
Abstract
Murine bone marrow-derived macrophages (M0) and M1- and M2-polarized macrophages are being widely used as a laboratory model for polarized macrophages related molecular mechanism analysis. Gene expression analysis based on reference gene normalization using RT-qPCR was a powerful way to explore the molecular mechanism. But little is known about reference genes in these cell models. So, the goal of this study was to identify reference genes in these types of macrophages. Candidate reference genes in murine bone marrow-derived and polarized macrophages were selected from microarray data using Limma linear model method and evaluated by determining the stability value using five algorithms: BestKeeper, NormFinder, GeNorm, Delta CT method, and RefFinder. Finally, the selected stable reference genes were validated by testing three important immune and inflammatory genes (NLRP1, IL-1β, and TNF-α) in the cell lines. Our study has clearly shown that Ubc followed by Eef1a1 and B2m respectively were recognized as the three ideal reference genes for gene expression analysis in murine bone marrow-derived and polarized macrophages. When three reference genes with strong different stability were used for validation, a large variation of a gene expression level of IL-1β, TNF-α and NLRP1 were obtained which provides clear evidence of the need for careful selection of reference genes for RT-qPCR analysis. Normalization of mRNA expression level with Ubc rather than Actb or Gusb by qPCR in macrophages and polarized macrophages is required to ensure the accuracy of the qPCR analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Macrophages derived from the hematopoietic system are found in all tissues of our body, these large phagocytes exhibit high plasticity and great functional diversity depending on different microenvironment [1, 2]. Not only do macrophages play pivotal roles in innate and adaptive immune responses [3], but they also have crucial roles in organ development, tissue repair and homeostasis, cancer progression and therapy [4,5,6,7]. Based on specifically microenvironmental stimuli, macrophages can be induced into two different phenotypic and functional cell types, classically activated macrophages (M1) and alternatively activated macrophages (M2) [8]. It is well acknowledged that macrophages exhibit phenotypic plasticity in vitro. IFN-γ or LPS induces M1 macrophages associated with a pro-inflammatory phenotype while IL-4 or IL-13 induces an alternative activation program in M2 macrophages associated with an anti-inflammatory and tissue remodelling phenotype [7, 9]. A large number of reports have generally indicated that macrophage M1/M2 polarization is a key determinant of multiple disease development and regression such as cancer, allergic and chronic inflammation, infection, metabolic diseases, atherosclerosis which might be served as novel diagnostic and therapeutic strategies [7, 10]. Most importantly, after different forms of macrophage polarization and activation, studies in the fields of different molecular changes of cytokines, chemokines and signalling molecules using gene expression analysis to understand the M1/M2 macrophage targeting molecular regulation mechanisms of diseases play a key role in macrophage biology and their behaviour [11].
RT-qPCR is one of the most widely used tools to detect and quantify mRNA levels because of its high sensitivity and accuracy with the potential for high throughput, which allows quantification of low mRNA concentrations [12]. RT-qPCR includes RNA extraction from samples, reverse transcription to prepare cDNA using purified RNA as template and quantitative PCR (qPCR, quantitatively measure the amplification of cDNA using fluorescent dyes). At present, there are a lot of superior quality kits for RNA extraction, cDNA preparation, and qPCR. Indeed, the three determinants (RNA and cDNA quantity, and qPCR efficiency) need to be normalized. However, these are not enough to compare RT-qPCR data directly. Normalization of RT-qPCR data and target gene expression level with suitable endogenous reference genes (RGs) is required because the selection of ideal RGs has an important impact on the experimental outcomes [13,14,15]. The suitable RGs should be stable in cells and tissues or under different experimental conditions. But increasing evidence has shown that RGs expression levels often vary under different experimental settings [14,15,16,17,18,19]. Thus, it is necessary to meticulously evaluate and validate the RGs expression for each experimental situation [20].
To date, there are many studies have reported on the selection of suitable RGs in different macrophage cell types [14, 15, 17,18,19]. However, no optimal RGs have been identified and validated for the mRNA level normalization between polarized macrophages. In view of the roles of polarized macrophages in physiology and pathology as well as the importance of studying their precise molecular regulatory mechanisms, our aim is to identify the most stable RGs in polarized macrophages from C57BL/6 mice using NormFinder [21], GeNorm [22], BestKeeper software [23], delta CT method [24] and RefFinder methods [25, 26].
