
Abstract
SSRs were successfully isolated from the Perilla crop in our current study, and used to analyze Perilla accessions from East Asia. Analyses of the clear genetic diversity and relationship for Perilla crop still remain insufficient. In this study, 40 new simple sequence repeat (SSR) primer sets were developed from RNA sequences using transcriptome analysis. These new SSR markers were applied to analyze the diversity, relationships, and population structure among 35 accessions of the two cultivated types of Perilla crop and their weedy types. A total of 220 alleles were identified at all loci, with an average of 5.5 alleles per locus and a range between 2 and 10 alleles per locus. The MAF (major allele frequency) per locus varied from 0.229 to 0.943, with an average of 0.466. The average polymorphic information content (PIC) value was 0.603, ranging from 0.102 to 0.837. The genetic diversity (GD) ranged from 0.108 to 0.854, with an average of 0.654. Based on population structure analysis, all accessions were divided into three groups: Group I, Group II and the admixed group. This study demonstrated the utility of new SSR analysis for the study of genetic diversity and population structure among 35 Perilla accessions. The GD of each locus for accessions of cultivated var. frutescens, weedy var. frutescens, cultivated var. crispa, and weedy var. crispa were 0.415, 0.606, 0.308, and 0.480, respectively. Both weedy accessions exhibited higher GD and PIC values than their cultivated types in East Asia. The new SSR primers of Perilla species reported in this study may provide potential genetic markers for population genetics to enhance our understanding of the genetic diversity, genetic relationship and population structure of the cultivated and weedy types of P. frutescens in East Asia. In addition, new Perilla SSR primers developed from RNA-seq can be used in the future for cultivar identification, conservation of Perilla germplasm resources, genome mapping and tagging of important genes/QTLs for Perilla breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Perilla frutescens (L.) Britton is a self-fertilizing crop that is widely cultivated in East Asia. The species includes two varieties on the basis of their morphology and dual uses. P. frutescens var. frutescens is used as an oil crop, whereas P. frutescens var. crispa is used as a Chinese medicine or vegetable crop (Nitta and Ohnishi 1999; Lee and Ohnishi 2001; Nitta et al. 2003). Today, the two varieties of P. frutescens are extensively cultivated and used in Korea and Japan. However, the Perilla crop probably originated from China (Li 1969; Makino 1961; Nitta 2001; Lee and Ohnishi 2001; Nitta et al. 2003). In East Asia, var. frutescens is extensively cultivated and used as both an oil crop and a leafy vegetable in Korea. In contrast, var. crispa is extensively cultivated and used in Japan, where its leaves are used for vegetables or pickles (Lee and Ohnishi 2001; Lee et al. 2002; Nitta et al. 2003). On the other hand, weedy plants were reported for two cultivated types of P. frutescens by Nitta and Ohnishi (1999), Lee and Ohnishi (2001) and Nitta et al. (2003). In East Asia, the weedy plants are grown naturally and commonly found in such habitats as roadsides, waste lands and around farming fields or farmhouses (Lee and Ohnishi 2001; Lee et al. 2002; Nitta et al. 2003, 2005). Thus, knowledge of genetic diversity and genetic relationships between two cultivated types of P. frutescens and their weedy types is essential for the long-term success of breeding programs and maximizes the use of the germplasm resources in East Asia.
As an advanced molecular marker technique, PCR-based molecular markers, such as RAPD (random amplified polymorphic DNA), AFLP (amplified fragment length polymorphism), and SSR (simple sequence repeat), have provided useful information regarding genetic diversity and genetic relationships in many crops (Schontz and Rether 1999; Prasad et al. 2000; Lee et al. 2002; Hamza et al. 2004; Xia et al. 2005, Sa et al. 2010). In Perilla crop, RAPD, AFLP and SSR analyses were used to analyze the genetic diversity and genetic relationships among two cultivated types of P. frutescens and their weedy types in East Asia (Nitta and Ohnishi 1999; Lee et al. 2002; Lee and Ohnishi 2003; Lee and Kim 2007; Park et al. 2008, Sa et al. 2013, 2015; Ma et al. 2017). Among the various types of DNA-based markers, SSRs are very abundant in both coding and non-coding regions of eukaryotic genomes and exhibit a highly variable number of repeats between individuals in a given population (Park et al. 2009). SSRs are the preferred choice for genetic studies because they are highly reproducible, polymorphic, generally codominant, and abundant in plant genomes (Powell et al. 1996; Park et al. 2009). In our previous study, SSRs were successfully isolated from Perilla crop (Kwon et al. 2005; Park et al. 2008) and used to analyze Perilla accessions from East Asia (Lee and Kim 2007; Lee et al. 2007; Park et al. 2008, Sa et al. 2013; Ma et al. 2017). However, analyses of the clear genetic diversity and genetic relationship for Perilla crop remain insufficient. Therefore the new SSR markers derived from EST sequence are more useful to study the germplasms population structure based on the molecular marker resources to characterize the morphology.
