
Abstract
Advanced glycation end products (AGEs) are formed from the non-enzymatic glycation reaction of reducing sugars or their metabolites with the free amino groups of several biomolecules and are known to play pathophysiological roles in various inflammatory diseases. In an earlier study, it was suggested that tumor necrosis factor-like weak inducer of apoptosis (TWEAK) has a unique role to regulate the tumor necrosis factor α (TNFα)-induced inflammatory response. In this study, we investigated the effect of the AGEs–TWEAK interaction on proinflammatory signaling responses in endothelial cells and the influence of AGEs on the cellular function of TWEAK in the inflammatory process. The effect of AGEs on the TWEAK/TNFα-induced gene expression of interleukin-8 (IL-8) was determined by real-time RT-PCR in endothelial-like EA.hy.926 cells. The pull-down assay was performed using recombinant His-tagged TWEAK and AGEs. The NF-κB activation was analyzed by Western blotting with canonical and non-canonical pathway-specific antibodies. AGEs dose-dependently inhibited TWEAK-induced IL-8 gene expression, whereas AGEs themselves had almost no effect on IL-8 expression. AGEs were found to bind directly to TWEAK in the pull-down assay. TNFα-induced IL-8 production and canonical NF-κB activation were suppressed by TWEAK pretreatment, whereas TWEAK-induced non-canonical NF-κB activation was enhanced by pretreatment. These effects induced by TWEAK pretreatment were abolished by the co-addition of AGEs. Our findings suggest that AGEs attenuate the function of TWEAK to regulate the TNFα-induced inflammatory responses, which provide important clues for understanding the significance of the AGEs–TWEAK interaction in inflammatory processes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Advanced glycation end products (AGEs) are known as heterogeneous glycation products produced non-enzymatically from reducing sugar or its metabolites with the free amino groups of various biomolecules. AGEs are classified into subtypes based on the derived molecules, and the properties are suggested to differ among the subtypes [1,2,3,4]. AGEs have been reported to be produced in vivo, and modifications by AGEs induce dysfunction in several functional biomolecules [5,6,7,8,9]. In addition, the accumulation of AGEs has been found to be correlated with natural aging and the severity of several diseases such as diabetes, cardiovascular diseases, and Alzheimer’s disease [10,11,12,13]. Although the mechanism by which AGEs contribute to the progression of these diseases is still unclear, the interaction between AGEs and their endogenous receptor, receptor for AGEs (RAGE), has been implicated in AGE-induced pathogenesis. It has been shown that several intracellular signaling pathways are activated via the AGEs–RAGE interaction [14]. These pathways eventually lead to the activation of nuclear factor-kappa B (NF-κB) complexes, which are responsible for the production of several proinflammatory cytokines including interleukin-8 (IL-8) [15], and the generation of reactive oxygen species [14]. These findings suggest the hypothesis that the AGEs–RAGE interaction induces “sterile” inflammation, and the prolongation of this phenomenon may lead to the inflammatory diseases described above. In addition to RAGE, however, there is little known about the existence of extracellular factors which interact with AGEs at present. Therefore, it remains unknown whether AGEs are related with the progression of inflammatory diseases other than through the AGEs–RAGE interaction.
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) is a recently identified multi-functional cytokine [16]. TWEAK is initially expressed as a transmembrane protein and is released as a soluble form by selective processing [17]. Several reports suggest that the changes in serum concentration of soluble TWEAK correlate with the severity of atherosclerosis, diabetes, heart failure, and some autoimmune diseases [18,19,20,21]. Fibroblast growth factor-inducible 14 (Fn14) has been identified as a receptor for TWEAK, and the TWEAK–Fn14 system is thought to be involved in the above noted diseases [22, 23]. In several cells, including endothelial cells, TWEAK has been reported to activate several cellular signaling pathways and to change the expression of various inflammation-related genes, suggesting the involvement of TWEAK in inflammation-related diseases [23]. Also, tumor necrosis factor α (TNFα) is well known to function as a master regulator of inflammatory responses [24], and an excess of TNFα is thought to cause some inflammatory diseases such as rheumatoid arthritis. Interestingly, TWEAK was reported to have roles in modulating TNFα-induced changes in cellular signaling processes [25], suggesting that TWEAK exerts its effects by regulating excessive inflammatory responses.
As noted above, there remains the possibility that AGEs function other than through AGEs–RAGE interactions. To investigate this possibility, we hypothesized that AGEs may interact with the extracellular modulators for inflammatory responses, such as TWEAK, and alter their functions. In the present study, we focused on the possibility that AGEs alter TWEAK-driven regulation against the inflammatory responses induced by TNFα. To test this possibility, we investigated the interaction between AGEs and TWEAK and examined some changes in TNFα-induced proinflammatory responses, including IL-8 expression, and NF-κB activation induced by this interaction in endothelial cells.
