
Abstract
Fiber length is one of the most important fiber quality traits in Upland cotton (Gossypium hirsutum L.), the most important fiber crop, and its improvement has been impeded in part by a lack of knowledge regarding its genetic basis. Introgressed backcross inbred lines (BILs) or near isogenic lines (NILs) differing in fiber length in the same genetic background, developed through advanced backcrossing between Upland cotton and extra-long staple cotton (G. barbadense L.), provide an important genomic resource for studying the molecular genetic basis of fiber length. In the present study, a long-fiber group and a short-fiber group, each with five BILs of Upland cotton, were selected from a BIL population between G. hirsutum and G. barbadense. Through a microarray-based comparative transcriptome analysis of developing fibers at 10 days postanthesis from the two groups, 1478 differentially expressed genes (DEGs) were identified. A total of 166 DEGs were then mapped to regions of fiber length quantitative trait loci (QTL), including 12 QTL hotspots and 2 QTL identified previously in the BIL population from which the two sets of BILs were selected. Several candidate genes possibly underlying the genetic control of fiber length differences between G. barbadense and G. hirsutum, including GhACX and GhKIF, were identified in this study. These results provide a list of positional candidate genes for the fine-scale mapping and map-based cloning of fiber length QTL, which will facilitate targeted gene transfer from G. barbadense to Upland cotton to further improve fiber quality.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton (Gossypium spp.) is an economically important industrial crop in the world, and its fibers represent one of the main naturally available raw materials for the textile industry, which contributes more than 10 billion dollars a year to the global economy. The Gossypium genus contains nine different genome groups comprising approximately 50 species [40]. Of the four Gossypium species that have been historically cultivated for the fiber [4], tetraploids G. hirsutum and G. barbadense currently dominate commercial cotton fiber production, with the former accounting for approximately 95% of worldwide cotton production [39], due to its high yield potential and broad environmental adaptability. G. barbadense accounts for the rest of world cotton fiber production, due to its superior fiber quality attributes. Combining the yield from G. hirsutum and the fiber quality from G. barbadense has been an ongoing goal of cotton breeders and geneticists; however, only limited success has been achieved thus far [47].
Cotton fibers are single-celled trichomes that eventually differentiate from single cells located in the ovule epidermis [2]. Fiber development consists of four overlapping stages: initiation, primary cell wall formation, secondary cell wall formation, and maturation. The quality and productivity of cotton fibers depend on two biological processes: fiber initiation to determine the number of fibers present on each ovule and fiber elongation to control the final length (and partially strength and fineness) of each fiber [16]. After initiation from − 1 to 3 days post anthesis (DPA), fibers are in the fastest elongation stage at 5–10 DPA, and cotton fiber elongation continues up to 20–25 DPA. Numerous studies have demonstrated that cotton fiber development is a complex biological process [24, 48, 49]. Therefore, understanding the mechanisms underlying fiber elongation through genetic and molecular analysis is essential for improving fiber quality.
Identification and characterization of quantitative trait loci (QTL) of cotton fiber quality have been a research focus for more than 2 decades [32,33,34]. As a result, a large number of QTL associated with fiber quality-related traits have been reported based on interspecific G. hirsutum × G. barbadense populations [5, 6, 8,9,10,11, 19,20,21,22, 28, 44,45,46]. Among the numerous cotton fiber quality QTL that have been mapped, more QTL have been mapped for fiber length than any for other fiber quality trait. Specifically, 281 QTL have been detected for fiber length based on 88 different QTL studies (as summarized in [34]). However, few studies involving validation of the efficacy or marker association of cotton fiber length QTL in advanced breeding generations have been published. Hence, the genes underlying fiber length QTL remain elusive. Cotton fiber cell elongation is a complex physiological process regulated by many genes [2]. For example, GhPIP2, GhHOX3, GhCaM7, GhPAG1, and GhCPC play an important role in controlling cotton fiber elongation [1, 23, 26, 35, 36, 42] and 81 genes were upregulated and 2553 ‘expansion-associated’ genes were downregulated in G. arboreum compared with their expression in Upland cotton at 7–10 DPA through microarray analysis, suggesting a trend of global gene repression during the fiber elongation stage in allotetraploids. In a recent RNA-seq study, 1551 differentially expressed genes (DEGs) were identified between two BILs differing in fiber length, but only eight DEGs were co-localized with 4 fiber length QTL identified in the BIL population containing the two BILs [25]. A comparative transcriptome study was also conducted in diploid cultivated cotton using isogenic fuzzy-lintless and normal fuzzy-linted diploid G. arboretum lines at two stages (0 and 10 DPA), using an Affymetrix cotton GeneChip genome array [12]. Numerous DEGs have been identified during the fiber elongation stage. However, these genes do not explain the molecular mechanism underlying the natural genetic differences between genotypes with different fiber length. Therefore, it is necessary to use more BILs to perform genome-wide analyses of DEGs to identify genes associated with cotton fiber quality traits, including fiber length.
In this study, RNA was extracted from developing fibers at 10 DPA in a long-fiber (LF) group and a short-fiber (SF) group, each including five backcross inbred lines (BILs), and was then compared for transcriptome analysis. Since the fibers of 10 DPA are in the fastest elongation phase after initiation, we chose to analyze the fibers of 10 DPA to identify DEGs that may be associated with determining the length of the cotton fibers at this important stage of fiber development. The aim of this study was to identify DEGs associated with fiber length QTL by a microarray analysis using the Affymetrix GeneChips and to gain insight into the potential molecular genetic differences in cotton fiber elongation between two groups of BILs differing in fiber length. This positional candidate gene approach based on the integration of Affymetrix GeneChips with QTL mapping and physical mapping represents one of the most efficient strategies to narrow the number of candidate genes for fiber length.