Recently, the transcriptome sequencing approach RNA-seq represents a powerful tool for transcriptional analysis, novel gene discovery, and development of molecular markers for non-model crops, such as Perilla (Mutz et al. 2012; Wang et al. 2015). This approach also facilitates rapid mining of SSR markers in non-model crops (Fukushima et al. 2015; Wang et al. 2015). In particular, SSR markers developed from RNA-seq can enable marker-assisted selection for Perilla breeding programs because these regions are coding sequences and may be close to or within functionally transcribed genes (Mutz et al. 2012; Fukushima et al. 2015). In our previous study, we sequenced and assembled one cultivated type (PF98095) of P. frutescens var. frutescens using transcriptome sequencing by RNA-seq (Tong et al. 2015) and obtained 15,991 SSR loci. This information will be useful for developing SSR primers in Perilla crop. In this study, we successfully isolated SSR primers from Perilla species, and these new Perilla SSR markers were used to analyze the genetic diversity, genetic relationships and population structure among two cultivated types of P. frutescens and their weedy types in East Asia.
Materials and methods
Plant materials and DNA extraction
To evaluate polymorphisms to identify new SSR markers in Perilla accessions, we used 35 accessions (15 accessions that consisted of five cultivated type var. frutescens, five weedy type var. frutescens and five weedy type var. crispa in Korea; 13 accessions that consisted of seven cultivated type var. frutescens, three weed type var. frutescens and three weedy type var. crispa in China; and seven accessions included two cultivated type var. frutescens and five cultivated type var. crispa in Japan) (Table 1). Total DNA was extracted from the leaf tissues of a representative individual plant for each accession following the Plant DNAzol Reagent protocol (GibcoBRL Inc., Grand Island, NY, USA).
SSR marker development
For construction of the transcriptome reference set in a previous study (Tong et al. 2015), de novo assembly of the PF98095 RNA-seq data was performed using Trinity software (http://TrinityRNASeq.sourceforge.net). The raw reads from NGS sequencing with a Phred quality score of at least 20 and a read length of at least 50 bp of HiSeq 2000 data were filtered before assembly. A Perl script MISA tool (http://pgrc.ipk-gatersleben.de/misa) was used to search microsatellite sites in the assembled transcriptome sequences of PF98095. The SSRs with di-, tri-, and tetra-nucleotide repeat units were identified. Based on the SSR flanking sequences, PRIMER 3 software was employed to design the primer pairs. As a result, we searched all unigenes in the cultivated type of var. frutescens (PF98095). We detected a total of 15,991 SSR loci. All SSRs can be classified by the number of repeat units. Di-nucleotide SSRs represent the largest number of SSRs (9910) followed by tri-nucleotide (5652) SSRs and tetra-nucleotide (429) SSRs. In this study, we first designed 198 SSR primer sets based on the di- and tri-nucleotide types and the number of repeat units.