Materials and methods
Materials
Recombinant human TWEAK, TNFα, Human IL-8 (CXCL8) mini ABTS ELISA development kits, and ABTS ELISA buffer kits were purchased from PeproTech (Rocky Hill, NJ, USA). Recombinant human IL-8 was from Shenandoah Biotechnology (Warwick, PA, USA). Cobalt-coated magnetic beads were from Life Technologies (Gaithersburg, MD, USA). Cobalt-coated agarose beads and In-Fusion HD cloning kits were from Clontech Laboratories (Mountain View, CA, USA). Protein G magnetic beads were from Bio-Rad Laboratories (Hercules, CA, USA). Horseradish peroxidase-linked anti-rat IgG antibody and Amersham Hybond PVDF were from GE Healthcare (Waukesha, WI, USA). Horseradish peroxidase-linked anti-rabbit IgG antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Horseradish peroxidase-linked anti-mouse IgG antibody, horseradish peroxidase-linked anti-His-tag antibody, and horseradish peroxidase-linked anti β-actin antibody were from Medical & Biological Laboratories (Nagoya, Japan). Anti-human IκBα and anti-human p100/p52 antibody were from Cell Signaling Technology (Beverly, MA, USA). Anti-human IL-8 antibody was from R&D systems (Minneapolis, MN, USA). Luminata Forte Western HRP Substrate was from Millipore (Billerica, MA, USA). pCold vectors, Primestar HS DNA Polymerase, and Ex Taq were from Takara Bio (Kusatsu, Japan). Total RNA purification kits were from GMbiolab (Taichung, Taiwan). ReverTra Ace qPCR RT Master Mix with gDNA Remover, Thunderbird qPCR mix, and Can Get Signal Immunoreaction Enhancer Solution were from Toyobo (Osaka, Japan). Pierce High Capacity Endotoxin Removal Spin Column was from Pierce Biotechnology (Rockford, IL, USA). ToxinSensor Gel Clot Endotoxin Assay single test kits were from GenScript (Piscataway, NJ, USA). Other chemicals used were of analytical grade and obtained from standard sources.
The glyceraldehyde-modified bovine serum albumin (AGE-2) was prepared as previously described [4, 5]. Briefly, 25 mg/ml BSA was incubated under sterile conditions with 100 mM glyceraldehyde in 200 mM sodium phosphate buffer (pH 7.4) at 37 °C for 7 days. Then the reaction mixture was dialyzed against phosphate-buffered saline (PBS) for 2 days at 4 °C and used as AGE-2 in this study. The endotoxin content in the AGE-2 preparation was confirmed to be less than 1 endotoxin unit (EU)/ml. A monoclonal antibody specific for AGE-2 was made in our laboratory using the standard cell fusion technique.
Construction of expression vectors
In order to construct N-terminally His-tagged human TNFα and TWEAK expression vectors, the cDNAs encoding TNFα and TWEAK were amplified by PCR with the cDNA prepared from human THP-1 cells and a synthetic TWEAK gene with a sequence optimized for E. coli codon usage constructed by Eurofins Genomics (Tokyo, Japan). The forward and reverse primers used for this purpose were as follows: TNFα, 5′- AGAGGTAATACCATATGCATCATCATCATCATCATAAAGGCCGCAAAACCCGTG -3′ and 5′- GTACCGAGCTCCATATTAATGAACCTGGAACAGACCGAAATAGG -3′; TWEAK, 5′- ATCATCATCATCATATGGTCAGATCATCTTCTCGAACC -3′ and 5′- GTACCGAGCTCCATATCACAGGGCAATGATCCCAAAG -3′. PCR was carried out with Primestar HS DNA Polymerase or Ex Taq. The obtained DNA fragments were subcloned into the corresponding sites of pCold II (for TNFα expression) or IV (for TWEAK expression) vectors using the In-Fusion HD cloning kit. The sequences of all constructs were verified by DNA sequencing (Eurofins Genomics).
Expression and purification of recombinant proteins and pull-down assays
The expression vectors were introduced into the E. coli BL21 (DE3). Expression of the recombinant proteins was induced according to the manufacturer’s instructions. Twenty-four hours after induction of recombinant protein expression, the cells were collected by centrifugation. The cells were then suspended in wash buffer (50 mM sodium phosphate, 300 mM NaCl, pH 8) or denaturing buffer (50 mM sodium phosphate, 300 mM NaCl, 6 M guanidine-HCl, pH 8), sonicated, and then centrifuged to remove insoluble cellular debris.
For the His-tagged recombinant protein-based pull-down assays, the cells suspended in the wash buffer were utilized. The endotoxin in the soluble fractions of the cells was removed by Endotoxin Removal Spin Column, and its amount was confirmed to be less than 1 EU/μg. For the pull-down assays, His-tagged recombinant proteins were purified using cobalt-coated magnetic beads according to the manufacturer’s instructions. These proteins bound to beads were incubated with AGE-2 in wash buffer at room temperature for 30 min. The beads were then washed in wash buffer, and proteins bound to the beads were lysed in 1 × Laemmli sample buffer (2% SDS, 5% 2-mercaptoethanol, 5% sucrose, 62.5 mM Tris–HCl, pH 6.8, and 0.002% bromophenol blue). The protein samples were analyzed by Western blotting. Prior to Western blotting, using SDS-PAGE followed by Coomassie Brilliant Blue staining, it was confirmed that the same amount of His-tagged recombinant proteins was used for each pull-down assay.
