
Abstract
GTPase activating proteins (GAPs) were initially considered as the inhibitors of cell signaling pathways because of their nature to activate the intrinsic GTPase activity of the RhoGTPases. But recent studies of dysregulated GAPs in many cancers such as glioblastoma, colorectal cancer, breast cancer, and renal cancer have elucidated the important roles of GAPs in carcinogenesis and GAPs have been shown to perform multiple nonconventional functions in different contexts. We have discussed the recent developments in the roles played by different types of srGAPs (SLIT-ROBO Rho GTPase-activating proteins) in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
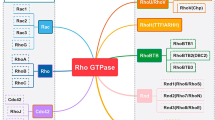
RhoGTPases (Rho G-proteins) are regulated by various factors and alternate between GTP-bound active state and GDP-bound inactive state. Rho family of small GTPases consists of 20 proteins which can be classified into subfamilies such as Rho (RhoA, RhoB, and RhoC), Rac (RhoG, Rac1, Rac2, and Rac3), and Cdc42 and some of the less characterized members are Rnd, RhoD, RhoBTB, RhoH, and RhoE [1]. RhoGAPs (Rho GTPase activating proteins) increase the slow intrinsic GTPase activity of RhoGTPases leading to the hydrolysis of GTP and inactivation of the pathway. There are more than 70 known RhoGAPs so far identified in eukaryotes and based on their homology of RhoGAP domains they have been further classified into subfamilies [2]. RhoGTPases are involved in the regulation of fundamental biological processes such as cell cycle progression, cell dynamics, intracellular membrane trafficking, cell growth, gene transcription, and apoptosis. RhoGTPases are also involved in gastrointestinal diseases [3] and regulate Wnt signaling in colorectal cancer [4, 5]. RhoGTPases couple environment to intracellular cell signaling pathways and the GDP-GTP cycle of RhoGTPases is controlled majorly by three classes of proteins (shown in Fig. 1).
-
(1)
GEFs (Guanine nucleotide exchange factor)—GEFs activate the Rho GTPase activity by replacing GDP for GTP. Some GEFs can activate multiple Rho GTPases but others are GTPase specific. GEFs are recruited by different adaptor proteins under the influence of upstream cell signaling and they interact with multiple effector proteins by their specific protein domains [6].
-
(2)
GAPs (GTPase activating proteins)—GAP proteins enhance the intrinsic GTPase activity of the Rho-GTPases that results in the inactivation of G proteins leading to termination of the cell signaling. GAPs have a conserved domain that changes the conformation of GTP-bound Rho proteins to orient the GTP for a better nucleophilic attack by water. GAPs also induce a GDP-like charge distribution. GAPs are generally very specific for their substrate Rho GTPases [7].
-
(3)
GDIs (Guanine nucleotide dissociation inhibitor)—GDIs are a family of small regulatory proteins that bind to GDP-bound Rho GTPases and prevent the exchange of GDP with GTP. They also prevent the localization of Rho GTPases to the plasma membrane. The inhibitory nature of GDIs can be removed by GDI displacement factors [8].

Rho-GDP (inactive)-Rho-GTP (active) cycling mediated by GEF, GAP, and GDI
Non-conventional roles of GAPs
GAPs were initially considered as signal terminators but they were later discovered to have a role in sensing the external cues and in Rho mediated signaling pathways. Later, they were found to have context-dependent functions that were independent of cell signaling pathways. Rac-GAPs were expected to act as a tumor suppressor as they induce the Rac mediated GTP hydrolysis but in-vivo studies could barely prove their tumor suppressor roles. Rac-GAPs have also been unexpectedly shown to act as an oncogene in certain types of cancers. The GAP proteins negatively regulate Rac mediated cell cycle and migration but the effects are variable depending on the epithelial or mesenchymal nature of the cancer cells. β2 chimaerin acts as a tumor inducer as well as a tumor suppressor depending on the context and cancer types [9, 10]. SrGAP2 and SrGAP3 are suppressed in human osteosarcoma and invasive ductal breast carcinoma hinting towards a tumor suppressor role of these proteins but the downregulation of SrGAP2 leads to more aggressive cancer by inducing cell migration