SSR analysis and silver-staining
SSR amplifications were conducted in a total volume of 20 µl consisting of 20 ng genomic DNA, 1 × PCR buffer, 0.5 µM of forward and reverse primers, 0.2 mM dNTPs, and 1 unit of Taq polymerase (Biotools, Madrid, Spain). The PCR profile consisted of an initial denaturation at 95 °C for 3 min followed by 36 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min 30 s with a final extension step of 5 min at 72 °C. After PCR, 5 µl of the final product were mixed with 10 µl of electrophoresis loading buffer (98% formamide, 0.02% BPH, 0.02% Xylene C, and 5 mM NaOH). After denaturing and immediate cooling, 2 µl from each sample was loaded onto a 6% denaturing (7.5 M urea) acrylamide-bis acrylamide gel (19:1) in 1 × Tris-borate-EDTA (TBE) buffer and then electrophoresed at 1800 V and 60 W for 130 min. The separated fragments were then visualized using a silver staining kit (Promega, Madison, WI, USA).
Data analysis
The number of alleles, allele frequency, major allele frequency (MAF), gene diversity (GD), and polymorphic information content (PIC) for 40 new SSR markers were calculated using the PowerMarker 3.25 program (Liu and Muse 2005). GD is defined as the probability that two randomly chosen alleles from the population are different. GD can be estimated at the lth locus as:
where f represents the inbreeding coefficient; Plu, the frequency of the uth allele; and n, the sample size.
PIC (Bostein et al. 1980) was calculated based on the following equation:
where Plu2 and Plv2 are the frequencies of the uth and vth alleles, respectively, of marker l.
The genetic similarities (GS) were calculated for each pair of accessions using the Dice similarity index (Dice 1945). The similarity matrix was used to construct an unweighted pair group method with arithmetic mean algorithm (UPGMA) dendrogram with the help of SAHN-clustering from NTSYSpc version 2.1 (Rohlf 1998). The population structure of 35 Perilla accessions was analyzed using the model-based program STRUCTURE 2.2 (Pritchard and Wen 2003). The membership coefficient for each individual in each subpopulation was run five times for each cluster (K), ranging from 1 to 10, using the admixture model with a burn-in of 100,000 and a replication of 100,000. Given that the estimated log probability of data [LnP(D)] overestimated the number of subgroups, we used the ad hoc criterion (ΔK) described by Evanno et al. (2005) to determine the most likely value of K. The run of the estimated numbers of subgroups demonstrating the maximum likelihood was used to assign Perilla accessions with membership probabilities ≥ 0.80 to subgroups. The Perilla accessions with membership probabilities < 0.80 were assigned to an admixed group (Wang et al. 2008).
Results
SSR identification and polymorphisms
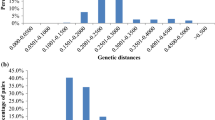
Among the newly developed 198 SSR primer pairs, 40 SSR primer pairs exhibited good amplification patterns and polymorphisms among 35 Perilla accessions (Table 2). However, the remaining 158 SSR primer pairs exhibited a monomorphic band (41) or ambiguous band pattern (58) and poor or no amplification (59) in the Perilla accessions. The new 40 SSR primer pairs were used to measure polymorphisms in terms of the number of alleles, MAF, GD, and PIC among 35 Perilla accessions, including two cultivated types of Perilla crop and their weedy types in East Asia. A total of 220 alleles were detected segregating in the 35 Perilla accessions with an average of 5.5 alleles per locus, ranging from 125 to 310 bp. The number of alleles per locus ranged from two (KNUPE-8 and KNUPE-38) to ten (KNUPE-13). The MAF per locus varied from 0.229 (KNUPE-6) to 0.943 (KNUPE-8), with an average of 0.466. The genetic diversity ranged from 0.108 (KNUPE-8) to 0.854 (KNUPE-6), with an average of 0.654. The average polymorphic information content value was 0.603, ranging from 0.102 (KNUPE-8) to 0.837 (KNUPE-6) (Table 2). Analysis of all alleles led to the identification of 50 alleles as rare (frequency < 0.05) and private alleles. We identified 160 alleles as intermediate (frequency 0.05–0.5) and 10 alleles as abundant (frequency > 0.5) (Fig. 1). In the analysis of the four groups of accessions (cultivated and weedy types of var. frutescens and of var. crispa) using the 40 SSR primers, the average number of alleles ranged from 3.1, 3.7, 2.0 and 3.0 for the cultivated and weedy types of var. frutescens, and for the cultivated and weedy types of var. crispa, respectively. The average gene diversity values were 0.415, 0.606, 0.308 and 0.480 for the cultivated and weedy types of var. frutescens, and for the cultivated and weedy types of var. crispa, respectively (Table 3).