For the AGE-2-based pull-down assays, the cells suspended in the denaturing buffer were utilized. His-tagged recombinant proteins in the soluble fractions of the cells were purified using cobalt-coated agarose beads according to the manufacturer’s instructions. The purity of the purified proteins was examined by SDS-PAGE followed by Coomassie Brilliant Blue staining. The purified fractions containing recombinant protein were dialyzed against PBS at 4 °C overnight. The endotoxin included in the fractions was then removed by Endotoxin Removal Spin Column, and its amount was confirmed to be less than 1 EU/μg. For the pull-down assays, protein G magnetic beads were incubated with anti-AGE-2 antibody at room temperature for 30 min. Anti-AGE-2 antibody bound to protein G magnetic beads was then incubated with AGE-2 followed by His-tagged recombinant proteins or IL-8 recombinant protein at room temperature for 1 h, respectively. The proteins bound to the beads were lysed in 1 × Laemmli sample buffer and were analyzed by Western blotting.
Cell culture and ELISA
EA.hy.926 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum at 37 °C in humidified air containing 5% CO2. To measure IL-8 secreted from the cells, culture medium was collected and centrifuged for 17,500×g at 4 °C for 10 min, and then the supernatants were diluted 20-fold with PBS containing 0.05% Tween-20 and 0.1% BSA. The concentration of IL-8 in the supernatant was measured by ELISA using Human IL-8 (CXCL8) mini ABTS ELISA development kits. The optical density of each sample at 405 nm was measured using Varioskan LUX Multimode Microplate Reader (Thermo Scientific, Rockford, IL, USA), and then the values of optical density were corrected by subtracting at 650 nm, according to the manufacturer’s instructions.
Western blotting
The cells were washed with ice-cold PBS and then directly lysed in 1 × Laemmli sample buffer. After sonication, protein samples were boiled for 5 min at 95 °C. The samples were separated by SDS-PAGE and electrotransferred to a hydrophobic polyvinylidene difluoride membrane. The membrane was blocked with Tris-buffered saline (pH 7.5) containing 3% dried milk and 0.1% Tween-20 and then incubated with primary antibodies in Can Get Signal Solution 1 at 4 °C overnight, followed by incubation with horseradish peroxidase-labeled secondary antibodies in Can Get Signal Solution 2 at room temperature for 1 h. Proteins were finally treated with a Luminata Forte Western HRP substrate and visualized with the aid of a LAS-1000plus lumino-imaging analyzer (Fuji Film, Tokyo, Japan). The densities of the bands detected in the Western blotting were calculated using ImageJ (version 1.49, U. S. National Institutes of Health).
Semi-quantitative real-time RT-PCR
Total RNAs were isolated from EA.hy.926 cells cultured under each condition using total RNA isolation kits. The concentration and purity of the extracted total RNA were assessed using the 260/280 nm absorbance ratio. cDNAs were prepared from 500 ng of total RNA using ReverTra Ace qPCR RT Master Mix with gDNA Remover. Real-time PCR was performed using Thunderbird qPCR mix with the aid of Thermal Cycler Dice Real-Time System Lite (Takara Bio). The forward and reverse primers used were as follows: human IL-8, 5′- ACTGAGAGTGATTGAGAGTGGAC -3′ and 5′- AACCCTCTGCACCCAGTTTTC -3′; human β-actin, 5′ - GGACTTCGAGCAAGAGATGG -3′ and 5′- AGGAAGGAAGGCTGGAAGAG -3′. The PCR conditions were as follows: denaturation at 95 °C for 5 s and annealing and extension at 60 °C for 30 s (40 cycles). The transcript level of IL-8 was normalized against the level of β-actin with the ddCT method [26].
Statistical analyses
The statistical analysis across multiple groups was determined using Dunnett’s test. The statistical differences between paired groups were determined using Student’s t test. Analyses were performed using R (version 3.1.3, The R Foundation for Statistical Computing). P values <0.05 were considered significant. All data were presented as the mean ± standard error.
Results
TWEAK induces expression of inflammatory cytokine IL-8 in EA.hy.926 cells, whereas AGE-2 induces negligible responses
To investigate whether AGEs alter TWEAK function to regulate TNFα-mediated cellular responses, we utilized the human endothelial-like EA.hy.926 cells that expressed Fn14, the specific receptor for TWEAK. Firstly, we examined the responsiveness of the cells to TWEAK. For this purpose, the cells were stimulated by 100 ng/ml of the recombinant soluble form of TWEAK and then changes in some gene expressions were examined by RT-PCR. As a result, the expression of proinflammatory cytokine IL-8 was found to be increased in the cells stimulated by TWEAK (data not shown). We therefore examined this response using semi-quantitative real-time RT-PCR in detail. Consistent with the results of the RT-PCR, the mRNA level of IL-8 was increased by the addition of 100 ng/ml TWEAK (Fig. 1a). In contrast, AGE-2 induced only a slight change in the mRNA level of IL-8, even when it was added at a concentration of 1000 μg/ml (Fig. 1b). So far, it has been reported that TWEAK functions to regulate TNFα-mediated cellular responses [25]. We therefore examined the effect of TWEAK on the TNFα-mediated IL-8 gene expression in EA.hy.926 cells. The results showed that TNFα strongly induces IL-8 gene expression, and co-addition of TWEAK potentiates this response (Fig. 1a).

