cells. Therefore, the effect of IDH1 mutation on mTOR signaling pathway could not assessed in IDH1 suppressed HUVEC and glioma cells.
As conclusion, IDH1 gene has critical roles in tumorigenesis of glioma cells. IDH1 mutation promotes glioma cell proliferation, invasion and migration. The level of IDH1 gene expression and mTOR signaling pathway associated genes were correlated. Therefore, we propose that overexpression of IDH1 gene as well as IDH1 mutation activates mTOR signaling pathway. Considering the previous studies both supporting the positive correlation between IDH1 and mTOR signaling pathway and/or showing the opposite evidences; we speculate that the role of mutant IDH1 mutation in glioma development and progression is probably dynamic and context dependent.
Data Availability
All data generated or analyzed during this study are included in this published article/as supplementary information files. Raw results data files can be shared upon request.
References
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al (2021) The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23(8):1231–1251 Epub 2021/06/30. doi: https://doi.org/10.1093/neuonc/noab106. PubMed PMID: 34185076; PubMed Central PMCID: PMCPMC8328013.
Mirchia K, Richardson TE (2020) Beyond IDH-Mutation: Emerging Molecular Diagnostic and Prognostic Features in Adult Diffuse Gliomas. Cancers (Basel) 12(7). doi: https://doi.org/10.3390/cancers12071817. PubMed PMID: 32640746; PubMed Central PMCID: PMCPMC7408495 Epub 2020/07/10
Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X et al (2014) IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res 74(12):3317–3331 Epub 2014/04/24. doi: https://doi.org/10.1158/0008-5472.CAN-14-0772-T. PubMed PMID: 24755473; PubMed Central PMCID: PMCPMC4885639.
Penard-Lacronique V, Bernard OA (2016) IDH1, histone methylation, and so forth. Cancer Cell 30(2):192–194
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19(1):17–30
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118(4):469–474 Epub 2009/06/26. doi: https://doi.org/10.1007/s00401-009-0561-9. PubMed PMID: 19554337.
Yang H, Ye D, Guan K-L, Xiong Y (2012) IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 18(20):5562–5571
Shi J, Sun B, Shi W, Zuo H, Cui D, Ni L et al (2015) Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumor Biology 36(2):655–662
Shi J, Zuo H, Ni L, Xia L, Zhao L, Gong M et al (2014) An IDH1 mutation inhibits growth of glioma cells via GSH depletion and ROS generation. Neurol Sci 35(6):839–845
Stupp R, Hegi ME, Gilbert MR, Chakravarti A (2007) Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol 25(26):4127–4136
Liu P, Cheng H, Roberts TM, Zhao JJ (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discovery 8(8):627–644
Regad T (2015) Targeting RTK signaling pathways in cancer. Cancers 7(3):1758–1784
Porta C, Paglino C, Mosca A (2014) Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol 4:64 Epub 2014/05/02. doi: https://doi.org/10.3389/fonc.2014.00064. PubMed PMID: 24782981; PubMed Central PMCID: PMCPMC3995050.
Karar J, Maity A (2011) PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci 4:51 Epub 20111202. doi: https://doi.org/10.3389/fnmol. 2011.00051. PubMed PMID: 22144946; PubMed Central PMCID: PMCPMC3228996.
Carbonneau M, Gagné LM, Lalonde M-E, Germain M-A, Motorina A, Guiot M-C et al (2016) The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun 7(1):1–12
Zhu H, Zhang Y, Chen J, Qiu J, Huang K, Wu M et al (2017) IDH1 R132H mutation enhances cell migration by activating AKT-mTOR signaling pathway, but sensitizes cells to 5-FU treatment as NADPH and GSH are reduced. PLoS ONE 12(1):e0169038
Batsios G, Viswanath P, Subramani E, Najac C, Gillespie AM, Santos RD et al (2019) PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci Rep 9(1):1–15
Shen X, Wu S, Zhang J, Li M, Xu F, Wang A et al (2020) Wild–type IDH1 affects cell migration by modulating the PI3K/AKT/mTOR pathway in primary glioblastoma cells. Mol Med Rep 22(3):1949–1957
Seidel S, Garvalov BK, Acker T (2015) Isolation and culture of primary glioblastoma cells from human tumor specimens. Stem Cell Protocols: Springer; p. 263 – 75
Asem MS, Buechler S, Wates RB, Miller DL, Stack MS (2016) Wnt5a Signaling in Cancer.Cancers (Basel). ; 8(9). Epub 20160826. doi: 10.3390/cancers8090079. PubMed PMID: 27571105; PubMed Central PMCID: PMCPMC5040981.