Materials and methods
Plant material
The interspecific backcross inbred population of 146 lines was developed at New Mexico State University [46]. For the current study, 10 BILs were then selected based on differences in fiber length and similarities in other traits (Supplementary Data 2).The results were analyzed using least significant difference (LSD) tests for significant differences with SAS software (SAS Institute Inc., 2009, USA). Five BILs (i.e. NMGA-062, NMGA-089, NMGA-017, NMGA-051 and NMGA-070, designated LF1, LF2, LF3, LF4, and LF5, respectively) were long-fiber (LF) with an average fiber length of 31.79 mm; and five other BILs (i.e., NMGA-095, NMGA-026, NMGA-144, NMGA-105, and NMGA-140, designated SF1, SF2, SF3, SF4 and SF5, respectively) were short-fiber (SF) with an average fiber length of 27.35 mm. The two group (LF vs. SF), differed significantly in fiber length (31.79 vs. 27.35 mm), but no significant differences in strength, micronaire, seed cotton yield, lint yield, lint percentage, boll weight and Verticillium wilt index value. The 10 lines, 5 LF and 5 SF BILs were planted in the Experimental Farm, the Cotton Research Institute of the Chinese Academy of Agricultural Sciences, Anyang, Henan Province, China (E 114°48′, N 36°06′) in the summer of 2009. The field plots were arranged using a randomized complete block design with three replications. The flowers appearing on each flowering day were marked, and the cotton boll samples were harvested before 10 am at 5, 10, 15, 20, and 25 DPA and placed immediately on ice [41]. To minimize experimental errors, all samples from each developmental stage were collected on the same day. The fibers of 5–25 DPA developing ovules were dissected, frozen in liquid nitrogen, and stored at − 80 °C until further processing.
RNA extraction
Total RNA was extracted from developing fibers at 10 DPA, from the BILs and from two parental lines, Giza75 and SG 747, through the improved cetyltrimethylammonium bromide (CTAB) method [27]. Agarose gel electrophoresis was used to detect the proportion of 28S and 18S rRNA in the total RNA to test its integrity. A DU®800 Nucleic Acid/Protein Analyzer (Beckman Coulter, Brea, CA, USA) was employed to determine the RNA concentration and the OD260/OD280 value.
Microarray hybridizations and data analysis
The commercially available Affymetrix GeneChip® Cotton Genome Microarray (Affymetrix Inc., Santa Clara, CA), including 239,777 probe sets representing 21,854 cotton transcripts from various EST databases, was used in this study. The extracted RNA was sent to Shanghai Seaview Biotech Co., Ltd. for GeneChip analysis. RNA labeling was performed using the Affymetrix GeneChip® 3′ IVT Express kit, and hybridization was performed according to the manufacturer’s protocols. After the chip was scanned with a high-resolution scanner, the data were analyzed. PARTEK software was used for analysis of variance (ANOVA) because it uses a robust multi-array averaging (RMA) method that calculates only the signal values of perfectly matched (PM) probes and considers the signal value of all samples. The RMA method is currently the mainstream gene chip algorithm being used.
Reverse transcription (RT) and quantitative PCR
The cDNA reaction was carried out using the iScript™ cDNA Synthesis kit (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions. The reaction performed in the absence of reverse transcriptase was used as a negative control for detecting genomic DNA contamination in the RNA samples. This reaction was then used as a qPCR reaction template to verify that no amplification occurred. After cDNA synthesis, the qPCR reaction was performed with a Bio-Rad CFX96 real-time PCR detection system using iTaq™ SYBR® Green Supermix (Bio-Rad Laboratories). The PCR conditions and the protocols for determining primer efficiency have been previously described [3]. Histone3 (GenBank Accession AF024716) was used as an internal control. Primer sequences are listed in Supplementary Data 1.
Chromosomal locations of DEGs within fiber length QTL regions
To locate the DEGs in the fiber length QTL anchored by simple sequence repeat (SSR) markers, we collected the tag names from the [“Guazuncho2” (G. hirsutum) × “VH8-4602” (G. barbadense)] map that was previously published [32, 33]. The marker sequence data were downloaded from the CottonGen database (https://www.cottongen.org/data/download/marker) (Yu et al. 2014). Secondly, using the marker and DEG sequences as the query and the genomic sequence [49] as the subject, the chromosomal locations of the marker and DEGs were identified. The chromosomal location of the QTL hotspot regions of the fiber lengths previously reported by Said et al. [32,33,34], and the 4 fiber length QTL reported by Yu et al. [46] were identified using the anchoring marker locations in the map. The candidate genes in the QTL hotspot regions or the loci were then identified and used in further studies. This analysis method of chromosomal locations of DEGs within fiber length QTL regions followed Wu et al. [41].
Results
Microarray and data analysis
The mature fiber length of LF group (i.e. NMGA-062, NMGA-089, NMGA-017, NMGA-051 and NMGA-070) and SF group (i.e., NMGA-095, NMGA-026, NMGA-144, NMGA-105, and NMGA-140) BILs averaged 31.79 versus 27.35 mm, respectively, as described previously [43] (see “Materials and methods”) and were selected for their differences in fiber length (FL) and similarities in other agronomic. Their highly similar genetic background was also evident in that they were identical in 96.7% of the SSR and SNP marks assayed (unpublished), which is suitable for the current comparative analysis. In this study, we selected 10 DPA developing fibers from the LF group and SF group to study the molecular aspects of fiber elongation, because the elongation rate of fiber at 10 DPA was shown to be highest with a significant difference in the FL between the LF group and SF group [25]. Affymetrix GeneChip Cotton Genome Array with multiple probes was used for a comparison each gene between the LF and SF BIL groups further minimized complications. In the current study, 1478 DEGs between the LF group and SF group in the 10 DPA fiber transcriptome were identified (> twofold change; Bonferroni-corrected P-value threshold < 0.05) (Supplementary Data 3). When the LF and SF groups were compared, 403 (27.27% of 1478) of the DEGs were upregulated and 1075 genes (72.73% of 1478) were downregulated in the LF group.