IL-8 gene expression stimulated by TWEAK, TNFα, AGE-2, and BSA. EA.hy.926 cells were treated for 6 h with molecules as follows: TWEAK (100 ng/ml); TNFα (50 ng/ml); TNFα (50 ng/ml) plus TWEAK (100 ng/ml); AGE-2 (0.001, 0.1, 10, and 1000 μg/ml); BSA (1000 μg/ml). Data are shown as ratios to the IL-8 gene expression level in the cells treated with vehicle as a control. *** P < 0.001 versus control by Dunnett’s test (n = 3)
AGE-2 inhibits the IL-8 gene expression induced by TWEAK but not by TNFα
We next examined the effect of AGE-2 on TWEAK-induced IL-8 gene expression. EA.hy.926 cells were stimulated by TWEAK in the presence of various concentrations of AGE-2 (0.001–1000 μg/ml) for 6 h, and then semi-quantitative real-time RT-PCR for IL-8 was carried out using total RNA from these cells. The results clearly showed that AGE-2 suppressed TWEAK-induced IL-8 gene expression in a dose-dependent manner (Fig. 2a). Similarly, this suppressive effect was found in the cells stimulated by TNFα plus TWEAK, but the suppressive rate of IL-8 gene expression in these cells was lower than that in the cells treated with TWEAK alone (Fig. 2b). On the other hand, AGE-2 did not alter the IL-8 gene expression induced by TNFα alone (Fig. 3c). These results suggested that the suppressive effect of AGE-2 was specific to TWEAK but not to TNFα.

Differences of IL-8 gene expression stimulated by TWEAK, TWEAK plus TNFα, and TNFα in the presence or absence of AGE-2. EA.hy.926 cells were treated for 6 h with molecules as follows: a TWEAK (100 ng/ml) together with or without AGE-2 (0.001, 0.1, 10, and 1000 μg/ml) or BSA (1000 μg/ml). b TNFα (50 ng/ml) plus TWEAK (100 ng/ml) together with or without AGE-2 (0.001, 0.1, 10, and 1000 μg/ml) or BSA (1000 μg/ml). c TNFα (50 ng/ml) together with or without AGE-2 (0.001, 0.1, 10, and 1000 μg/ml) or BSA (1000 μg/ml). Data are shown as ratios to the IL-8 gene expression level in the cells treated with TWEAK (a), TNFα plus TWEAK (b), or TNFα (c) as a control. *** P < 0.001 versus control by Dunnett’s test (n = 3)

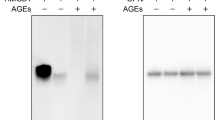
Interaction between AGE-2 and TWEAK/TNFα. Essentially the same amount of His-tagged TWEAK (His-TWEAK) or His-tagged TNFα (His-TNFα) immobilized on cobalt-coated beads was incubated with 1000 μg/ml AGE-2. The amounts of AGE-2 bound to the beads (Lanes 1–3) or AGE-2 alone (500 ng, Lane 4) were visualized by Western blotting using anti-AGE-2 antibody
AGE-2 directly binds to TWEAK
To clarify the suppressive effect of AGE-2 on TWEAK-induced IL-8 gene expression in more detail, we next examined whether AGE-2 directly interacts with TWEAK. Recombinant N-terminally His-tagged TWEAK or TNFα were obtained using the E. coli expression system. These His-tagged proteins were immobilized on cobalt-coated beads, and then incubated with AGE-2. After the incubation, the amounts of AGE-2 bound to the His-tagged TWEAK or TNFα were examined by Western blotting using anti-AGE-2 antibody. As shown in Lane 4 of Fig. 3, the AGE-2 used in the present study was exhibited as a complex molecular species with broad molecular weights. A similar blotting pattern was obtained only in the sample eluted from the His-tagged TWEAK-immobilized beads (Fig. 3, Lane 1). In contrast, AGE-2 molecules were hardly detectable in the eluate from the His-tagged TNFα-immobilized beads (Fig. 3, Lane 2). These results raised the possibility that AGE-2 selectively bound to TWEAK and may have suppressed the function of TWEAK.
AGE-2 inhibits the priming effect of TWEAK to suppress TNFα-induced IL-8 gene expression and NF-κB activation
To further explore the pathophysiological significance of the suppressive effect of AGE-2 on TWEAK function, we examined the effect of AGE-2 on the regulatory function of TWEAK in TNFα-stimulated cellular responses. In an earlier study, it was described that pretreatment by TWEAK suppressed TNFα-stimulated cellular responses [25]. We therefore examined whether this effect occurs in EA.hy.926 cells. The cells were pretreated with TWEAK for 30 h and then these primed cells were stimulated by TNFα for 6 h in the presence of TWEAK. As a result, TNFα-stimulated IL-8 gene expression showed about 20% reduction by TWEAK pretreatment, as compared with no pretreatment (Fig. 4). Furthermore, when the cells were primed by TWEAK together with AGE-2, the priming effect of TWEAK was almost completely abolished (Fig. 4). These results showed that the priming by TWEAK suppressed TNFα-stimulated IL-8 gene expression in EA.hy.926 cells, and AGE-2 was able to reverse the priming effect of TWEAK.