Methods
Murine bone marrow-derived macrophage isolation
The murine bone marrow-derived macrophages were isolated by a previously reported method with slight modification [27]. Briefly, bone marrow was flushed from fresh femurs and tibiae of 8–10-week-old healthy wild type C57BL/6 mice obtained from Nanjing medical university (Nanjing, China) after CO2 euthanasia. The single-cell suspended BM cells were cultured in RPMI-1640 medium (Invitrogen) containing 10% fetal bovine serum, 50 nM β-mercaptoethanol, and 50 ng/mL M-CSF (Peprotech) at the concentration of 2 × 106 cells/mL at 37 °C under 5% CO2. Bone marrow macrophage medium was refreshed on day 3 and mature M0-macrophages were generated on day 7.
All animal care and procedures were per the ethical standards approved by Jiangsu Society for Animal Welfare, China (Acceptance number: XZMC20130226) and Science and Technology Department of Jiangsu Province, China (Acceptance number: SYXK(SU)-2015-0030 and SCXK(SU)-2015-0009).
Polarization of murine macrophages
M1-polarization was induced by 100 ng/mL LPS (Sigma) and 20 ng/mL recombinant murine IFNγ (Peprotech), whereas M2-polarization by 20 ng/mL IL-4 (Peprotech) overnight as previously described [28]. There were eight repeats for each cell type including M0 macrophage.
Characterization of murine bone marrow-derived macrophage and polarization
It was reported in a previous study that macrophage markers were CD11b and F4/80 [29]. M1 macrophage markers were CD80 and CD86, and M2 macrophage markers were CD206 and CD163 [30]. In our study, macrophages were stained with CD11b-FITC, F4/80-APC, CD80-PE or CD206-FITC. Using flow cytometry, M0, M1-Mφ, and M2-Mφ cells were stained with CD11b+ F4/80+, F4/80+ CD80+CD206low, and F4/80+CD80low CD206+ respectively [31, 32].
RT-qPCR
RNA was isolated from M0, M1 and M2-polarized macrophages using the TRIZOL-chloroform extraction method [20]. Genomic DNA was removed using RNase-free DNase I (Beyotime). The extracted RNA concentration and purity were calculated by NanoDrop 2000 spectrophotometer (Thermo USA). The integrity was also evaluated using 1% Agarose gel electrophoresis. All cDNA samples were synthesized from 500 ng isolated RNA samples using PrimeScript RT Reagent Kit (Perfect Real Time) from Takara and kept at − 20 °C until ready for use. Melting temperatures of all genes were designed at 60 °C. Primers of reference genes and target genes were selected in consideration of different intracellular biological functions, purchased from Thermo scientific and the detail information was listed in Table 1. The specificity and efficiency of all reference gene primers were checked [20]. RT-qPCR was done using light cycler®480 and SYBR Green master mix. 10 μL of 2 × supermix SYBR Green 1 Master, 1 μL forward and reverse primers (10 nM), 8 μL of distilled water and 1 μL of cDNA were all included in the PCR reaction tube. The RT-qPCR runs were comprised of 1 min- predenaturation at 95 °C, followed by 40 cycles of three-step PCR including denaturing phase (95 °C for 20 s), annealing phase (60 °C for 15 s) and extension phase (72 °C for 15 s). After PCR, the melting curve and the CT values were obtained from the Light Cycler software. Samples were measured in technical duplicates.
Selection of candidate reference genes from microarray data
The microarray data having been deposited into the NCBI Gene Expression Omnibus database (GEO ID: GSE69607) was discussed in our study [33]. In this database, the same M0, M1, and M2 from C57BL/6J mice were used for microarray analysis. The data were normalized with the RMA algorithm using GenePattern software. The differential expression analysis in this microarray experiment was provided by a freely available bioinformatics tool Limma linear model method [34]. The expression levels of 14 commonly used reference genes were compared and the cut-off criteria to identify the differentially expressed candidate reference genes in M0, M1 and M2 were set to P-value < 0.05 implemented in Limma and log2|Fold change| ≥ 0.585 [35]. On the contrary, the candidate stable reference gene was identified as P-value > 0.05 and log2|Fold change| < 0.585.
Statistical analysis of the stability of the reference genes
Four software; geNorm [22], NormFinder [21] and bestkeeper [23] together with the comparative delta CT method [24] were used to assess the reference genes stability. The basic principles of the four software and How the most reference genes filtered by the software were described by our previous study [20]. RefFinder-comprehensive ranking method were used for over ranking the above four methods according the introduction of previous research [36, 37]
Validation of reference genes
After reference genes selection, gene expression based on the selected most stable reference genes, the least stable reference genes, and the most usually used reference genes (Actb) were detected to validate the reference genes using the 2−ΔΔCt methods.