Histogram of allele frequency for all 220 alleles in the 35 accessions of cultivated types of Perilla crop and their weedy types
Cluster analysis and population structure
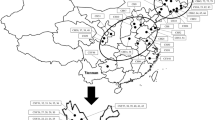
The phylogenetic tree constructed using UPGMA revealed that the 35 Perilla accessions clustered into three major groups (Fig. 2). Group I included 13 accessions of cultivated var. frutescens and one accession of weedy var. frutescens. Group II included three accessions of weedy var. frutescens and one accession of cultivated var. frutescens. Group III included 17 accessions, which consisted of eight accessions of weedy var. crispa, five accessions of cultivated var. crispa and four accessions of weedy var. frutescens (Fig. 2). In addition, to understand the population structure among the 35 Perilla accessions, we used a model-based approach via STRUCTURE software to divide into each accession into their subgroups. It proved difficult to choose K values calculated from five replicate sets ranging from 1 to 10; thus, we used the ad hoc measure ΔK suggested by Evanno et al. (2005). Based on this criteria, all 35 Perilla accessions were divided into two main groups at K = 2 (Fig. 3). However, some Perilla accessions were admixed within these two groups. Following the membership probability threshold of 0.8, all accessions were divided into three groups; Group I, Group II and the admixed group. As a result, Group I only included 13 accessions of cultivated var. frutescens. Group II included 18 Perilla accessions that consisted of five accessions of weedy var. frutescens, five accessions of cultivated var. crispa and eight accessions of weedy var. crispa. The admixed group included four accessions, which consisted of one accession of cultivated var. frutescens and three accessions of weedy var. frutescens (Fig. 4).

UPGMA dendrogram based on the new Perilla SSR markers. The accessions of cultivated types of Perilla crop and their weedy types are presented in Table 1. Open circle: Cultivated var. frutescens, filled circle: Weedy var. frutescens, open squared: Cultivated var. crispa, filled squared: Weedy var. crispa

Magnitude of ΔK as a function of K. The peak value of ΔK was revealed at K = 2, suggesting two genetic clusters in Perilla accessions

Population structure of 35 accessions of cultivated types of Perilla crop and their weedy types based on the new 40 Perilla SSR markers for K = 2
Discussion
Evaluation of the genetic diversity and relationships for plant genetic resources is an essential prerequisite for the conservation and development of crop species. Of the protocols that facilitate the assessment of molecular diversity, SSR or microsatellite marker is the preferred system since it detects a large number of DNA polymorphisms with relatively simple technical complexity (Park et al. 2009). SSRs have therefore become the preferred molecular marker system for analysis in crop genetics. Recently, the utilization of SSR markers has become well established for the assessment of genetic diversity, relationships, and population structure in Perilla crop (Lee and Kim 2007; Lee et al. 2007; Park et al. 2008, Sa et al. 2013, 2015; Ma et al. 2017). However, few SSR markers have been developed in previous studies of Perilla (Kwon et al. 2005; Park et al. 2008), and these SSR markers are insufficient to correctly assess genetic diversity and relationships in Perilla crop. The results presented herein using new SSR primers in Perilla crop will certainly provide a clue to expand our understanding of the genetic diversity in Perilla species from East Asia. The high costs of developing SSR primers have limited their utilization in minor crops. The primer sequences for SSR loci designed for the population presented herein and the resulting genetic information will be highly useful in Perilla crop germplasm preservation based on the molecular marker resources to characterize morphology in East Asia and other genetic studies, such as molecular breeding programs.
In this study, we demonstrated the successful application of new Perilla SSR markers in a study of the genetic diversity and population structure among accessions of cultivated types of Perilla crop and their weedy types in East Asia. According to our results, a total of 220 alleles with 40 SSRs were detected segregating in the 35 Perilla accessions from China, Korea and Japan, which yielded an average of 5.5 alleles per locus (Table 2). This value appears to be low or high when compared to the effective number of alleles per SSR locus in other major crops, such as the 6.8 in rice (Ni et al. 2002), 5.9 in maize (Jochen et al. 2005), and 3.6 in barley (Hamza et al. 2004). Thus, the number of alleles in our study reflect the utility of new Perilla EST-SSR markers in determining unique genetic profiles of individual genotypes of Perilla crop that should prove useful in future genetic and breeding studies.