The changes of TNFα-induced IL-8 gene expression in EA.hy.926 cells pretreated with TWEAK together with or without AGE-2. Initially, the cells were pretreated for 30 h with or without molecules as follows: TWEAK (100 ng/ml); AGE-2 (1000 μg/ml); BSA (1000 μg/ml). Then the cells were treated for 6 h with or without molecules as follows: TNFα (50 ng/ml), TWEAK (100 ng/ml), AGE-2 (1000 μg/ml), BSA (1000 μg/ml). Data are shown as ratios to the IL-8 gene expression level in the cells treated with TNFα alone for 6 h as a control. ** P < 0.01; *** P < 0.001 versus control by Dunnett’s test (n = 3)
In addition, under the same conditions, we examined the changes in NF-κB activities. The cells were lysed using Laemmli sample buffer and then the extracted proteins were analyzed by Western blotting. In Fig. 5a, the protein level of IκBα, a key regulator of the canonical NF-κB pathway, was up-regulated in the cells which were primed by TWEAK followed by TNFα-stimulation. Because IκBα is degraded in the course of canonical NF-κB activation, this up-regulation indicates suppression of the canonical NF-κB pathway. In contrast, the protein level of p52, a key regulator of the non-canonical NF-κB pathway, was up-regulated in the cells which were primed by TWEAK followed by TNFα-stimulation (Fig. 5b), indicating activation of the non-canonical NF-κB pathway. These changes in NF-κB activities were suppressed by the co-addition of AGE-2 (Figs. 5a, b). Taken together, similar to the changes in IL-8 gene expression shown in Fig. 4, the priming effect of TWEAK on NF-κB activations was also reversed by addition of AGE-2.

Activation of TNFα-stimulated NF-κB pathway in the cells pretreated with TWEAK together with or without AGE-2. EA.hy.926 cells were pretreated under the same conditions described in Fig. 4. After the pretreatment, the cells were treated for 7.5 min (a) or 6 h (b) with or without molecules as follows: TNFα (50 ng/ml), TWEAK (100 ng/ml), AGE-2 (1000 μg/ml), BSA (1000 μg/ml). An aliquot of total protein (10 μg) extracted from each cell was analyzed using Western blotting, and the densities of the bands were calculated. The density values, which represent the protein content of IκBα (a) or p52 (b), were normalized against the value of β-actin. Data are shown as ratios to the IκBα (a) or p52 (b) protein level in the cells treated with TNFα alone as a control. Each Western blot is representative of three independent experiments.*P < 0.05; **P < 0.01; ***P < 0.001 versus control by Dunnett’s test (n = 3)
Furthermore, to support the above findings, we measured IL-8 production in the cell culture medium using ELISA. In Fig. 6, compared with the cells stimulated by TNFα alone, the levels of IL-8 protein were increased in the media of the cells stimulated by TNFα together with TWEAK for 18 h. In contrast, pretreatment with TWEAK suppressed TNFα-stimulated IL-8 production (Fig. 6). On the other hand, the lowest IL-8 protein level was observed under the condition with priming by TWEAK together with AGE-2, indicating discordance between IL-8 gene expression and protein secretion (Figs. 4, 6).

IL-8 protein secreted from the TNFα-stimulated cells pretreated with TWEAK together with or without AGE-2. EA.hy.926 cells were pretreated under the same conditions described in Fig. 4. After the pretreatment, the cells were treated for 18 h with or without molecules as follows: TNFα (50 ng/ml), TWEAK (100 ng/ml), AGE-2 (1000 μg/ml), BSA (1000 μg/ml). Then the culture medium was analyzed using IL-8 ELISA. *P < 0.05 versus IL-8 protein concentration in the culture medium of the cells treated with TNFα alone by Dunnett’s test (n = 3)
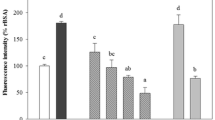
To clarify the cause of this discordance, we examined the influence of AGE-2 on the IL-8 ELISA. Serial dilutions of standard IL-8 proteins were prepared with or without 1000 μg/ml of AGE-2, which was the same concentration as that used in the cell culture analysis in Fig. 6, and then these samples were evaluated on the IL-8 ELISA. The results indicated that the level of IL-8 was underestimated by ELISA about 20% by the co-existence of 1000 μg/ml of AGE-2 (Fig. 7).