Results
Characterization of murine bone marrow-derived macrophage phenotype
Macrophage progenitors adhered to the cell dish and could not be washed away on day 3. The murine bone marrow-derived macrophages (M0) were induced using 50 ng/mL M-CSF-contained medium for 7 days. During the 7 days culture, the mature M0-macrophages were observed as adherent flattened cells with larger size (Fig. 1a). M1-Mφ were induced with 20 ng/mL LPS and 20 ng/mL IFNγ overnight. The morphological features were similar to M0–Mφ (Fig. 1a). M2–Mφ were induced with 20 ng/mL IL-4 overnight causing the cells to be rounded and more loosely attached (Fig. 1a), which is similar to M2–Mφ.

Characterization of murine bone marrow-derived macrophage phenotype Morphological and cell surface marker expression (CD11b-FITC, F4/80-APC, CD80-PE or CD206-FITC.) changes observed by microscopic (a) and flow cytometric analyses (b, c)
In our study, flow cytometric analysis was employed to identify and quantify bone marrow-derived macrophages and their polarized forms using CD11b-FITC, F4/80-APC, CD80-PE or CD206-FITC antibodies. The results demonstrated that M0, M1, and M2 cells respectively amounted to 99.8%, 99.2% and 92% of the total number of cells stained (Fig. 1b). We also compared MFI of CD80-PE and CD206-FITC in M1 and M2 cells. The results showed that the CD80 expression level in M1 was higher than in M2 while CD206 expression level in M2 is higher than in M1 (Fig. 1c) which suggested that M1 was CD80 + CD206low and M2 was CD80low CD206+,consistent with previous reports [31, 32].
Selection and characteristics of candidate reference genes
To identify candidate RGs, we compared the gene expression levels of 14 commonly used RGs. Genes with low expression levels were excluded (as the underline value shown in Table 2). After filtration, 12 genes except Tbp and Tfrc were selected as the candidate reference genes for qPCR. Six genes including Actb, Eef1a1, B2m, Rplp0, Hmbs, and Ppia were the most stable RGs based on the database of a published microarray data and a Limma linear model method, which suggested that the candidate stable reference gene should be identified at P-value > 0.05 and log2|Fold change| < 0.585 [34, 35]. All data was shown in Table 2.
Primer expression level and specificity detection
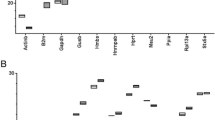
To further screen stable reference genes for qPCR analysis, we designed and synthesized 12 primers with high efficiency [20]. Firstly, we detected the expression levels and specificity of the 12 RGs using SYBR Green I Master [38]. As shown in Fig. 2a, different genes had different CT values between 14.2 and 31.9 in different samples. Actb had the highest expression level while Hmbs expression level was the lowest. The result is similar to the above microarray data. The specificity of all the primers was high with unique single-peak amplification according to the melting curve as shown in (Fig. 2b).

Primer specificity and expression level analysis. a Ranges of Ct values of the 12 pre-selected reference genes in M0, M1, and M2 Plotted as boxes are the ranges of Ct values, with the included horizontal line identifying the mean (n = 8). The blue boxes represent M0 and the green boxes represent M1, and the red boxes represent M2. Bars represent the mean ± SD. *p < 0.05. b The images represent the melting curve of the 12 reference genes amplicons after the RT-qPCR reactions from one of 8 independent experiments. (Color figure online)
Identification of the most stable RGs
Bestkeeper
In our experiment, the mean Ct values ranged from 15.68 (Actb) to 29.7 (Hmbs) in all M0, M1 and M2 samples (Table 3). STD [±CP], CV% and Pearson coefficient [R] are usually used to evaluate gene stability and the STD [±CP] is the index of the bestkeeper, most commonly used to compare the stability of the selected RGs [23]. Genes with the lowest STD [±CP] values have less variation and vice versa in gene expression. Our results showed that the gene stability was different in M0, M1 and M2 macrophages. In M0, Rplp0, Hprt1 and Ywhaz were ranked as the most stable RGs. Gapdh, Hmbs and Eef1a1 were stably expressed in M1 while Alsasl, Gapdh, and Eef1a1 were the most stable in M2. In all the cell types, Rplp0 with (STD [± CP] = 0.51) and Hprt1with (STD [± CP] = 0.55) followed by Ywhaz with (STD [± CP] = 0.59) were highly stable, while Gusb with (STD [± CP] = 2.26) having the highest value, indicating it had the lowest stability (as shown in Table 4).