A cluster analysis of the gene expression data from the 1478 DE genes (DEGs) separated all the five LF lines from two of the five SF lines (Fig. 1). Specifically, NMGA-062 (LF1), NMGA-089 (LF2), NMGA-017 (LF3), and NMGA-070 (LF5) of LF group had the highest similarity in gene expression patterns, followed by the high similarity of gene expression between NMGA-026 (SF2), NMGA-144 (SF3), NMGA-105 (SF4), and NMGA-140 (SF5) of SF group. The result demonstrates the reliability of the selected BILs and the grouping method used in this study and provides great confidence in the results obtained. However, one of the five LF lines, i.e., NMGA-051 (LF4) was group together with the SF group, indicating that high levels of similarity in phenotypes (here lint yield, lint percent, boll weight, Verticillium wilt index, uniformity) may not always be reflected by high levels of similarity in gene expressions. The DEGs identified between the two BIL groups may indicated a common genetic regulatory mechanism for genes associated with fiber development (more specifically, fiber length-related genes) in cotton.

Clustering of differentially expressed genes. LF1:NMGA-062; LF2:NMGA-089; LF3:NMGA-017; LF4:NMGA-051; LF5:NMGA-070; SF1:NMGA-095; SF2:NMGA-026; SF3:NMGA-144; SF4:NMGA-105; SF5:NMGA-140
Candidate DEGs within the fiber length QTL regions identified in the BIL population
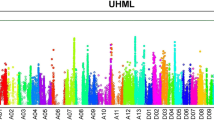
The chromosomal distribution of the DEG identified in this study can be determined by completing the G. hirsutum (AD) genomic sequencing [24, 49]. According to published data for the cotton genome, 420 of the 1478 DEGs were mapped to chromosome locations from At_chr1 to At_chr13 in the At subgenome, 727 were mapped from Dt_chr1 to Dt_chr13 in the Dt subgenome, and the remaining 121 DEGs were mapped to unmapped scaffolds. Co-localization of the QTLs with the identified 1478 DEGs will help identify candidate genes for fiber length (Supplementary Data 4). We mapped 166 DEGs with the 4 fiber length QTLs [46] and 13 fiber length QTL hotspots, as determined by a previous meta-analysis [33] (Supplementary Data 5 and Fig. 2). The 166 genes were distributed in A01 (5 DEGs), A03 (12), A04 (4), A05 (2), A07 (4), A11 (10), A12 (9), D05 (54), D08 (30), D11 (3) and D12 (33). Of the 166 DEGs, 145 were divided into 32 GO terms based on their biological process, cellular component, and molecular function (Fig. 3). Among the cellular component category, the DEGs were further classified as intracellular (42 genes, 25.30% of 166 DEGs), partially intracellular (41 genes, 24.70%), and organelle (36 genes, 21.69%). For the biological processes, the organic substance metabolic process (56 genes, 33.73%), the primary metabolic process (54 genes, 32.53%), and the cellular metabolic process (49 genes, 29.52%) were associated with a higher number of identified DEGs. In the molecular function category, organic cyclic compound binding (34 genes, 20.48%), heterocyclic compound binding (34 genes, 20.48%), and ion binding (30 genes, 18.07%) were the three most abundant subclasses, followed by transferase activity, hydrolase activity, small molecule binding, and protein binding. The KOBAS was used to further analyze the 166 DEGs identified by the microarray analysis to identify the metabolic pathways in which they function. The KOBAS mapped 49 of the 166 DEGs to 31 KEGG pathways. The pathways with the highest 8 unigene representation were purine metabolism (8, 25.81%), thiamine metabolism (5, 16.13%), and cysteine and methionine metabolism (3, 16.13%) (Fig. 4).


Mapping of DEGs with fiber length QTL hotspots in G. hirsutum

Distribution of the gene ontology functional classifications of 166 differentially expressed genes

Significantly enriched pathways of 166 differentially expressed genes identified using KOBAS
A gene expression enrichment analysis was performed on the 44 genes upregulated in the LF group and the 122 genes upregulated in the SF group. The 44 upregulated DEGs in the LF group were divided into 39 GO terms based on their biological process, cellular component, and molecular function, respectively (Fig. 5). For the biological processes, the organic substance metabolic process (25 genes, 56.82% of the upregulated 44 DEGs) and the primary metabolic process (24 genes, 54.55%) were associated with the highest number of DEGs identified in the GO terminology. In the molecular function category, organic cyclic compound binding (11 genes, 25.00%) and heterocyclic compound binding (11 genes, 25.00%) were the two most abundant subclasses, followed by ion binding, transferase activity, small molecule binding, carbohydrate derivative binding, and hydrolase activity. Among the cellular component category, the DEGs were further divided into partial intracellular (11 genes, 25.00%) and intracellular (11 genes, 25.00%). Based on their biological process, cellular component, and molecular function (Fig. 6), the 122 upregulated DEGs in the SF group were also classified into 33 GO terms. For the biological processes, the organic substance metabolic process (31 genes, 25.41% of the 122 DEGs) and the primary metabolic process (30 genes, 24.59%) represented the majority of the DEGs. In the molecular function category, organic cyclic compound binding (23 genes, 18.85%) and heterocyclic compound binding (23 genes, 18.85%) were the two most abundant subclasses, followed by ion binding, hydrolase activity, transferase activity, protein binding, small molecule binding and oxidoreductase activity. Among the cellular component, DEGs were further classified into intracellular (31 genes, 25.41%) and partially intracellular (30 genes, 24.59%).