Effect of AGE-2 on IL-8 ELISA. Serial dilutions of standard IL-8 proteins were prepared without (white bar) or with (black bar) 1000 μg/ml of AGE-2 and were analyzed by IL-8 ELISA. ***P < 0.001; **P < 0.01; *P < 0.05 by Student’s t test at each concentration of IL-8
IL-8 also binds to AGE-2
To confirm the interfering effect of AGE-2 on the IL-8 ELISA measurement in more detail, pull-down assays using AGE-2 and IL-8 were carried out. AGE-2 immobilized on the beads was incubated with IL-8, His-tagged TWEAK, or His-tagged TNFα. After the incubation, proteins bound to AGE-2 were examined by Western blotting using anti-His-tag antibody or anti-IL-8 antibody, respectively. As shown in Fig. 8a, TWEAK, but not TNFα, was shown to bind to AGE-2, indicating the same results described in Fig. 3. Similarly, IL-8 also bound to AGE-2 under the same condition (Fig. 8b), suggesting that this interaction between IL-8 and AGE-2 was responsible for the underestimation in the IL-8 ELISA measurement.

Interaction between IL-8 and AGE-2. Anti-AGE-2 antibody immobilized on beads was incubated with AGE-2 followed by His-tagged TNFα (25 ng/μl, His-TNFα), His-tagged TWEAK (25 ng/μl, His-TWEAK), or IL-8 (10 ng/μl). Molecules bound to beads were analyzed by Western blotting using anti-His-tag antibody (a) or anti-IL-8 antibody (b), respectively
Discussion
AGEs are known to play pathophysiological roles in various inflammatory diseases [10,11,12,13]. TWEAK was reported to regulate TNFα-induced cellular responses [25]. However, at present, very little is actually known about the existence of extracellular factors influenced by AGEs. In this study, we investigated the effect of the AGEs–TWEAK interaction on proinflammatory signaling responses and the influence of AGEs on the cellular function of TWEAK.
Initially, we examined whether the presence of AGE-2 alters the proinflammatory IL-8 gene expression induced by TWEAK. The results clearly showed that AGE-2 suppressed the expression of IL-8 in a dose-dependent manner (Fig. 2a). Earlier studies indicated that the serum concentration of AGE-2 is 10–20 μg/ml in non-alcoholic steatohepatitis or diabetes patients [27, 28], and this range of AGE-2 was shown enough to interfere with the TWEAK function (Fig. 2a). On the other hand, local accumulation of AGEs was observed in pathological conditions [29], suggesting that some local environment may be exposed to more high concentrations of AGE-2. Therefore, we selected 1000 μg/ml of AGE-2 as the highest concentration of AGE-2 which has little effect on EA.hy.926 cells in the subsequent investigations.
According to the finding described above, AGE-2 appears to regulate the TWEAK-induced inflammatory response. However, the magnitude of IL-8 gene expression induced by TWEAK is relatively small compared with that induced by TNFα (Fig. 1a). This difference may be accounted for by the difference in the class of NF-κB activation pathway induced by TWEAK or TNFα, which has already been suggested [23, 30]. Namely, TNFα activates the canonical NF-κB pathway, resulting in IL-8 production, whereas TWEAK mainly activates the non-canonical pathway. As described above, TWEAK weakly induces inflammatory cytokine IL-8 in EA.hy.926 cells. On the other hand, TWEAK was also reported to function as the modulator of TNFα-stimulated cellular signaling [25, 31, 32]. In these reports, the pretreatment with TWEAK attenuated the TNFα-induced cellular responses. TWEAK and TNFα-induced signaling pathways share the same signal transduction molecule, TNF receptor-associated factor 2 (TRAF2). Therefore, in the cells pretreated with TWEAK, TRAF2 has been consumed for TWEAK-induced signal transduction, and this depletion of TRAF2 attenuates subsequent TNFα-induced signaling. The physiological significance of this molecular event seems to protect cells from TNFα-induced excessive inflammatory reactions. In the present study, TNFα-stimulated IL-8 gene expression is additionally increased by concomitant stimulation with TWEAK, while it was significantly suppressed by pretreatment with TWEAK in EA.hy.926 cells, suggesting a similar molecular event occurred (Fig. 4). In IL-8 gene expression experiments, the effect of the TWEAK pretreatment was completely abolished by AGE-2 co-existing with TWEAK (Fig. 4). However, similar abolishment was not observed in IL-8 protein measurement by ELISA, exhibiting a significant discordance for IL-8 gene expression (Fig. 6). As shown in Fig. 7, this difference between mRNA expression and protein production of IL-8 was thought to be caused by AGE-2 co-existing in the ELISA samples. If we tentatively re-calculate the results shown in Fig. 6 by assuming the data shown in Fig. 7 as a standard curve, the amount of secreted IL-8 from EA.hy.926 cells pretreated with TWEAK in the presence of AGE-2 was estimated to be increased about 1.2-fold; this estimation supports the experimental data regarding the changes of IL-8 gene expression shown in Fig. 4. Taken together, our findings suggest that the co-existence of AGE-2 led to dysfunction in the regulatory action of TWEAK on TNFα-mediated inflammatory responses.