GeNorm analysis
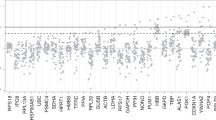
GeNorm grades the candidate gene stability according to the calculated M-value. M-value ≤ 1.5 was considered a stable expression of RGs. Genes with the lowest M-value were considered as the most stable genes. As seen in Table 5, most of the genes were stable in M0, M1, and M2 except Gusb, which meant that it is not suitable as a control gene for qPCR analysis. The results obtained from the GeNorm resembled those got from BestKeeper. After a combination, the two most stable genes were Ubc and B2m. All pairwise variation was shown in Fig. 3b to determine the optimal number of the candidate genes. According to [22], he recommends a cut-off value of 0.15, that genes with a V ≤ 0.15 should be included. Therefore, based on the V value, the two most stable RGs (V2/V3) of this dataset would be adequate for accurate normalization (Tables 6, 7, 8).

Graphical presentation of stability value by GeNorm. a Showing the ranking of the12 reference by geNorm software, with the most stable toward the right and least toward the left. b Determination of the minimal number of reference genes by pairwise variation (Vn/n + 1). It was shown the determination of the optimal number of housekeeping genes by pairwise variation. A cut-off value of 0.15 was set. The V value defined the pair-wise variation between two sequential normalization factors. The two most stably expressed reference genes may be accurate for qRT-PCR normalization. More reference genes for qRT-PCR normalization will not increase the stability of reference genes
NormFinder
The principle of NormFinder ranking stability of RGs is based on stability values. Gene with the lowest stability value is considered to remain constant, while those with the highest stability values are the least stable RGs. After NormFinder software analysis, Ubc (Stability value = 0.076 and 0.504) in M0 and M1 cells, Gapdh (Stability value = 0.084) in M2 cells were the most stable genes. After the combination of all the data in the three cell lines, Eef1a1 (Stability value = 0.422) was considered to be the most stably expressed RG (Fig. 4).

Relative gene expression levels of IL-1β, TNF-αand NLRP1gene using reference genes with different stability based on the 2−ΔΔCt method. For normalization, the most (Ubc) and least consistently expressed reference genes (Gusb) and the traditionally used reference gene Actb which ranked in the midway were used for the calculation of ΔCt. Bars represent the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001
The comparative delta-Ct method
The delta-Ct method determines the stability of RGs according to the average of STDEV within the samples [39]. Those with lower STDEV value between samples is considered as the most stable RGs. Consistent with NormFinder analysis, the delta-Ct method also identified the Eef1a1 and Ubc as the most stable RGs. Meanwhile, Gusb was recognized as the least stable.
The overall ranking of reference gene stability
We co-ranked the four methods and calculated the geometric mean using the RefFinder-comprehensive ranking method to screen the most stable RGs [14, 26]. The geometric mean values were used to rank stable genes. Finally, it ranked Ubc and Eef1a1 as the two most stable, and Gusb and Rplp0 were considered as the least stable RGs in all M0, M1 and M2 cells.
Validation of reference genes
To validate the performance of the above selected RGs, we determined the gene expression levels of three genes from the inflammatory signaling pathways, NLRP1, IL-1β and TNF-α using the most and least stable reference and Actb according to their stability in M0, M1, and M2 cells. The results showed strong variation in fold changes when different reference genes were used in M1 and M2 cells, which indicated that normalization of RT-qPCR data with an unstably expressed RG could impact upon the significance of gene expression study.
Discussion
RT-qPCR is most usually used to detect gene expression levels and predict possible gene functions in different experimental conditions. Recently, more and more researchers have found out that the traditional RGs such as Actb and Gapdh are not always stable in a lot of experimental situations making them unsuitable to be used as reference genes to normalize RT-qPCR results [20]. Moreover, an increasing number of RT-qPCR studies were validated by most reference gene selection although there are a great number of studies still lacking reliable RGs selection for quantitative gene expression analysis. Macrophages with polarization plasticity (M0, M1, and M2) belonging to the immune system play a key role in many normal physiological conditions and disease processes. They largely perform their functions through the regulation of encoding cytokines or expression of other genes. Some studies have reported on the selection of suitable RGs for the accurate quantitation and normalization in different macrophage cell types such as in rat osteoblasts [14], PMA-induced THP-1 macrophages [18], J774A1 macrophage cell line [16], LPS-stimulated murine macrophages [15], and differentiating osteoblasts, osteoclasts and macrophages under different biological conditions [17]. To our knowledge, selection and validation of stable RGs in polarized macrophages have not been performed. So, in this study, we aimed to validate reference genes in a well-established primary macrophage model, BMDMs (M0) and their polarized forms (M1 and M2 cells).