In the analysis of the four groups of accessions (cultivated and weedy types of var. frutescens and of var. crispa) using the 40 SSR primers, the average gene diversity values were 0.415 and 0.606 for the cultivated and weedy types of var. frutescens, respectively, and 0.308 and 0.480 for the cultivated and weedy types of var. crispa, respectively (Table 3). Although the number of samples was limited, the obtained results using new SSR primers implied that the weedy types of var. frutescens and var. crispa maintain increased variability compared with the cultivated types in East Asia (Table 3). Perilla crop is widely distributed and cultivated in East Asia. This information regarding genetic diversity between the two cultivated types of Perilla crop, and their weedy types may be useful for the preservation of germplasm resources and crop conservation in East Asia. On the other hand, according to our results, the clustering patterns and population structure did not permit clear discrimination of the two cultivated types of P. frutescens and their weedy types and did not clearly relate to the geographical location of these Perilla accessions in the UPGMA dendrogram. However, if classification is considered at the level of sub-clusters, the overall pattern of the two cultivated types of P. frutescens and their weedy types in the dendrogram and population structure were consistent with the cultivated or weedy types of P. frutescens. In particular, most accessions of cultivated var. frutescens are clearly distinct from accessions of weedy var. frutescens and cultivated and weedy types of var. crispa, but most accessions of weedy type of var. frutescens and cultivated and weedy types of var. crispa were not clearly identified. The diffusion for accessions of cultivated and weedy types of Perilla crop in East Asia might occur from China to Korea and Japan via multiple routes, such as the previous route reported by Lee and Ohnishi (2003).
What is the origin of the weedy types of cultivated types of Perilla crop? Weedy types have been reported in several crops, e.g., rice, barley, oat and sorghum (Hancock 1992; Ladizinsky 1998). In these crops, the weedy types have been considered either wild ancestor(s) of the crop or escaped form(s) from cultivated crops. Nitta and Ohnishi (1999) first suggested that the weedy type of Perilla crop in Japan probably originated from hybrids between cultivated types of var. frutescens and var. crispa or may represent a form that escaped from cultivation. Lee et al. (2002) and Lee and Ohnishi (2003) suggested that the weedy types of Perilla crop in East Asia are the key taxon in understanding the origin of cultivated types of var. frutescens and of var. crispa. In the dendrogram and population structure presented in our results, most accessions of the weedy type of var. frutescens and cultivated and weedy types of var. crispa were closely related to each other. Although we cannot offer a clear explanation regarding this clustering given the small number of Perilla samples examined, some weedy accessions of var. frutescens and var. crispa might be considered a wild form of Perilla crop because the wild type Perilla crop is not yet known. However, some of these accessions may be derived from either voluntarily escaped seeds from cultivation or hybrids between weedy and cultivated types. However, natural hybrids and the outcrossing rate in Perilla species have not been reported to date to our knowledge. The previous studies of Lee and Ohnishi (2001) and Sa et al. (2012) reported that the cultivated type of var. frutescens might be differentiable from the weedy type of var. frutescens but that the cultivated and weedy types of var. crispa were not sufficiently differentiable. The accessions of cultivated type of var. frutescens exhibited greater variation in seed characteristics than the weedy type of var. frutescens and the cultivated and weedy types of var. crispa. Namely, most accessions of cultivated var. frutescens had a seed sizes greater than 2 mm and comparatively high seed germination rates, whereas weedy var. frutescens and cultivated and weedy types of var. crispa exhibited seed sizes of < 2 mm and low seed germination rates. Thus, var. crispa may not be considered a complete domesticated form. Therefore, further analyses are necessary to clarify the taxonomic position for these weedy Perilla samples. Our study results using new Perilla SSR primers validate the proposal that the weedy types of Perilla species are the key taxon in understanding the origin of cultivated type of var. frutescens and of var. crispa. The new Perilla SSR primers described in this study should facilitate the evaluation of genetic diversity and population structure and could be used for cultivar identification, conservation of Perilla germplasm resources, genome mapping and tagging of important genes/QTLs for Perilla breeding programs in the future.