while downregulation of srGAP3 leads to Rac1 dependent, anchorage independent cell proliferation [11, 12]. FilGAP (ArhGAP, a Rac-GAP) is suppressed in renal tumors and it is correlated with poor patient survival. The overexpression of ArhGAP24 inhibits G1/S cell cycle transition, reduces invasion, and induces apoptosis in renal cell cancer [13]. In breast cancer, ArhGAP24 has pro-metastatic as well as anti-metastatic roles [14, 15]. RASAL2 mediated inhibition of ArhGAP24 is shown to induce invasion by increased Rac activity and high RASAL2 is correlated with poor outcomes in triple-negative breast cancer patients [16, 17]. The complexity of Rac-GAP function is also revealed by a tumorigenic function of RacGAP1 (MgcRacGAP) as its expression is directly linked to the aggressiveness of human cancer cells [18]. It has been recently shown to act as a driver for metastasis in uterine carcinosarcoma [19]. Overexpression of Rac/Cdc42 GAP CdGAP is correlated with poor prognosis in breast cancer and it is highly expressed in basal breast cancer subtypes [20]. CdGAP is important for TGFβ and Neu/ErbB2 regulated cell migration and invasion independent of GAP activity [21]. CdGAP forms a complex with Zeb2 and represses E cadherin leading to enhanced epithelial-mesenchymal transition [22]. In another captivating example, overexpression of P190B RhoGAP in the mammary gland leads to Rac1 activation and erbB2 mediated tumor growth and metastasis [23]. ARHGAP4, a Rho-GAP, depletion leads to increased cell proliferation and migration, and Septin 9 acts as a negative regulator of ARHGAP4 [24]. The tumor-promoting activity of GAP can be considered as independent of enhancing the GTPase activity of Rac and these GAPs can have important non-conventional roles to play in different contexts [25]. IQGAP acts as a scaffolding protein to assemble the GTPases, GEFs, and effector proteins in a multistep process, and the binding of IQGAP1 with Rac-GEF Tiam1 and Rac 1 to activate Rac1 is a well-studied example [26]. The overexpression of Rac-GAP β2-chimaerin in breast cancer epithelial cells leads to the reduction of E-cadherin and hence induces the detachment of cell–cell contacts. In MMTV-Neu/ErbB2 mice, inhibition of β2-chimaerin leads to accelerated cancer onset but a delayed tumor progression. In Her2 positive breast cancer, β2-chimaerin and E-cadherin gene expressions are inversely related and the patients having a lower expression of β2-chimaerin have lower relapse-free survival but cancer metastasis develops at similar times. Hence, in-vivo experiments support the dual function of β2-chimaerin as it suppresses the tumor initiation but at the same time also supports the progression of the tumor [27]. Suppression of RhoA-GAP, DLC1 induces tumor growth, and RhoGAPs ARHGAP11A and RACGAP1 are highly expressed in the basal-like subtype of breast cancer. In contrast to DLC1, both of these Rho GAPs behave as an inducer of cancer rather than a tumor inhibitor. Epigenetic regulation of ARHGAP28 increases the Rho activity in highly metastatic colon cancer cells [28, 29]. Different RhoGAPs play unique and context-dependent roles based on their spatial position, regulation, and cancer subtypes [30].
RhoGAPs are regulated by lipids, phosphorylation, protein degradation, and protein–protein interactions and they are involved in multiple biological and physiological functions such as cell differentiation, endocytosis, migration, exocytosis, cytokinesis, tumor suppression, angiogenesis, and neuronal morphogenesis. Dysregulation of genes encoding RhoGAPs may lead to the development of diseases such as cancer, Bardet-Biedl syndrome, MLS, and X-linked mental retardation. More than 70 RhoGAP are known across yeast to humans and the human genome is known to encode 59–70 proteins having RhoGAP domain. There are 20 different genes known to encode for RhoGAPs domain containing proteins. There are 22 mammalian genes that encode Rho proteins, and 5 Rho proteins are identified in yeast, ten in worms, and 11 in fly genomes. The number of RhoGAPs is 2–3 times higher than the number of Rho GTPases. Some GAPs display broad specificity, while others are specific to a single Rho GTPase. For some of the GAPs, there are also differences in in-vitro and in-vivo specificity.