Reference genes refer to the genes whose expression level does not change under specific experimental conditions which are selected from housekeeping genes with different functions. The large-scale gene expression data such as microarray can be used to identify condition-stable RGs [40, 41]. First, we compared 14 reference genes based on published microarray data of M0, M1, and M2 cells and found out that the expression levels of Tbp and Tfrc were too low to be used as suitable RGs in these three cell lines. The expression levels of Alas1 and Hprt1 in M0, M1, and M2 were different, suggesting that they do not qualify as RGs in polarized macrophages. But the stability of these two genes needs further RT-qPCR verification to confirm, so we designed 12 reference gene primers except Tbp and Tfrc to evaluate their gene stability in M0, M1, and M2 cells. Gapdh and Actb are the two most widely used reference genes [42], but it has been proven that they are not the most stable RGs, even under certain experimental conditions they appeared to be the most unstable genes [16]. In this study, these two genes were also used and our results revealed that the expression stability of Actb in M0, M1 and M2 cells is strikingly similar to Gapdh even though they are not the most stable ones. We have realized that this result corroborates another previous study [15]. These findings also suggested the necessity of evaluating reference genes used for qPCR evaluation, even if they are traditionally used reference genes.
To our knowledge, a lot of software such as RefFinder, NormqPCR, RefGenes, OLIVER, GeNorm, BestKeeper, and NormFinder had been developed for stable reference gene selection. The grading of RG stability may vary due to the discrepancies of the above methods. There is no one computational program or statistical tool universally accepted to analyze gene expressions for specific experiments. A lot of studies including our previous study [20], usually employ several applets to determine gene expression stability. NormFinder [21], GeNorm [22], BestKeeper software [23] along with the delta CT [24] and RefFinder method [25, 26] are the most commonly used method to rank the stability of reference gene expression. These five methods were explored in our study to determine the most stable reference genes in M0, M1 and M2 cells. After over ranking of these methods, our results revealed that Ubc followed by Eef1a1 and B2m respectively were the three most stable RGs. This result is peculiar compared to other studies which selected reference genes in a different type of macrophages [14,15,16,17]. In rat osteoblasts [14], the researcher compared 31 reference genes in bone marrow osteoblasts, calvarial osteoblasts, and UMR-106 osteoblasts evaluated byΔCt method, DataAssist™, NormFinder and BestKeeper methods. They revealed that Eif2b1was the most stable reference gene, and Rps17 was the least stable gene. In PMA-induced THP-1 macrophages [18], using GeNorm, NormFinder and BestKeeper, the author found that β-actin and RPL37A were the two most stable reference genes But in J774A1 macrophage cell line [16] indifferent culture condition, the most stability gene was β-actin and B2M. Alexandre S Stephens showed that the most stable gene in bone marrow macrophage were Hprt1/B2m by GeNorm which is similar to our result [17]. In our result we showed that in M0 cells the most stable gene was Hprt1/Ywhaz. However in LPS stimulated BMDMs, the Hnrnpab and Stx5a were better [15]. In LPS treated RAW 264.7 cells and peritoneal macrophages [15], the stability of reference genes were various due to the software selected, the cell type, the candidate reference gene selected and the culture conditions. And there was no comparable to our data. And all of the results suggested, the most stable RG is different in various macrophage cell types. The reasons were including peculiarities of cells, the analytical method, the candidate reference gene selected and different treatment conditions. These indicate that reference gene selection in macrophages of the different experimental conditions is necessary with over-ranking methods and enough candidate reference gene selected. This requires us to carry out routine internal reference gene screening on macrophages in different experimental conditions, to ensure the accuracy and reliability of qPCR results.
As far as we know, this is the first attempt to select and validate suitable RGs for RT-qPCR analysis in M0, M1 and M2 murine macrophages. Since distinct experimental settings in macrophages require individual validation of internal control genes and more and more methods were used for stable genes selection, we believe that another promising candidate reference gene stably expressed in macrophage and polarized macrophages may exist. Most importantly, reference genes selected from a large number of housekeeping genes by high-throughput technologies was needed and this alternative approach can be robust. Several studies had already shown that the NCBI GEO database is available for the discovery of novel reference genes [15, 40, 41] and RNA-seq has been more widely used for selecting stable reference genes from a large number of reference genes [43,44,45]. But reference genes selected by the above high-throughput technologies in M0, M1 and M2 murine macrophages were still needed to be validated.