Ross FA, MacKintosh C, Hardie DG (2016) AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J 283(16):2987–3001. doi: https://doi.org/10.1111/febs.13698
Leung AK, Robson WL (2007) Tuberous sclerosis complex: a review. J Pediatr Health Care 21(2):108–114. doi: https://doi.org/10.1016/j.pedhc.2006.05.004. PubMed PMID: 17321910
Inoki K, Corradetti MN, Guan KL (2005) Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. ;37(1):19–24. doi: https://doi.org/10.1038/ng1494. PubMed PMID: 15624019
Yuan S, Li J, Diener DR, Choma MA, Rosenbaum JL, Sun Z (2012) Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proceedings of the National Academy of Sciences. ;109(6):2021-6
Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer Cell 12(1):9–22. doi: https://doi.org/10.1016/j.ccr.2007.05.008. PubMed PMID: 17613433
Li L, Yuan H, Weaver CD, Mao J, Farr GH 3, Sussman DJ et al (1999) Axin and Frat1 interact with dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF-1. EMBO J 18(15):4233–4240. doi: https://doi.org/10.1093/emboj/18.15.4233. PubMed PMID: 10428961; PubMed Central PMCID: PMCPMC1171499
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X et al (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126(5):955–968. doi: https://doi.org/10.1016/j.cell.2006.06.055. PubMed PMID: 16959574
Manning BD, Cantley LC (2003) Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 28(11):573–576
Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K et al (2014) Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res 20(11):2898–2909
Birner P, Pusch S, Christov C, Mihaylova S, Toumangelova-Uzeir K, Natchev S et al (2014) Mutant IDH1 inhibits PI3K/Akt signaling in human glioma. Cancer 120(16):2440–2447
Cui D, Ren J, Shi J, Feng L, Wang K, Zeng T et al (2016) R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/β-catenin signaling. Int J Biochem Cell Biol 73:72–81
Hu H, Wang Z, Liu Y, Zhang C, Li M, Zhang W et al (2015) Genome-wide transcriptional analyses of Chinese patients reveal cell migration is attenuated in IDH1-mutant glioblastomas. Cancer Lett 357(2):566–574 Epub 20141212. doi: https://doi.org/10.1016/j.canlet.2014.12.018. PubMed PMID: 25511738.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324(5924):261–265
Lu J, Li D, Zeng Y, Wang H, Feng W, Qi S et al (2019) IDH1 mutation promotes proliferation and migration of glioma cells via EMT induction. J BUON 24(6):2458–2464 PubMed PMID: 31983120
Liu Y, Lu Y, Li A, Celiku O, Han S, Qian M et al (2020) mTORC2/Rac1 pathway predisposes cancer aggressiveness in IDH1-mutated glioma. Cancers 12(4):787
Yan B, Hu Y, Ma T, Wang Y (2018) IDH1 mutation promotes lung cancer cell proliferation through methylation of Fibulin-5. Open Biol. ;8(10). Epub 20181010. doi: https://doi.org/10.1098/rsob.180086. PubMed PMID: 30305430; PubMed Central PMCID: PMCPMC6223204
Wang JB, Dong DF, Wang MD, Gao K (2014) IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pac J Cancer Prev 15(1):427–432 PubMed PMID: 24528069
Li S, Chou AP, Chen W, Chen R, Deng Y, Phillips HS et al (2013) Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neurooncology 15(1):57–68
Avsar T, Sursal A, Turan G, Yigit BN, Altunsu D, Cantasir K et al (2020) Development of a Rapid and Sensitive IDH1/2 Mutation Detection Method for Glial Tumors and a Comparative Mutation Analysis of 236 Glial Tumor Samples. Mol Diagn Ther 24(3):327–338
Pusch S, Schweizer L, Beck AC, Lehmler JM, Weissert S, Balss J et al (2014) D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol Commun 2:19 Epub 20140214. doi: https://doi.org/10.1186/2051-5960-2-19. PubMed PMID: 24529257; PubMed Central PMCID: PMCPMC3937031.
Acknowledgements
This study was funded by Bahcesehir University (BAU), Scientific Research Projects Council, (Project number BAP.2018 − 2.01), and The Scientific and Technological Research Council of Turkey (TUBITAK, project number 118S539).
Funding
This study was funded by Bahcesehir University (BAU), Scientific Research Projects Council, (Project number BAP.2018 − 2.01), and The Scientific and Technological Research Council of Turkey (TUBITAK, project number 118S539).
Author information
Authors and Affiliations
Contributions
TA and TBK conceived and designed the study and wrote the manuscript. TA, TBK, MDO and GT participated in data acquisition, analysis and interpretation of data. TK provided the tumor samples and patient information and participated in critical discussions. Also, all authors and acknowledged contributors have read and approved the manuscript and state that the content of this manuscript, in part or in full, has not been published elsewhere in any form. Authors stated their consent for participation and publication of the study.
Corresponding author
Ethics declarations
Conflict of interests
The authors have no personal, financial or institutional interest in any of the methods, materials, or devices described in this article. Authors declare no conflict of interests.
Ethical Standards
This study was approved by Ethical Committee of Bahcesehir University, School of Medicine. All the procedures were performed regarding ethical adherence. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent: Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.


Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Avsar, T., Kose, T.B., Oksal, M.D. et al. IDH1 mutation activates mTOR signaling pathway, promotes cell proliferation and invasion in glioma cells. Mol Biol Rep 49, 9241–9249 (2022). https://doi.org/10.1007/s11033-022-07750-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07750-1