srGAPs and their cellular functions
RhoGAP family proteins, srGAP1 (Slit-Robo GAP1), srGAP2, srGAP3, and p115 contain N-terminal FCH (Fes/CIP4 (Cdc42 interacting protein4) homology) domain, a central RhoGAPs domain, and an SH3 domain in their highly conserved structures. srGAP1 protein is encoded by the SRGAP1 gene in humans. srGAP1 is known to be active in-vivo on Cdc42 and RhoA proteins while srGAP3 is primarily known to be active in-vitro on Rac1 and Cdc42. P115 causes the loss of stress fibers and downregulates RhoA. p115 binds with MEKK1 and provides a link between MEKK signaling and cytoskeleton maintenance inside the cell. srGAP1 interacts with Slit-Robo receptor protein through its SH3 domain. The contractile adherens junctions connect the cells of epithelial tissues to form a monolayer and the contractile force is more focused on E-cadherin-based adherens junction called zonula adherens. The interaction between actomyosin and zonula adherens regulates the cell–cell rearrangement during the process of morphogenesis. The scaffold protein cortactin and cellular signals such as the RhoA pathway regulates the assembly of F-actin and non-muscle Myosin II at the contractile adherens junction. The dephosphorylation of tyrosine residues of cortactin induces the recruitment of rho antagonist srGAP1 at zonula adherens leading to downregulation of RhoA signaling and contractibility. srGAP1 is present in a higher amount in sub-confluent epithelial cells adherens junction compared to confluent culture. srGAP1 RNAi is shown to restores the RhoA signaling and contractility in sub-confluent cell culture conditions [31]. srGAP1 has also been shown to act as a podocyte specific RhoGAP and it prevents the podocyte foot process effacement. srGAP1 ensures spatial restriction of the Rho GTPase protein activity of RAC1 and maintains morphologic plasticity in pathological conditions [32].
srGAPs and cancer
srGAP1 is involved in papillary thyroid carcinoma susceptibility. Linkage analysis and association studies had identified the SRGAP1 gene as a linkage peak candidate gene [33]. Slit2-Robo1 has been shown to inhibit the cell migration in colorectal cancer and its downstream molecule srGAP1 mediates the Slit2 anti-migration function through the inhibition of Cdc42. srGAP1 is remarkably low in colorectal cancer tissues and its diminished expression is correlated with high TNM stage, lymphatic invasion, poor survival, and poor differentiation [34]. SRGAP1 is shown to be highly expressed in gastric cancer cell lines and its knockdown inhibits cell proliferation, reduces colony formation, and suppresses cell invasion and migration. SRGAP1 knockdown was also shown to inhibit the Wnt signaling pathway and it was directly targeted by tumor suppressor miRNAs, miR-340 and miR-124. miR-340 and SRGAP1 were inversely correlated in samples of primary gastric cancer and overexpression of SRGAP1 restored the anticancer effects of miR-340 [35]. The expression of srGAP1 and srGAP2 is increased in hepatocellular carcinoma (HCC) relative to normal tissues as observed in Oncomine and TCGA datasets and srGAP2 is highly expressed at mRNA and protein levels. SRGAP2 level has also been found to be associated with HCC stages. srGAP2 has also been linked to cellular metabolic signaling [36]. srGAP3 has been shown to control the cytoskeleton (actin and microtubule) dynamics through downregulation of Rac. Depletion of srGAP3 by RNAi mediated inhibition leads to anchorage independent and Rac dependent growth of partially transformed human mammary epithelial cells and the expression of srGAP3 is found to be lower in most of the breast cancer cell lines. srGAP3 might be playing a tumor suppressor role through inhibition of Rac1. TET1 positively regulates srGAP3 and srGAP3 is needed for TET-mediated neuronal differentiation of Neuro2a cells. This link can be a useful target in neuroblastoma management [37]. The fusion of SRGAP3 and RAF1 genes is involved in posterior fossa pilocytic astrocytomas [38]. srGAP3 is involved in radiotherapy induced radio-resistance in murine squamous cell carcinoma [39]. The fusion of the SRGAP3-RAF1 genes is shown to activate the MAPK signaling pathway and phosphoinositide-3 kinase/mammalian target of rapamycin (PI3K/mTOR) signaling pathways in pediatric low grade gliomas [40].