Distribution of the gene ontology functional classifications of 44 differentially expressed genes

Distribution of the gene ontology functional classifications of 122 differentially expressed genes
Since the 10 BILs were selected by analyzing the G. hirsutum × G. barbadense BIL population [46], we concentrated on the 4 fiber length QTLs identified in the BILs to identify fiber length-related DEGs. Of the 4 fiber length QTLs, two were found to colocalize with 6 DEGs, as shown in Supplementary Data 6. The 2 QTLs for fiber length were qFL-08A-c12-1 on At_chr 12 and qFL-08A-c21-1 on Dt_chr11. Each QTL region carried three DEGs related to fiber length. The qFL-08A-c12-1 on At_chr 12 had three co-localized DEGs encoding a kinesin-like protein, a probable lipid phosphate phosphatase beta protein, and an unknown protein. The qFL-08A-c21-1 QTL on Dt_chr11 also exhibited three co-localized DEGs encoding a peroxisomal acyl-coenzyme a oxidase 1-like protein, the dual-specificity protein phosphatase 8 (DSP8) protein, and an unknown protein.
Mapping more DEGs with the previously reported fiber length QTL hotspots in the G. hirsutum genome
Since only 2 fiber length QTL were mapped with DEGs in the BIL population, including the 10 lines used in this study, we also used the 13 fiber length QTL hotspots previously reported from a meta-analysis [33] to identify more fiber length-related genes. A total of 160 DEGs were found, and a total of 12 QTL hotspot fibers were co-localized. The 12 fiber length QTL hotspots with DEGs were mapped onto 10 chromosomes, including 7 At chromosomes and 3 Dt chromosomes (At_chr1, At_chr3, At_chr4, At_chr5, At_chr7, At_chr11, At_chr12, Dt_chr5, Dt_chr8, Dt_chr12).
Four of these 12 fiber length QTL hotspots were mapped to two pairs of homeologous chromosomes (At_chr5 vs. Dt_chr5 and At_chr12 vs. Dt_chr12). Fiber length (FL)_QTL_Hotspot(4) was identified at 0.19–2.08 cM on At_chr5, and this hotspot contained 2 DEGs encoding the uncharacterized protein LOC105770639 and an unknown protein. FL_QTL_Hotspot(4) at 0.04–8.96 cM on Dt_chr5 contained 33 DEGs, including genes encoding proteins such as spx domain-containing protein 1-like, nad kinase 1-like, monogalactosyldiacylglycerol synthase chloroplastic-like, glucan endo-1, 3-beta-glucosidase 14-like isoform ×1, vacuolar sorting-associated protein 4, calcineurin b-like protein 10, and jasmonate ZIM-domain protein isoform 1. FL_QTL_Hotspot(6) at 8.96–16.75 cM on Dt_chr5 contained 21 DEGs, including genes encoding proteins such as pleiotropic drug resistance protein 1-like, plac8 family protein, 1-aminocyclopropane-1-carboxylate oxidase, beta-galactosidase 1-like, casein kinase i isoform delta-like, and calmodulin-binding protein. Since the three hotspots were not located in the same regions of the two homeologous chromosomes (At_chr5 vs. Dt_chr5), no common DEGs were identified.
Similarly, no common DEGs were identified for the QTLs on the other two pairs of homeologous chromosomes. FL_QTL_Hotspot(4) was identified at 70.04–76.00 cM on At_chr12, and it contained five DEGs encoding for heat shock factor protein 5, AP2 ERF and B3 domain-containing transcription factor rav1-like, the ubiquitin-conjugating enzyme E2 2, the probable signal peptidase complex subunit 1, and an unknown protein. As a comparison, FL_QTL_Hotspot(6) was identified at 25.00–45.88 and 50.67–59.04 cM on Dt_chr12, and it contained 33 DEGs, including genes encoding nucleotidyltransferase family protein isoform 1, skp1 ask-interacting protein 16 isoform 1, and fasciclin-like arabinogalactan protein 19, among others.
In addition, in the two above mentioned At subgenome chromosomes, FL_QTL_Hotspot(10) ranged from 17.66 to 36.80 cM on At_chr1 and contained five genes, including genes encoding the glycine-rich RNA-binding protein mitochondrial-like, mfp1 attachment factor 1-like, the n-lysine methyltransferase SEE1, the ribonuclease p protein subunit p25-like protein isoform x1, and an unknown protein. FL_QTL_Hotspot(6) ranged from 2.82 to 9.85 cM on At_chr3 and contained 6 genes, including genes encoding CBL-interacting serine threonine-protein kinase 11, a haloacid dehalogenase-like hydrolase superfamily protein, the inner membrane protein PPF-chloroplastic isoform × 1, an expansin protein, a protein from the glutaredoxin family and an unknown protein. FL_QTL_Hotspot(7) ranged from 35.00 to 81.57 cM on At_chr3 and contained six genes, including genes encoding for the male gametophyte defective 2 isoform 1, the E3 ubiquitin-protein ligase prt1-like, the protein salt inducible-like partial, the E3 ubiquitin-protein ligase rnf182-like, the probable calcium-binding protein cml27 and the NEDD8-activating enzyme E1 regulatory subunit. FL_QTL_Hotspot(5) ranged from 40.41 to 53.33 cM on At_chr4 and contained 4 genes, including genes encoding for the transducin family protein wd-40 repeat family isoform partial, the protein gpr107-like, the protein tip41-like, and the kinesin-like protein kif22. FL_QTL_Hotspot(4) ranged from 4.69 to 9.28 cM on At_chr7 and contained 4 genes, including genes encoding for the protein cyclin-h1-1 isoform × 2, the homeobox-leucine zipper protein hat5-like, the 40 s ribosomal protein s7, and the ribosomal protein s21 family protein isoform 1.
One additional Dt subgenome chromosome carried fiber length QTL hotspots. FL_QTL_Hotspot(7) was located at 32.00-58.05 cM on Dt_chr8, and it contained 30 DEGs encoding for proteins such as expansin-like b1, sucrose synthase 1, the snare-interacting protein KEULE, the nuclear pore complex protein NUP98a, the protein trichome birefringence-like 43, and the bromo-adjacent domain-containing protein, among others.