Here we performed RT-qPCR using 2−ΔΔCt methods to determine mRNA level of three target genes NLRP1, IL-1β and TNF-α based on three reference genes with different levels of stability. Interestingly, the gene expression levels in M1 and M2 were significantly different when RGs with different stability were used, which means that it is really necessary to select and validate suitable RGs before RT-qPCR analysis. But unfortunately, only a few researchers provided evidence for RG selection. Hence forth, our results will be very helpful in guiding investigators to select the appropriate RGs to accurately quantify mRNA in studies involving M0, M1 and M2 murine macrophages. Lastly, what we want to achieve more is to encourage researchers to ascertain RGs stability before gene expression analysis for every experimental setting.
Conclusion
Our study has clearly shown that Ubc followed by Eef1a1 were recognized as the two ideal RGs for mRNA level analysis in M0, M1, and M2 cells. Our results showed strong variation in fold changes of NLRP1, IL-1β, and TNF-α when reference genes with different stability were used in M1 and M2 cells. Our study indicates that it is necessary to carefully select RGs for gene expression normalization by RT-qPCR study.
Data availability
The data and materials used to support the findings in this study are included in the article.
Abbreviations
- BMDM or M0:
-
Bone marrow-derived macrophage
- RT-qPCR:
-
Real-time quantitative PCR
- M1:
-
Classically activated macrophages
- M2:
-
Alternatively activated macrophages
- RG:
-
Reference gene
- STDEV:
-
Standard deviation
References
Davies LC, Taylor PR (2015) Tissue-resident macrophages: then and now. Immunology 144(4):541–548. https://doi.org/10.1111/imm.12451
Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496(7446):445–455. https://doi.org/10.1038/nature12034
Navegantes KC, de Souza GR, Pereira PAT, Czaikoski PG, Azevedo CHM, Monteiro MC (2017) Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med 15(1):36. https://doi.org/10.1186/s12967-017-1141-8
Vannella KM, Wynn TA (2017) Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol 79:593–617. https://doi.org/10.1146/annurev-physiol-022516-034356
Isidro RA, Appleyard CB (2016) Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol 311(1):G59–73. https://doi.org/10.1152/ajpgi.00123.2016
Gordon S, Martinez-Pomares L (2017) Physiological roles of macrophages. Pflugers Arch 469(3–4):365–374. https://doi.org/10.1007/s00424-017-1945-7
Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT, Sahebkar A (2018) Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233(9):6425–6440. https://doi.org/10.1002/jcp.26429
Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, Li J (2014) Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal 26(2):192–197. https://doi.org/10.1016/j.cellsig.2013.11.004
Ying W, Cheruku PS, Bazer FW, Safe SH, Zhou B (2013) Investigation of macrophage polarization using bone marrow derived macrophages. J Vis Exp. https://doi.org/10.3791/50323
Koh YC, Yang G, Lai CS, Weerawatanakorn M, Pan MH (2018) Chemopreventive effects of phytochemicals and medicines on M1/M2 polarized macrophage role in inflammation-related diseases. Int J Mol Sci. https://doi.org/10.3390/ijms19082208
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Investig 122(3):787–795. https://doi.org/10.1172/JCI59643
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25(2):169–193
Appukuttan B, Ashander LM, Ma Y, Smith JR (2018) Selection of reference genes for studies of human retinal endothelial cell gene expression by reverse transcription-quantitative real-time polymerase chain reaction. Gene Rep 10:123–134. https://doi.org/10.1016/j.genrep.2017.11.009
Abuna RPF, Oliveira FS, Ramos JIR, Lopes HB, Freitas GP, Souza ATP, Beloti MM, Rosa AL (2018) Selection of reference genes for quantitative real-time polymerase chain reaction studies in rat osteoblasts. J Cell Physiol. https://doi.org/10.1002/jcp.26886
Tanaka A, To J, O'Brien B, Donnelly S, Lund M (2017) Selection of reliable reference genes for the normalisation of gene expression levels following time course LPS stimulation of murine bone marrow derived macrophages. BMC Immunol 18(1):43. https://doi.org/10.1186/s12865-017-0223-y