This hypothesis agrees with the earlier study that genetic ablation or antibody neutralization of TWEAK augmented endotoxin-induced proinflammatory cytokine production in immune cells [33]. Furthermore, similar to our findings, TWEAK was reported to suppress TNFα-stimulated IL-6 and IL-8 production in fibroblast-like synoviocytes, suggesting that TWEAK may have a role in inhibiting the progression of TNFα-activated rheumatoid arthritis [34]. In addition to rheumatoid arthritis, TNFα is well known to play a crucial role in the formation of several chronic inflammatory diseases, including atherosclerosis caused by endothelial dysfunction [35]. As shown in the present study, if TWEAK has a protective role in inhibiting TNFα-induced endothelial dysfunction, AGEs accumulated during the inflammatory process may impair this protective role of TWEAK and are considered to be involved in the exacerbation of endothelial dysfunction. The pathophysiological involvement of TWEAK in several inflammatory diseases has been suggested [23], but the relationship between TWEAK and TNFα-induced cellular dysfunction is rarely investigated. Therefore, to generalize our findings describing the impairment of TWEAK functions by AGE-2, it remains a challenge for future investigation to elucidate the possible involvement of TWEAK in TNFα-induced chronic inflammatory diseases.
Figure 5 shows the effect of the AGE-2–TWEAK interaction on TNFα-stimulated NF-κB activation. Similar to an earlier study [25], TWEAK pretreatment suppressed TNFα-stimulated canonical NF-κB activation in EA.hy.926 cells, and the co-existence of AGE-2 inhibited this effect (Fig. 5a). Analogous to other proinflammatory cytokines, IL-8 gene expression is known to be controlled under the canonical NF-κB pathway [23]. Therefore, our findings showing the effect of AGE-2 on the regulation of IL-8 production may be applied to other proinflammatory cytokine responses in several types of cells. In addition, it was also shown that AGE-2 suppressed TWEAK-mediated activation of the non-canonical NF-κB pathway (Fig. 5b). As described above, the canonical NF-κB pathway is known to induce the expression of some genes related to inflammatory reactions, whereas little is known about the target genes of non-canonical pathway activation. Therefore, the importance of TWEAK-induced non-canonical activations is poorly understood at present, and thus further investigations are needed to elucidate the physiological significance of alteration in this pathway mediated by AGE-2.
The pull-down assay indicated that AGE-2 specifically interfered with TWEAK function by direct binding between AGE-2 and TWEAK (Figs. 3, 8). Several endogenous molecules are known to interact with AGEs. Many of these molecules were classified as cell surface receptors, such as RAGE and CD36 [36,37,38], and were known to play roles as ligands in intracellular signaling or scavengers for AGEs, whereas there is little known about the possible interaction of AGEs with extracellular biomolecules. Our findings indicated the role of AGEs to alter the functions of TWEAK or IL-8 through direct interaction with them. These findings suggested that some other extracellular signaling biomolecules, including cytokines and/or hormones, may also interact with AGEs to interfere with their functions. To date, the pathophysiological functions of AGEs have been discussed mainly through the AGEs–RAGE interaction [7,8,9, 14]. Therefore, if AGEs alter the functions of signaling biomolecules involved in inflammatory responses, our hypothesis based on direct interaction between AGEs and extracellular biomolecules will provide novel mechanisms by which AGEs evoke inflammatory diseases.
Conclusions
In the present study, we demonstrated that AGE-2 attenuated TWEAK-induced down-regulation of TNFα-stimulated proinflammatory IL-8 production and NF-κB activation. In addition, this effect was suggested to be caused by direct binding between AGE-2 and TWEAK. Our findings provide a new clue to elucidate the pathophysiological roles of AGEs for prevention and treatment of some inflammatory diseases.