Candidate DEG expression analysis
Two upregulated and four downregulated DEGs identified in the LF group by microarray analysis that co-localized with fiber length QTLs were further analyzed using qRT-PCR. Developing fibers at 5, 10, 15, 20, and 25 DPA were analyzed. The six genes included kinesin-like protein (GenBank no. DT054207.1, designated GhKIF), probable lipid phosphate phosphatase beta (GenBank no. DW502593.1, designated GhPAP2), peroxisomal acyl-coenzyme a oxidase 1-like (GenBank no. DT462539.1, designated GhACX), dual-specificity protein phosphatase dsp8 (GenBank no. DT051818.1, designated GhDSP8), and unknown genes (GenBank no. DT052644.1, and GenBank no. DT455687.1) (Fig. 7).

Expression profile of GhKIF, GhACX, Unknown1, GhPAP2, GhDSP8, and Unknown2 in developing fibers at 5–25 days postanthesis (DPA). a Kinesin-like protein (GenBank no. DT054207.1, designated GhKIF); b Peroxisomal acyl-coenzyme a oxidase 1-like (GenBank no. DT462539.1, designated GhACX); cUnknown1 (GenBank no. DT052644.1); d Probable lipid phosphate phosphatase beta (GenBank no. DW502593.1, designated GhPAP2); e dual-specificity protein phosphatase dsp8 (GenBank no. DT051818.1, designated GhDSP8); fUnknown2 (GenBank no. DT455687.1). Long-fiber group, LF1:NMGA-062; LF2:NMGA-089; LF3:NMGA-017; LF4:NMGA-051; and LF5:NMGA-070. Short-fiber group, SF1:NMGA-095; SF2:NMGA-026; SF3:NMGA-144; SF4:NMGA-105; SF5:NMGA-140
The expression of GhKIF in 25 DPA fibers was higher than that in the other fiber developmental stages, and the expression in the LF group was downregulated at 5–20 DPA compared with that in the SF group (Fig. 7a). For the GhACX gene, mRNA expression was the lowest in 5 DPA fibers and then linearly increased at 5–20 DPA in SF group. Furthermore, its expression was highest in 5 DPA fibers and then linearly decreased at 10–15 DPA, then increased at 20–25 DPA in the LF lines (Fig. 7b). The expression of the Unknown1 gene at the mRNA level was lower in the 5 DPA fibers compared to the other fiber developmental stages and in the LF group compared with the SF group at 5–15 DPA (Fig. 7c). The expression of GhPAP2 in the SF group was upregulated in fibers including developing fibers at 0 DPA, which indicated that this gene is negatively correlated with fiber development (Fig. 7d). The expression of GhDSP8 in the 20 DPA fibers was higher than that in the other fiber developmental stages, and the expression of GhDSP8 in the LF group was lower than that in the SF group at 5 and 15–25 DPA (Fig. 7e). The expression of the Unknown2 gene at the mRNA level was reduced in the 25 DPA fibers compared to the other fiber developmental stages in the LF group (Fig. 7F). In terms of the Unknown2 gene, the expression at the mRNA level was highest in the SF lines in the 25 DPA fibers.. Interestingly, a completely opposite trend was observed in the 5–25 DPA fibers in the LF and SF groups. The expression of GhACX and GhDSP8 genes were up-regulated in the LF group based on microarray analyses, but which is no significant difference between LF group and SF group in the qRT-PCR results. The results indicated that the differential expression of the six genes between the LF group and SF group was relative congruence between the microarray and qRT-PCR results. Although the results indicated that the expression of GhKIF and GhACX was down-regulated in the LF group at 5–25 DPA, but the expression of GhKIF and GhACX was higher and higher during fiber development, which maybe plays an important role in fiber development.
Discussion
In the present study, we performed a microarray-based comparative transcriptome analysis of 10 DPA fibers between two groups of BILs based on differences in fiber length and similarities in other traits (long fiber-LF vs. short fiber-SF). A total of 1478 differentially expressed genes (DEGs) were identified that may be involved in fiber elongation. We mapped 166 DEGs with 2 fiber length QTL and 12 fiber length QTL hotspots that were previously reported [33, 46]. Most importantly, six genes with two FL QTL were identified in a BIL population from the two groups of introduction lines, which is the focus of the current study. The mining of DEGs in these QTL and QTL hotspots regions helps to identify the key candidate genes for FL. Our chip-based comparative DEG analysis, combined with physical mapping, FL QTL and gene mapping, identified several candidate genes that possibly underlie the genetic control of FL differences between G. barbadense and G. hirsutum. To the best of our knowledge, this fiber quality DEG study is the first to use gene-chip technology and genotyping to detect candidate genes for FL, which is the most effective strategy based on QTL mapping and physical mapping of a sequenced genome for narrowing down the number of candidate genes. This study paves the way for the identification of candidate genes for fiber traits in cotton in the future.
In the current study, 1478 DEGs between the “long” and “short” groups of BILs were identified in the 10 DPA fiber transcriptomes, 166 DEGs were mapped with 2 fiber length QTL and 12 fiber length QTL hotspots that were previously reported from the two groups of BILs selected for this study. Most importantly, six genes with 2 FL QTLs were identified in a population from the two groups of introduction lines. In our study, the 2 QTL for fiber length were qFL-08A-c12-1 on At_chr 12 and qFL-08A-c21-1 on Dt_chr11. The qFL-08A-c12-1 QTL on At_chr 12 included three co-localized DEGs, encoding a kinesin-like protein, a probable lipid phosphate phosphatase beta protein, and an unknown protein. The qFL-08A-c21-1 QTL on Dt_chr11 also included three co-localized DEGs, encoding a peroxisomal acyl-coenzyme a oxidase 1-like protein, the dual-specificity protein phosphatase 8 (DSP8) protein, and an unknown protein [25] found that 8 DEGs were co-localized with 4 FL QTLs identified in the BIL population containing the two BILs. The 4 FL QTL (qFL-07X-c5-1, qFL-07W-c11-1, qFL-08A-c21-1, and qFL-08A-c12-1) were mapped onto A05, A11, D11, and A12, respectively. The QTL regions of qFL-08A-c21–1 and qFL-08A-c12–1 were consistent between the results of Li et al. and our findings, but none of the same genes were identified. This study provide a glimpse into the relationship of DEGs with the genetic variation of fiber length in cotton.