srGAP2 have a metastasis suppressor role in osteosarcoma as evident from a study in murine osteosarcoma cell lines K12 and K7M2s. In early stage recurrence of triple-negative breast cancer cell adhesion/motility related gene SRGAP2 has been shown as one of the upregulated genes [41]. Neural stem cells renewal and differentiation are tightly regulated by SRGAP2 and FAM72 master genes and their dysregulation might lead to the transformation of cancer stem cells into glioblastoma multiforme [42]. The SNP (rs2580520) which is located at a predicted enhancer region of the SRGAP2 gene is frequently associated with a highly increased chance of breast cancer in Chinese women as observed in a study of recessive genetic model [43]. srGAP1 is downregulated in U87-IM3 and U251-IM3 glioblastoma multiforme cells and miR145 is shown to target and downregulate the srGAP1 [44]. miR-145 acts as a tumor suppressor by targeting Sox9 and adducin 3 in human glioma cells [45]. miR-145 induces invasion in glioblastoma multiforme and enhances the chemosensitivity of glioblastoma stem cells to demethoxycurucumin [46]. srGAP1 is shown to have a higher expression in prostate cancer cell lines that are castration-resistant while its expression is very low in androgen-sensitive prostate as well as normal prostate epithelial cells. srGAP1 is relatively overexpressed in castration-resistant prostate cancer tissues and androgen-sensitive LNCaP cells under androgen deprivation or Wnt stimulated condition show induced expression of srGAP1 [47]. srGAP2 localizes at the protrusions of the plasma membrane and cytoplasm and their knockdown leads to decreased cell–cell adhesion and increased cell migration but the effect on cell proliferation is negligible. Protein arginine methyltransferase 5 binds to the N terminal of srGAP2 and methylates Arg-927 and hence plays an active role in cell migration and dissemination through regulation of membrane protrusion [48]. The methylation mutant srGAP2 can’t localize to the plasma membrane and disturbs the F-BAR domain-mediated homodimerization of the srGAP2 and hence the arginine methylation can be involved in cell spreading and migration mediated by cell protrusions.
Conclusion
srGAPs play many important nonconventional roles especially in carcinogenesis and abnormal cellular pathology. The functions of srGAPs can be context-dependent and they might act as a tumor suppressor or oncogene depending on the upstream or downstream pathways and various regulatory factors that interact with them. Novel srGAP target molecules and their interacting partners are getting explored and many surprising and unexpected roles of srGAPs have been revealed. Elucidating the detailed mechanism and functions of srGAPs in cancer can provide novel target molecules for the treatment of advanced and metastatic cancers in the future.
Data availability
Not applicable.
References
Wennerberg K, Der CJ (2004) Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci 117:1301–1312. https://doi.org/10.1242/jcs.01118
Tcherkezian J, Lamarche-Vane N (2007) Current knowledge of the large RhoGAP family of proteins. Biol Cell 99:67–86. https://doi.org/10.1042/BC20060086
Pradhan R, Ngo PA, Martínez-Sánchez LDC et al (2021) Rho GTPases as key molecular players within intestinal mucosa and GI diseases. Cells 10:66. https://doi.org/10.3390/cells10010066
Rodrigues P, Macaya I, Bazzocco S et al (2014) RHOA inactivation enhances Wnt signalling and promotes colorectal cancer. Nat Commun 5:5458. https://doi.org/10.1038/ncomms6458
Kishore C, Sundaram S, Karunagaran D (2019) Vitamin K3 (menadione) suppresses epithelial-mesenchymal-transition and Wnt signaling pathway in human colorectal cancer cells. Chem Biol Interact 309:108725. https://doi.org/10.1016/j.cbi.2019.108725
Sprang S (2001) GEFs: master regulators of G-protein activation. Trends Biochem Sci 26:266–267. https://doi.org/10.1016/S0968-0004(01)01818-7
Scheffzek K, Ahmadian MR (2005) GTPase activating proteins: structural and functional insights 18 years after discovery. Cell Mol Life Sci 62:3014–3038. https://doi.org/10.1007/s00018-005-5136-x