References
Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Buenting C, Heitmann KVH (1997) Orally absorbed reactive glycation products, glycotoxins. An environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci U S A 94:6474–6479. doi:10.1073/pnas.94.12.6474
Cerami C, Founds H, Nicholl I et al (1997) Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci 94:13915–13920. doi:10.1073/pnas.94.25.13915
Takeuchi M, Yamagishi S (2004) TAGE (toxic AGEs) hypothesis in various chronic diseases. Med Hypotheses 63:449–452. doi:10.1016/j.mehy.2004.02.042
Takahashi H, Mori S, Wake H et al (2009) Advanced glycation end products subspecies-selectively induce adhesion molecule expression and cytokine production in human peripheral blood mononuclear cells. J Pharmacol Exp Ther 330:89–98. doi:10.1124/jpet.109.150581.potential
Takeuchi M, Makita Z, Bucala R et al (2000) Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol Med 6:114–125
Henning C, Smuda M, Girndt M et al (2011) Molecular basis of maillard amide-advanced glycation end product (AGE) formation in vivo. J Biol Chem 286:44350–44356. doi:10.1074/jbc.M111.282442
Monnier VM, Cerami A (1981) Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science 211:491–493
Kaji Y, Usui T, Oshika T et al (2000) Advanced glycation end products in diabetic corneas. Invest Ophthalmol Vis Sci 41:362–368
Saito M, Marumo K (2010) Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int 21:195–214
Aso Y, Inukai T, Tayama K et al (2000) Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol 37:87–92
Kanauchi M, Tsujimoto N, Hashimoto T (2001) Advanced glycation end products in nondiabetic patients with coronary artery disease. Diabetes Care 24:1620–1623
Li SY, Du M, Dolence EK et al (2005) Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 4:57–64. doi:10.1111/j.1474-9728.2005.00146.x
Srikanth V, Maczurek A, Phan T et al (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32:763–777. doi:10.1016/j.neurobiolaging.2009.04.016
Xie J, Méndez JD, Méndez-Valenzuela V et al (2013) Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal 25:2185–2197
Lawrence T (2009) The nuclear factor NF-kappa B pathway in inflammation. Cold Spring Harb Perspect Biol 1:1–10
Wiley SR, Winkles JA (2003) TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev 14:241–249. doi:10.1016/S1359-6101(03)00019-4
Chicheportiche Y, Bourdon PR, Xu H et al (1997) TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 272:32401–32410. doi:10.1074/jbc.272.51.32401
Blanco-Colio LM, Martín-Ventura JL, Muñóz-García B et al (2007) Identification of soluble tumor necrosis factor-like weak inducer of apoptosis (sTWEAK) as a possible biomarker of subclinical atherosclerosis. Arterioscler Thromb Vasc Biol 27:916–922. doi:10.1161/01.ATV.0000258972.10109.ff
Simón-Muela I, Llauradó G, Chacón MR et al (2015) Reduced circulating levels of TWEAK are associated with gestational diabetes mellitus. Eur J Clin Invest 45:27–35
Ptaszynska-Kopczynska K, Marcinkiewicz-Siemion M, Lisowska A et al (2016) Alterations of soluble TWEAK and CD163 concentrations in patients with chronic heart failure. Cytokine 80:7–12. doi:10.1016/j.cyto.2016.02.005
Bertin D, Stephan D, Khrestchatisky M, Desplat-Jégo S (2013) Is TWEAK a biomarker for autoimmune/chronic inflammatory diseases? Front Immunol 4:1–5. doi:10.3389/fimmu.2013.00489
Wiley SR, Cassiano L, Lofton T et al (2001) A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 15:837–846. doi:10.1016/S1074-7613(01)00232-1
Wajant H (2013) The TWEAK-Fn14 system as a potential drug target. Br J Pharmacol 170:748–764. doi:10.1111/bph.12337
Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ 10:45–65
Wicovsky A, Salzmann S, Roos C et al (2009) TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell Death Differ 16:1445–1459. doi:10.1038/cdd.2009.80
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. doi:10.1006/meth.2001.1262
Hyogo H, Yamagishi S, Iwamoto K et al (2007) Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol 22:1112–1119. doi:10.1111/j.1440-1746.2007.04943.x
Tahara N, Yamagishi S, Takeuchi M et al (2012) Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [18F]fluorodeoxyglucose positron emission tomography. Diabetes Care 35:2618–2625. doi:10.2337/dc12-0087
Sugimoto K, Nishizawa Y, Horiuchi S, Yagihashi S (1997) Localization in human diabetic peripheral nerve of N(epsilon)-carboxymethyllysine-protein adducts, an advanced glycation endproduct. Diabetologia 40:1380–1387
Saitoh T, Nakayama M, Nakano H et al (2003) TWEAK induces NF-κB2 p100 processing and long lasting NF-κB activation. J Biol Chem 278:36005–36012
Vince JE, Chau D, Callus B et al (2008) TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNF?? J Cell Biol 182:171–184. doi:10.1083/jcb.200801010
Salzmann S, Lang I, Rosenthal A et al (2013) TWEAK inhibits TRAF2-mediated CD40 signaling by destabilization of CD40 signaling complexes. J Immunol 191:2308–2318. doi:10.4049/jimmunol.1202899
Maecker H, Varfolomeev E, Kischkel F et al (2005) TWEAK attenuates the transition from innate to adaptive immunity. Cell 123:931–944. doi:10.1016/j.cell.2005.09.022
Yamana J, Morand EF, Manabu T et al (2012) Inhibition of TNF-induced IL-6 by the TWEAK-Fn14 interaction in rheumatoid arthritis fibroblast like synoviocytes. Cell Immunol 272:293–298. doi:10.1016/j.cellimm.2011.09.004
Steyers CM, Miller FJ (2014) Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci 15:11324–11349. doi:10.3390/ijms150711324
Schmidt AM (1992) Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem 267:14987–14997
Febbraio M, Podrez EA, Smith JD et al (2000) Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 105:1049–1056. doi:10.1172/JCI9259
Ohgami N, Nagai R, Ikemoto M et al (2001) Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J Biol Chem 276:3195–3202. doi:10.1074/jbc.M006545200
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science [Grant Nos. 15K08605 and 15H06785] and the Mitsui Sumitomo Insurance Welfare Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Watanabe, M., Toyomura, T., Wake, H. et al. Advanced glycation end products attenuate the function of tumor necrosis factor-like weak inducer of apoptosis to regulate the inflammatory response. Mol Cell Biochem 434, 153–162 (2017). https://doi.org/10.1007/s11010-017-3045-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3045-6