Six DEGs identified in the LF group through microarray analysis that co-localized with fiber length QTL were further analyzed using qRT-PCR. Specifically, the gene expression levels of peroxisomal acyl-coenzyme a oxidase 1-like GhACX and the dual-specificity protein phosphatase GhDSP8 were higher in the LF group than in the SF group at 10 DPA, indicating that these genes are positively correlated with fast fiber elongation development. The gene expression levels of the kinesin-like protein GhKIF, the probable lipid phosphate phosphatase beta GhPAP2, and two unknown genes were lower in the LF group than in the SF group at 10 DPA. The peroxisomal acyl-coenzyme a oxidase 1-like GhACX gene was located at the c21_qFL-08A-c21-1 QTL for fiber length. Acyl-CoA oxidase (ACX) catalyzes the conversion of acyl-CoA to 2-trans-enoyl-CoA in the first reaction of the b-oxidation pathway [14, 17]. An acyl-CoA oxidase (ACX) that is involved in the oxidation of fatty acids has been cloned from Arabidopsis thaliana [13]. Fatty acid metabolism-related genes have also been reported to be involved in fiber development, as rapid fiber cell elongation requires related organelles and membranes, which are provided by fatty acid metabolism [31, 38]. Dong et al. found that the transcript of GhACX was most abundant in seeds at 25 DPA, indicating that it plays an important role in the fatty acid metabolism during cotton seed formation [7]. In this study, the expression level of the peroxisomal acyl-coenzyme a oxidase 1-like gene was relatively high in the developing fibers at 25 DPA and was upregulated in the LF group, indicating that the gene was preferentially expressed during the fiber development period and was positively correlated. The GhKIF expression levels were higher in the SF group than in the LF group at 25 DPA. Previous studies have identified more than a dozen kinesins that are abundantly expressed during fiber development in Upland cotton [15, 18, 29, 30, 37]. Wu et al. found that GhKIF11 expression levels were higher in the LY (low yield) group than in the HY (high yield) group at 15 and 20 DPA, but this gene was not located in any of the QTL regions for lint yield and yield traits [41]. This gene is located at the c12_qFL-08A-c12-1 QTL for fiber length. Therefore, GhKIF11 is likely one of the candidate genes for fiber length QTL.
Through a comparative genome-wide gene expression analysis of two groups of BILs with contrasting fiber lengths and the co-localization of DEGs with fiber length QTL, 1478 DEGs were narrowed to 166 genes in the QTL regions. Several candidate genes possibly underlying the genetic control of fiber length differences between G. barbadense and G. hirsutum, including GhACX and GhKIF, were likely the candidate genes for fiber length QTL. These results provide a list of candidate genes for fine mapping of fiber length QTL and mapping-based cloning, which will facilitate targeted gene transfer from island cotton to upland cotton to further improve fiber quality.
Abbreviations
- LF:
-
Long-fiber
- SF:
-
Short-fiber
- LFP:
-
Long-fiber parent
- SFP:
-
Short-fiber parent
- DEGs:
-
Differentially expressed genes
- BILs:
-
Backcross isogenic lines
- DPA:
-
Days post-anthesis
- QTLs:
-
Quantitative trait loci
References
Arpat AB, Waugh M, Sullivan JP, Gonzales M, Frisch D, Main D, Wood T, Leslie A, Wing RA, Wilkins TA (2004) Functional genomics of cell elongation in developing cotton fibers. Plant Mol Biol 54:911–929
Basra AS, Malik CP (1984) Development of the cotton fiber. Int Rev Cytol 89:65–113
Bennett J, Hondred D, Register JC (2015) Keeping qRT-PCR rigorous and biologically relevant. Plant Cell Rep 34:1–3
Brubaker CL, Paterson AH, Wendel JF (1999) Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome 42(2):184–203
Chee PW, Draye X, Jiang CX, Decanini L, Dehnonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum and Gossypium barbadense (cotton) by a backcross-self approach: I. Fiber elongation. Theor Appl Genet 111(4):757–763
Chee PW, Draye X, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of phenotypic variation between Gossypium hirsutum and Gossypium barbadense (cotton) by a backcross-self approach: III. Fiber length. Theor Appl Genet 111(4):772–781
Dong J, Wei LB, Yan H, Guo WZ (2013) Molecular cloning and characterization of three novel genes related to fatty acid degradation and their responses to abiotic stresses in Gossypium hirsutum L. J Integ Agric. 12(4):582–588
Draye X, Chee PW, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Wayne SC, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum. and G. barbadense (cotton) by a backcross-self approach: II. Fiber fineness. Theor Appl Genet 111(4):764–771
Fang L, Tian R, Li XH, Chen JD, Wang S, Wang P, Zhang TZ (2014) Cotton fiber elongation network revealed by expression profiling of longer fiber lines introgressed with different Gossypium barbadense chromosome segments. BMC Genom 15:838–853