References
Bostein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Genet 32:314–331
Dice LR (1945) Measures of the amount of ecologic association between species. Ecology 26:297–302
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fukushima A, Nakamura M, Suzuki H, Saito K, Yamazaki M (2015) High-throughput sequencing and de novo assembly of red and green forms of the Perilla frutescens var. crispa Transcriptome. PLoS ONE 10(6):e0129154. https://doi.org/10.1371/journal.pone.0129154
Hamza S, Hamida WB, Rebaϊ A, Harrabi M (2004) SSR-based genetic diversity assessment among Tunisian winter barley and relationship with morphological traits. Euphytica 135:107–118
Hancock JF (1992) Plant evolution and the origin of crop species. Prentice Hall, Englewood Cliffs
Jochen CR, Hamrit S, Heckenberger M, Schipprack W, Maurer HP, Bohn M, Melchinger AE (2005) Genetic structure and diversity of European flint maize populations determined with SSR analyses of individuals and bulks. Theor Appl Genet 111:906–913
Kwon SJ, Lee JK, Kim NS, Yu JW, Dixit A, Cho EG, Park YJ (2005) Isolation and characterization of SSR markers in Perilla frutescens Britt. Mol Ecol Notes 5:454–456
Ladizinsky G (1998) Plant evolution under domestication. Chapman & Hall Published, Raleigh
Lee JK, Kim NS (2007) Genetic diversity and relationships of cultivated and weedy types of Perilla frutescens collected from East Asia revealed by SSR markers. Korean J Breed Sci 39:491–499
Lee JK, Ohnishi O (2001) Geographic differentiation of morphological characters among Perilla crops and their weedy types in East Asia. Breed Sci 51:247–255
Lee JK, Ohnishi O (2003) Genetic relationships among cultivated types of Perilla frutescens and their weedy types in East Asia revealed by AFLP markers. Genet Resour Crop Evol 50:65–74
Lee JK, Nitta M, Kim NS, Park CH, Yoon KM, Shin YB, Ohnishi O (2002) Genetic diversity of Perilla and related weedy types in Korea determined by AFLP analyses. Crop Sci 42:2161–2166
Lee JK, Kwon SJ, Park BJ, Kim MJ, Park YJ, Ma KH, Lee SY, Kim JH (2007) Analysis of genetic diversity and Relationships of cultivated and weedy types of Perilla frutescens collected from Korea by using microsatellite markers. Korean J Genet 29:81–89
Li HL (1969) The vegetables of ancient China. Econ Bot 23:235–260
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Ma SJ, Sa KJ, Hong TK, Lee JK (2017) Genetic diversity and population structure analysis in Perilla frutescens from Northern areas of China based on simple sequence repeats. Genet Mol Res 16(3):gmr16039746. https://doi.org/10.4238/gmr16039746
Makino T (1961) Makino’s new illustrated flora of Japan. Hokuryukan, Tokyo (in Japanese)
Mutz KO, Heilkenbrinker A, Lonne M, Walter JG, Stahl F (2012) Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol 24:22–30
Ni J, Colowit PM, Mackill DJ (2002) Evaluation of Genetic Diversity in Rice Subspecies Using SSR Markers. Crop Sci 42:601–607
Nitta M (2001) Origin Perilla crops and their weedy type. Ph.D. Thesis, Kyoto University, Kyoto, p. 78
Nitta M, Ohnishi O (1999) Genetic relationships among two Perilla crops, shiso and egoma, and the weedy type revealed by RAPD markers. Jpn J Genet 74:43–48
Nitta M, Lee JK, Ohnishi O (2003) Asian Perilla crops and their weedy forms: their cultivation, utilization and genetic relationships. Econ Bot 57:245–253
Nitta M, Lee JK, Kang CW, Katsuta M, Yasumoto S, Liu D, Nagamine T, Ohnishi O (2005) The distribution of Perilla species. Genet Resour Crop Evol 52:797–8044
Park YJ, Dixit A, Ma KH, Lee JK, Lee MH, Chung CS, Nitta M, Okuno K, Kim TS, Cho EG, Rao VR (2008) Evaluation of genetic diversity and relationships within an on-farm collection of Perilla frutescens (L.) Britt. using SSR markers. Genet Resour Crop Evol 55:523–535