Ferraz FB, Fernandez JH (2016) Selection and validation of reference house-keeping genes in the J774A1 macrophage cell line for quantitative real-time PCR. Genet Mol Res 15(1):15017720. https://doi.org/10.4238/gmr.15017720
Stephens AS, Stephens SR, Morrison NA (2011) Internal control genes for quantitative RT-PCR expression analysis in mouse osteoblasts, osteoclasts and macrophages. BMC Res Notes 4:410. https://doi.org/10.1186/1756-0500-4-410
Maess MB, Sendelbach S, Lorkowski S (2010) Selection of reliable reference genes during THP-1 monocyte differentiation into macrophages. BMC Mol Biol 11:90. https://doi.org/10.1186/1471-2199-11-90
Taylor DL, Thomson PC, de Silva K, Whittington RJ (2007) Validation of endogenous reference genes for expression profiling of RAW264.7 cells infected with Mycobacterium avium subsp. paratuberculosis by quantitative PCR. Vet Immunol Immunopathol 115(1–2):43–55. https://doi.org/10.1016/j.vetimm.2006.10.007
Ju W, Smith AO, Sun T, Zhao P, Jiang Y, Liu L, Zhang T, Qi K, Qiao J, Xu K, Zeng L (2018) Validation of housekeeping genes as reference for reverse-transcription-qPCR analysis in busulfan-injured microvascular endothelial cells. Biomed Res Int 2018:4953806. https://doi.org/10.1155/2018/4953806
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Can Res 64(15):5245–5250. https://doi.org/10.1158/0008-5472.CAN-04-0496
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3(7):0034
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–excel-based tool using pair-wise correlations. Biotechnol Lett 26(6):509–515
Rao X, Huang X, Zhou Z, Lin X (2013) An improvement of the 2(-∆∆CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinform Biomath 3(3):71–85
Panina Y, Germond A, Masui S, Watanabe TM (2018) Validation of common housekeeping genes as reference for qPCR gene expression analysis during iPS reprogramming process. Sci Rep 8(1):8716. https://doi.org/10.1038/s41598-018-26707-8
Zyzynska-Granica B, Koziak K (2012) Identification of suitable reference genes for real-time PCR analysis of statin-treated human umbilical vein endothelial cells. PLoS ONE 7(12):e51547. https://doi.org/10.1371/journal.pone.0051547
Weischenfeldt J, Porse B (2008) Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. https://doi.org/10.1101/pdb.prot5080
Luo Y, Shao L, Chang J, Feng W, Liu YL, Cottler-Fox MH, Emanuel PD, Hauer-Jensen M, Bernstein ID, Liu L, Chen X, Zhou J, Murray PJ, Zhou D (2018) M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood Adv 2(8):859–870. https://doi.org/10.1182/bloodadvances.2018015685
Zhang X, Goncalves R, Mosser DM (2008) The isolation and characterization of murine macrophages. Curr Protoc Immunol. https://doi.org/10.1002/0471142735.im1401s83
Zhou Y, Yoshida S, Kubo Y, Yoshimura T, Kobayashi Y, Nakama T, Yamaguchi M, Ishikawa K, Oshima Y, Ishibashi T (2017) Different distributions of M1 and M2 macrophages in a mouse model of laser-induced choroidal neovascularization. Mol Med Rep 15(6):3949–3956. https://doi.org/10.3892/mmr.2017.6491
Glass EB, Masjedi S, Dudzinski SO, Wilson AJ, Duvall CL, Yull FE, Giorgio TD (2019) Optimizing mannose "click" conjugation to polymeric nanoparticles for targeted siRNA delivery to human and murine macrophages. ACS Omega 4(16):16756–16767. https://doi.org/10.1021/acsomega.9b01465
Laban H, Weigert A, Zink J, Elgheznawy A, Schurmann C, Gunther L, Abdel Malik R, Bothur S, Wingert S, Bremer R, Rieger MA, Brune B, Brandes RP, Fleming I, Benz PM (2018) VASP regulates leukocyte infiltration, polarization, and vascular repair after ischemia. J Cell Biol 217(4):1503–1519. https://doi.org/10.1083/jcb.201702048
Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M (2015) Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 10(12):e0145342. https://doi.org/10.1371/journal.pone.0145342
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47. https://doi.org/10.1093/nar/gkv007
Liu Y, Niu M, Yao C, Hai Y, Yuan Q, Liu Y, Guo Y, Li Z, He Z (2015) Fractionation of human spermatogenic cells using STA-PUT gravity sedimentation and their miRNA profiling. Sci Rep 5:8084. https://doi.org/10.1038/srep08084