Guo LX, Shi YZ, Gong JW, Liu AY, Tan YN, Gong WK, Li JW, Chen TT, Shang HH, Ge Q, Lu QW, Sun J, Yuan YL (2018) Genetic analysis of the fiber quality and yield traits in G. hirsutum background using chromosome segments substitution lines (CSSLs) from Gossypium barbadense. Euphytica 214:82
Guo WZ, Cai CP, Wang CB, Han ZG, Song XL, Wang K, Niu XW, Wang C, Lu KY, Shi B, Zhang TZ (2007) A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium. Genetics 176:527–541
Hande AS, Katageri IS, Jadhav MP, Adiger S, Gamanagatti S, Padmalatha KV, Dhandapani G, Kanakachari M, Kumar PA, Reddy VS (2017) Transcript profiling of genes expressed during fibre development in diploid cotton (Gossypium arboreum L.). BMC Genom 18:675
Hayashi H, De Bellis L, Ciurli A, Kondo M, Hayashi M, Nishimura M (1999) A novel acyl-CoA oxidase that can oxidize short-chain acyl-CoA in plant peroxisomes. J Biol Chem 274:12715–12721
Hiltunen JK, Mursula AM, Rottensteiner H, Wierenga RK, Kastaniotis AJ, Gurvitz A (2003) The biochemistry of peroxisomal b-oxidat ion in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 27(1):35–64
Ji SJ, Lu YC, Feng JX, Wei G, Li J, Shi YH, Fu Q, Liu D, Luo JC, Zhu YX (2003) Isolation and analyses of genes preferentially expressed during early cotton fiber development by subtractive PCR and cDNA array. Nucl Acids Res 31(10):2534–2543
John ME (1996) Structural characterization of genes corresponding to cotton fiber mRNA, E6: reduced E6 protein in transgenic plants by antisense gene. Plant Mol Biol 30(2):297–306
Kim MC, Kim TH, Park JH, Moon BY, Lee CH, Cho SH (2007) Expression of rice acyl-CoA oxidase isoenzymes in response to wounding. J Plant Physiol 164(5):665–668
Kong ZS, Ioki M, Braybrook S, Li S, Ye ZH, Lee YRJ, Hotta T, Chang A, Tian J, Wang GD, Liu B (2015) Kinesin-4 functions in vesicular transport on cortical microtubules and regulates cell wall mechanics during cell elongation in plants. Mol Plant 8:1011–1023
Lacape JM, Claverie M, Vidal RO, Carazzolle MF, Guimaraes Pereira GA, Ruiz M, Pre M, LIewellyn D, AI-Ghazi Y, Jacobs J, Dereeper A, Huquet S, Giband M, Lanaud C (2012) Deep sequencing reveals differences in the transcriptional landscapes of fibers from two cultivated species of cotton. PLoS ONE 7(11):e48855
Lacape JM, Llewellyn D, Jacobs J, Arioli T, Becker D, Calhoun S, AI-Ghazi Y, Liu SM, Palai O, Georges S, Giband M, Assuncao H, Barroso PAV, Claverie M, Gawryziak G, Jean J, Vialle M, Viot C (2010) Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum x G. barbadense RIL population. BMC Plant Biol 10:132
Lacape JM, Nguyen TB, Courtois B, Belot JL, Giband M, Gourlot JP, Gawryziak G, Roques S, Hau B (2005) QTL analysis of cotton fiber quality using multiple Gossypium hirsutum × Gossypium barbadense backcross generations. Crop Sci 45(1):123–140
Lacape JM, Nguyen TB, Thibivilliers S, Bojinov B, Courtois B, Cantrell RG, Burr B, Hau B (2003) A combined RFLP-SSR-AFLP map of tetraploid cotton based on a Gossypium hirsutum × Gossypium barbadense backcross population. Genome 46(4):612–626
Li DD, Ruan XM, Zhang J, Wu YJ, Wang XL, Li XB (2013) Cotton plasma membrane intrinsic protein 2 s (PIP2s) selectively interact to regulate their water channel activities and are required for fiber development. New Phytol 199(3):695–707
Li FG, Fan GY, Lu CR, Xiao GH, Zou CS, Kohel RJ, Ma ZY, Shang HH, Ma XF, Wu JY, Ling XM, Huang G, Percy RG, Liu K, Yang WH, Chen WB, Du XM, Shi CC, Yuan YL, Ye WW, Liu X, Zhang XY, Liu WQ, Wei HL, Wei SJ, Huang GD, Zhang XL, Zhu SJ, Zhang H, Sun FM, Wang XF, Liang J, Wang JH, He Q, Huang LH, Wang J, Cui JJ, Song GL, Wang KB, Xu X, Yu JZ, Zhu YX, Yu SX (2015) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33(5):524–529
Li XH, Wu M, Liu GY, Pei WF, Zhai HH, Yu JW, Zhang JF, Yu SX (2017) Identification of candidate genes for fiber length quantitative trait loci through RNA-Seq and linkge and physical mapping in cotton. BMC Genom 18:427
Liu BL, Zhu YC, Zhang TZ (2015) The R3-MYB gene GhCPC negatively regulates cotton fiber elongation. PLoS ONE 10(2):e0116272
Pang MX, Stewart JM, Zhang JF (2013) A mini-scale hot borate method for the isolation of total RNA from a large number of cotton tissue samples. Afr J Biotechnol 10(68):15430–15437
Paterson AH, Saranga Y, Menz M, Jiang CX, Wright RJ (2003) QTL analysis of genotype x environment interactions affecting cotton fiber quality. Theor Appl Genet 106(3):384–396
Preuss ML, Delmer DP, Liu B (2003) The cotton kinesin-like calmodulin-binding protein associates with cortical microtubules in cotton fibers. Plant Physiol 132(1):154–160
Preuss ML, Kovar DR, Lee YR, Staiger CJ, Delmer DP, Liu B (2004) A plant-specific kinesin binds to actin microfilaments and interacts with cortical microtubules in cotton fibers. Plant Physiol 136(4):3945–3955
Qin YM, Hu CY, Pang Y, Kastaniotis AJ, Hiltunen JK, Zhu YX (2007) Saturated very-long-chain fatty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell 19(11):3692–3704
Said JI, Knapka JA, Song MZ, Zhang JF (2015) Cotton QTLdb: a cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum x G. barbadense populations. Mol Genet Genom 290(4):1615–1625
Said JI, Lin ZX, Zhang XL, Song MZ, Zhang JF (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom 14:776–798