Park YJ, Lee JK, Kim NS (2009) Simple sequence repeat polymorphisms (SSRPs) for evaluation of molecular diversity and germplasm classification of minor crops. Molecules 14:4546–4569
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalski A (1996) The comparison of RFLP, RAPD, AFLP and SSR (SSR) markers for germplasm analysis. Mol Breed 2:225–238
Prasad M, Varshney RK, Roy JK, Balyan HS, Gupta PK (2000) The use of microsatellites for detecting DNA polymorphism, genotype identification and genetic diversity in wheat. Theor Appl Genet 100:584–592
Pritchard JK, Wen W (2003) Documentation for STRUCTURE software: Version 2
Rohlf FJ (1998) NTSYS-pc: numerical taxonomy and multivariate analysis system. Version: 2.02. Exter Software, Setauket
Sa KJ, Park JY, Park KJ, Lee JK (2010) Analysis of genetic diversity and relationships among waxy maize inbred lines in Korea using SSR markers. Genes Genomics 32:375–384
Sa KJ, Kim JA, Lee JK (2012) Comparison of seed characteristics between the cultivated and the weedy types of Perilla species. Hort Environ Biotechnol 53(4):310–315
Sa KJ, Choi SH, Ueno M, Park KC, Park YJ, Ma KH, Lee JK (2013) Identification of genetic variations of cultivated and weedy types of Perilla species in Korea and Japan using morphological and SSR markers. Genes Genomics 35:649–659
Sa KJ, Choi SH, Ueno M, Lee JK (2015) Genetic diversity and population structure in cultivated and weedy types of Perilla in East Asia and other countries as revealed by SSR markers. Hortic Environ Biotechnol 56(4):524–534
Schontz D, Rether B (1999) Genetic variability in foxtail millet, Setaria italica (L.) P. Beauv.: identification and classification of lines with RAPD markers. Plant Breed 118:190–192
Tong W, Kwon SJ, Lee JS, Choi IK, Park YJ, Choi SH, Sa KJ, Kim BW, Lee JK (2015) Gene set by de novo assembly of Perilla species and expression profiling between P. frutescens (L.) var. frutescens and var. crispa. Gene 559:155–163
Wang R, Yu Y, Zhao J, Shi Y, Song Y, Wang T, Li Y (2008) Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor Appl Genet 117:1141–1153
Wang L, Wang Z, Chen J, Liu C, Zhu W, Wang L, Meng L (2015) De novo transcriptome assembly and development of novel microsatellite markers for the traditional chinese medicinal herb, Veratrilla baillonii franch (Gentianaceae). Evol Bioinform 11(S1):39–45. https://doi.org/10.4137/EBO.S20942
Xia XC, Reif JC, Melchinger AE, Frisch M, Hoisington DA, Beck K, Pixley K, Warburton ML (2005) Genetic diversity among CIMMYT maize inbred lines investigated with SSR markers: II. Subtropical, tropical midaltitude, and highland maize inbred lines and their relationships with elite U.S. and European maize. Crop Sci 45:2573–2582
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (#2016R1D1A1B01006461).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kyu Jin Sa declares that he has no conflict of interest. Ik-Young Choi declares that he has no conflict of interest. Kyong-Cheul Park declares that he has no conflict of interest. Ju Kyong Lee declares that he has no conflict of interest.
Research involving human and animal participants
This article does not contain any studies with human subjects or animals performed by any of the above authors.
Rights and permissions
About this article

Cite this article
Sa, K.J., Choi, IY., Park, KC. et al. Genetic diversity and population structure among accessions of Perilla frutescens (L.) Britton in East Asia using new developed microsatellite markers. Genes Genom 40, 1319–1329 (2018). https://doi.org/10.1007/s13258-018-0727-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-0727-8