Johnson JL, Erickson JW, Cerione RA (2009) New insights into how the Rho guanine nucleotide dissociation inhibitor regulates the interaction of Cdc42 with membranes. J Biol Chem 284:23860–23871. https://doi.org/10.1074/jbc.M109.031815
Gutierrez-Uzquiza A, Colon-Gonzalez F, Leonard TA et al (2013) Coordinated activation of the Rac-GAP β2-chimaerin by an atypical proline-rich domain and diacylglycerol. Nat Commun 4:1849. https://doi.org/10.1038/ncomms2834
Caloca MJ, Fernandez N, Lewin NE et al (1997) β2-chimaerin is a high affinity receptor for the phorbol ester tumor promoters *. J Biol Chem 272:26488–26496. https://doi.org/10.1074/jbc.272.42.26488
Lahoz A, Hall A (2013) A tumor suppressor role for srGAP3 in mammary epithelial cells. Oncogene 32:4854–4860. https://doi.org/10.1038/onc.2012.489
Marko TA, Shamsan GA, Edwards EN et al (2016) Slit-robo GTPase-activating protein 2 as a metastasis suppressor in osteosarcoma. Sci Rep 6:39059. https://doi.org/10.1038/srep39059
Xu G, Lu X, Huang T, Fan J (2016) ARHGAP24 inhibits cell cycle progression, induces apoptosis and suppresses invasion in renal cell carcinoma. Oncotarget 7:51829–51839. https://doi.org/10.18632/oncotarget.10386
Uehara S, Saito K, Asami H, Ohta Y (2017) Role of ARHGAP24 in ADP ribosylation factor 6 (ARF6)-dependent pseudopod formation in human breast carcinoma cells. Anticancer Res 37:4837–4844
Dai X, Geng F, Dai J, et al (2018) Rho GTPase activating protein 24 (ARHGAP24) regulates the anti-cancer activity of sorafenib against breast cancer MDA-MB-231 cells via the signal transducer and activator of transcription 3 (STAT3) signaling pathway. Med Sci Monit 24:8669–8677. https://doi.org/10.12659/MSM.911394
Koh S-B, Ross K, Isakoff SJ et al (2021) RASAL2 confers collateral MEK/EGFR dependency in chemoresistant triple-negative breast cancer. Clin Cancer Res 27:4883–4897. https://doi.org/10.1158/1078-0432.CCR-21-0714
Feng M, Bao Y, Li Z et al (2014) RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J Clin Invest 124:5291–5304. https://doi.org/10.1172/JCI76711
Imaoka H, Toiyama Y, Saigusa S et al (2015) RacGAP1 expression, increasing tumor malignant potential, as a predictive biomarker for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis 36:346–354. https://doi.org/10.1093/carcin/bgu327
Mi S, Lin M, Brouwer-Visser J et al (2016) RNA-seq identification of RACGAP1 as a metastatic driver in uterine carcinosarcoma. Clin Cancer Res 22:4676–4686. https://doi.org/10.1158/1078-0432.CCR-15-2116
del Maldonado M, Dharmawardhane S (2018) Targeting rac and Cdc42 GTPases in cancer. Cancer Res 78:3101–3111. https://doi.org/10.1158/0008-5472.CAN-18-0619
He Y, Northey JJ, Primeau M et al (2011) CdGAP is required for transforming growth factor β- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Oncogene 30:1032–1045. https://doi.org/10.1038/onc.2010.477
He Y, Northey JJ, Pelletier A et al (2017) The Cdc42/Rac1 regulator CdGAP is a novel E-cadherin transcriptional co-repressor with Zeb2 in breast cancer. Oncogene 36:3490–3503. https://doi.org/10.1038/onc.2016.492
McHenry PR, Sears JC, Herrick MP et al (2010) P190B RhoGAP has pro-tumorigenic functions during MMTV-Neu mammary tumorigenesis and metastasis. Breast Cancer Res 12:R73. https://doi.org/10.1186/bcr2643
Kang N, Matsui TS, Liu S et al (2020) Comprehensive analysis on the whole Rho-GAP family reveals that ARHGAP4 suppresses EMT in epithelial cells under negative regulation by Septin9. FASEB J 34:8326–8340. https://doi.org/10.1096/fj.201902750RR
Kazanietz MG, Caloca MJ (2017) The rac GTPase in cancer: from old concepts to new paradigms. Cancer Res 77:5445–5451. https://doi.org/10.1158/0008-5472.CAN-17-1456
Casado-Medrano V, Baker MJ, Lopez-Haber C et al (2018) The role of Rac in tumor susceptibility and disease progression: from biochemistry to the clinic. Biochem Soc Trans 46:1003–1012. https://doi.org/10.1042/BST20170519
Casado-Medrano V, Barrio-Real L, García-Rostán G, et al (2016) A new role of the Rac-GAP β2-chimaerin in cell adhesion reveals opposite functions in breast cancer initiation and tumor progression. Oncotarget 7:28301–28319. https://doi.org/10.18632/oncotarget.8597