Said JI, Song MZ, Wang HT, Lin ZX, Zhang XL, Fang DD, Zhang JF (2015) A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol Genet Genom 290(3):1003–1025
Shan CM, Shangguan XX, Zhao B, Zhang XF, Chao LM, Yang CQ, Wang LJ, Zhu HY, Zeng YD, Guo WZ, Zhou BL, Hu GJ, Guan XY, Chen ZJ, Wendel JF, Zhang TZ, Chen XY (2014) Control of cotton fiber elongation by a homeodomain transcription factor GhHOX3. Nat Commun 21(5):5519
Tang WX, Tu LL, Yang XY, Tan JF, Deng FL, Hao J et al (2014) The calcium sensor GhCaM7 promotes cotton fiber elongation by modulating reactive oxygen species (ROS) production. New Phytol 202(2):509–520
Tiwari SC, Wilkins TA (1995) Cotton (Gossypium hirsutum) seed trichomes expand via diffuse growing mechanism. Can J Bot 73(5):746–757
Wanjie SW, Welti R, Moreau RA, Chapman KD (2005) Identification and quantification of glycerolipids in cotton fibers: reconciliation with metabolic pathway predictions from DNA databases. Lipids 40(8):773–785
Wendel JF (2000) Genome evolution in polyploids. Plant Mol Biol 42(1):225–249
Wendel JF, Cronn RC (2003) Polyploidy and the evolutionary history of cotton. Adv Agron 78(2):139–186
Wu M, Zhang LY, Li XH, Xie XB, Pei WF, Yu JW, Yu SX, Zhang JF (2016) A comparative transcriptome analysis of two sets of backcross inbred lines differing in lint–yield derived from a Gossypium hirsutum × Gossypium barbadense population. Mol Genet Genom 291(4):1749–1767
Yang ZR, Zhang CJ, Yang XJ, Liu K, Wu ZX, Zhang XY, Wu Z, Xun QQ, Liu CL, Lu LL, Yang ZE, Qian YY, Xu ZZ, Li CF, Li J, Li FG (2014) PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol 203(2):437–448
Yu J, Jung S, Cheng CH, Ficklin SP, Lee T, Zheng P, Jones D, Percy RG, Main D (2014) CottonGen: a genomics, genetics and breeding database for cotton research. Nucleic Acids Res 42:1229–1236
Yu JW, Yu SX, Fan SL, Song MZ, Zhai HH, Li XX, Zhang JF (2012) Mapping quantitative trait loci for cottonseed oil, protein and gossypol content in a Gossypium hirsutum x Gossypium barbadense backcross inbred line population. Euphytica 187(2):191–201
Yu JW, Yu SX, Gore M, Wu M, Zhai HH, Li XL, Song MZ, Zhang JF (2013) Identification of quantitative trait loci across interspecific F2, F2:3 and testcross populations for agronomic and fiber traits in tetraploid cotton. Euphytica 191(3):375–389
Yu JW, Zhang K, Yu SX, Fan SL, Song MZ, Zhai HH, Wu M, Li XL, Fan SL, Song MZ, Yang DG, Li YH, Zhang JF (2013) Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum x Gossypium barbadense backcross inbred line population. Theor Appl Genet 126(1):275–287
Zhang JF, Percy RG, McCarty JC (2014) Introgression genetics and breeding between Upland and Pima cotton: a review. Euphytica 198(1):1–12
Zhang M, Zheng XL, Song SQ, Zeng QW, Hou L, Li DM, Zhao J, Wei Y, Li XB, Luo M, Xiao YH, Luo XY, Zhang JF, Xiang CB, Pei Y (2011) Spatiotemporal manipulation of auxin biosynthesis in cotton ovule epidermal cells enhances fiber yield and quality. Nat Biotechnol 29(5):453–458
Zhang TZ, Hu Y, Jiang WK, Fang L, Guan XY, Chen JD, Zhang JB, Saski CA, Scheffler BE, Stelly DM, Hulse-Kemp AM, Wan Q, Liu BL, Liu CX, Wang S, Pan MQ, Wang YK, Wang D, Ye WX, Chang LJ, Zhang WP, Song QX, Kirkbride RC, Chen XY, Dennis E, LIewellyn DJ, Peterson DG, Thaxton P, Jones DC, Wang Q, Xu XY, Zhang H, Wu HT, Zhou L, Mei CF, Chen SQ, Tian Y, Xiang D, Li XH, Ding J, Zuo QY, Tao LN, Liu YC, Li J, Lin Y, Hui YY, Cao ZS, Cai CP, Zhu XF, Jiang Z, Zhou BL, Guo WZ, Li RQ, Chen ZJ (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol 33(5):531–537
Acknowledgements
The research was sponsored by grants from the National Key Research and Development Program of China (Grant Nos. 2018YFD0100300 and 2016YFD0101400), the National Natural Science Foundation of China (Grant No. 31621005), and the National Research and Development Project of Transgenic Crops of China (Grant No. 2016ZX08005005). The research was also supported in part by the New Mexico Agricultural Experiment Station.
Author information
Authors and Affiliations
Contributions
JFZ, SXY and JWY conceived the study. MW, LYL, GYL, XHL, WFP and XLL performed the experiments. MW wrote the manuscript. JWY and JFZ edited the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2019_4589_MOESM2_ESM.xlsx
Supplementary Data 2. Variance analysis of fiber quality, the yield trait, boll size, and the lint percentage. (XLSX 11 KB)
11033_2019_4589_MOESM3_ESM.xls
Supplementary Data 3. Microarray results for the differentially expressed genes (DEGs) identified in the long- and short-fiber groups. (XLS 322 KB)
11033_2019_4589_MOESM5_ESM.xlsx
Supplementary Data 5. Mapping of DEGs with the 4 fiber length QTLs and 13 fiber length QTL hotspots in the genome of G. hirsutum. (XLSX 67 KB)
Rights and permissions
About this article

Cite this article
Wu, M., Li, L., Liu, G. et al. Differentially expressed genes between two groups of backcross inbred lines differing in fiber length developed from Upland × Pima cotton. Mol Biol Rep 46, 1199–1212 (2019). https://doi.org/10.1007/s11033-019-04589-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-04589-x