Velada I, Ragonezi C, Arnholdt-Schmitt B, Cardoso H (2014) Reference genes selection and normalization of oxidative stress responsive genes upon different temperature stress conditions in Hypericum perforatum L. PLoS ONE 9(12):e115206. https://doi.org/10.1371/journal.pone.0115206
Khan S, Roberts J, Wu SB (2017) Reference gene selection for gene expression study in shell gland and spleen of laying hens challenged with infectious bronchitis virus. Sci Rep 7(1):14271. https://doi.org/10.1038/s41598-017-14693-2
Feng X, Xiong Y, Qian H, Lei M, Xu D, Ren Z (2010) Selection of reference genes for gene expression studies in porcine skeletal muscle using SYBR green qPCR. J Biotechnol 150(3):288–293. https://doi.org/10.1016/j.jbiotec.2010.09.949
Silver N, Best S, Jiang J, Thein SL (2006) Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol 7:33. https://doi.org/10.1186/1471-2199-7-33
Zhao DJ, Guo K, Kang L (2012) Identification of condition-specific reference genes from microarray data for locusts exposed to hypobaric hypoxia. FEBS Open Bio 2:235–240. https://doi.org/10.1016/j.fob.2012.08.001
Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC, Lin CH, Whang-Peng J, Hsu SL, Chen CH, Huang CY (2007) Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8:140. https://doi.org/10.1186/1471-2164-8-140
Gu YR, Li MZ, Zhang K, Chen L, Jiang AA, Wang JY, Li XW (2011) Evaluation of endogenous control genes for gene expression studies across multiple tissues and in the specific sets of fat- and muscle-type samples of the pig. J Anim Breed Genet 128(4):319–325. https://doi.org/10.1111/j.1439-0388.2011.00920.x
Hoang VLT, Tom LN, Quek XC, Tan JM, Payne EJ, Lin LL, Sinnya S, Raphael AP, Lambie D, Frazer IH, Dinger ME, Soyer HP, Prow TW (2017) RNA-seq reveals more consistent reference genes for gene expression studies in human non-melanoma skin cancers. PeerJ 5:e3631. https://doi.org/10.7717/peerj.3631
Carmona R, Arroyo M, Jimenez-Quesada MJ, Seoane P, Zafra A, Larrosa R, Alche JD, Claros MG (2017) Automated identification of reference genes based on RNA-seq data. Biomed Eng 16(Suppl 1):65. https://doi.org/10.1186/s12938-017-0356-5
Zhan C, Zhang Y, Ma J, Wang L, Jiang W, Shi Y, Wang Q (2014) Identification of reference genes for qRT-PCR in human lung squamous-cell carcinoma by RNA-Seq. Acta Biochim Biophys Sin 46(4):330–337. https://doi.org/10.1093/abbs/gmt153
Funding
This study was supported in part by National Natural Science Foundation of China under Grant [Nos. 81700178, 31872795 and 81570096]; Natural Science Foundation of Jiangsu Province under Grant [No. BK20170259]; Jiangsu Provincial Key Research and Development Program under Grant [No. BE2018637]; Jiangsu Province’s Key Provincial Talents Program under Grant [No. ZDRCA2016054]; China Postdoctoral Science Foundation Grant [No. 2018M632380]; and Jiangsu Postdoctoral Science Foundation under Grant [No. 1701064B] for purchasing reagents, testing fee, and publication charge.
Author information
Authors and Affiliations
Contributions
Conceptualization, AOS; Data curation, WJ and AOS; Formal analysis, KQ; Funding acquisition, WJ; Investigation, WJ, TS, WL, KQ and LZ; Methodology, TS, WL, YB, SYA and KQ; Project administration, WL; Resources, JQ and LZ; Software, TS, WL, YB and SYA; Supervision, LZ; Validation, WJ, KX, JQ and LZ; Visualization, JQ and LZ; Writing—original draft, WJ; Writing—review & editing, AOS, SYA, KX, JQ and LZ.
Corresponding authors
Ethics declarations
Conflict of interest
All authors have no dispute of interest to disclose.
Consent to publish
All authors approved the submission of the manuscript.
Ethics approval and consent to participate
All animal care and procedures were per the ethical standards, approved by Jiangsu Society for Animal Welfare, China (Acceptance number: XZMC20130226) and Science and Technology Department of Jiangsu Province, China (Acceptance number: SYXK(SU)-2015-0030 and SCXK(SU)-2015-0009).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ju, W., Sun, T., Lu, W. et al. Reference gene selection and validation for mRNA expression analysis by RT-qPCR in murine M1- and M2-polarized macrophage. Mol Biol Rep 47, 2735–2748 (2020). https://doi.org/10.1007/s11033-020-05372-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05372-z