Kasuya K, Nagakawa Y, Hosokawa Y et al (2016) RhoA activity increases due to hypermethylation of ARHGAP28 in a highly liver-metastatic colon cancer cell line. Biomedical Reports 4:335–339. https://doi.org/10.3892/br.2016.582
Chandra K (1969) Epigenetic regulation and promising therapies in colorectal cancer. Curr Mol Pharmacol 14:1–15
Lawson CD, Ridley AJ (2018) Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol 217:447–457. https://doi.org/10.1083/jcb.201612069
Liang X, Kiru S, Gomez GA, Yap AS (2018) Regulated recruitment of SRGAP1 modulates RhoA signaling for contractility during epithelial junction maturation. Cytoskeleton 75:61–69. https://doi.org/10.1002/cm.21420
Rogg M, Maier JI, Dotzauer R et al (2021) SRGAP1 controls small rho gtpases to regulate podocyte foot process maintenance. J Am Soc Nephrol 32:563–579. https://doi.org/10.1681/ASN.2020081126
He H, Bronisz A, Liyanarachchi S et al (2013) SRGAP1 is a candidate gene for papillary thyroid carcinoma susceptibility. J Clin Endocrinol Metab 98:E973–E980. https://doi.org/10.1210/jc.2012-3823
Feng Y, Feng L, Yu D et al (2016) srGAP1 mediates the migration inhibition effect of Slit2-Robo1 in colorectal cancer. J Exp Clin Cancer Res 35:191. https://doi.org/10.1186/s13046-016-0469-x
Huang T, Zhou Y, Zhang J et al (2018) SRGAP1, a crucial target of miR-340 and miR-124, functions as a potential oncogene in gastric tumorigenesis. Oncogene 37:1159–1174. https://doi.org/10.1038/s41388-017-0029-7
Li Y, Qiao L, Bai Y et al (2021) Identification of SRGAP2 as a potential oncogene and a prognostic biomarker in hepatocellular carcinoma. Life Sci 277:119592. https://doi.org/10.1016/j.lfs.2021.119592
Gao J, Ma Y, Fu H-L et al (2016) Non-catalytic roles for TET1 protein negatively regulating neuronal differentiation through srGAP3 in neuroblastoma cells. Protein Cell 7:351–361. https://doi.org/10.1007/s13238-016-0267-4
Forshew T, Tatevossian RG, Lawson ARJ et al (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181. https://doi.org/10.1002/path.2558
Nojiri K, Iwakawa M, Ichikawa Y et al (2009) The proangiogenic factor ephrin-A1 is up-regulated in radioresistant murine tumor by irradiation. Exp Biol Med 234:112–122. https://doi.org/10.3181/0806-RM-189
Jain P, Fierst TM, Han HJ et al (2017) CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 36:6348–6358. https://doi.org/10.1038/onc.2017.276
Tsai C-H, Chiu J-H, Yang C-W et al (2015) Molecular characteristics of recurrent triple-negative breast cancer. Mol Med Rep 12:7326–7334. https://doi.org/10.3892/mmr.2015.4360
Ho NTT, Rahane CS, Pramanik S et al (2021) FAM72, glioblastoma multiforme (GBM) and beyond. Cancers 13:1025. https://doi.org/10.3390/cancers13051025
Jiang Y, Shen H, Liu X et al (2011) Genetic variants at 1p112 and breast cancer risk: a two-stage study in Chinese women. PLoS One 6:e21563. https://doi.org/10.1371/journal.pone.0021563
Koo S, Martin G, Toussaint LG (2015) MicroRNA-145 promotes the phenotype of human glioblastoma cells selected for invasion. Anticancer Res 35:3209–3215
Rani SB, Rathod SS, Karthik S et al (2013) MiR-145 functions as a tumor-suppressive RNA by targeting Sox9 and adducin 3 in human glioma cells. Neuro Oncol 15:1302–1316. https://doi.org/10.1093/neuonc/not090
Qian C, Wang B, Zou Y et al (2019) MicroRNA 145 enhances chemosensitivity of glioblastoma stem cells to demethoxycurcumin. Cancer Manag Res 11:6829–6840. https://doi.org/10.2147/CMAR.S210076
Yokoyama NN, Sakai T, Sun Z et al (2014) Abstract 3314: Co-regulation of srGAP1 by Wnt and androgen receptor signaling in castration resistant prostate cancer. Cancer Res 74:3314–3314. https://doi.org/10.1158/1538-7445.AM2014-3314
Guo S, Bao S (2010) srGAP2 arginine methylation regulates cell migration and cell spreading through promoting dimerization. J Biol Chem 285:35133–35141. https://doi.org/10.1074/jbc.M110.153429
Acknowledgements
Not applicable.
Funding
There was no funding for this work.
Author information
Authors and Affiliations
Contributions
VJ wrote the original manuscript, CK edited the final manuscript and supervised the writing. Both the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
VJ declares that she has no conflict of interest. CK declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
