
Abstract
Swertia chirayita is a high-value medicinal herb exhibiting antidiabetic, hepatoprotective, anticancer, antiediematogenic and antipyretic properties. Scarcity of its plant material has necessitated in vitro production of therapeutic metabolites; however, their yields were low compared to field grown plants. Possible reasons for this could be differences in physiological and biochemical processes between plants grown in photoautotrophic versus photoheterotrophic modes of nutrition. Comparative transcriptomes of S. chirayita were generated to decipher the crucial molecular components associated with the secondary metabolites biosynthesis. Illumina HiSeq sequencing yielded 57,460 and 43,702 transcripts for green house grown (SCFG) and tissue cultured (SCTC) plants, respectively. Biological role analysis (GO and COG assignments) revealed major differences in SCFG and SCTC transcriptomes. KEGG orthology mapped 351 and 341 transcripts onto secondary metabolites biosynthesis pathways for SCFG and SCTC transcriptomes, respectively. Nineteen out of 30 genes from primary metabolism showed higher in silico expression (FPKM) in SCFG versus SCTC, possibly indicating their involvement in regulating the central carbon pool. In silico data were validated by RT-qPCR using a set of 16 genes, wherein 10 genes showed similar expression pattern across both the methods. Comparative transcriptomes identified differentially expressed transcription factors and ABC-type transporters putatively associated with secondary metabolism in S. chirayita. Additionally, functional classification was performed using NCBI Biosystems database. This study identified the molecular components implicated in differential modes of nutrition (photoautotrophic vs. photoheterotrophic) in relation to secondary metabolites production in S. chirayita.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant based medicines occupy roughly 25% of the current drug market, playing a significant role in the global healthcare sector [1,2,3]. The World Health Organization (WHO) estimated that the current demand for plant-based herbal raw material is around US $14 billion/year [4], growing at an annual rate of 15–25%, and expected to reach US $5 trillion by 2050 [5].
Swertia chirayita (S. chirayita) commonly known as ‘Chirata’, belongs to family Gentianaceae. It is endemic to temperate Himalayas (Kashmir to Bhutan, and Kashia hills), and inhabited at an altitude of 1200–3000 m [6]. The pronounced medicinal importance of this medicinal herb has been described in Indian pharmaceutical codex, the American and the British pharmacopoeias, and in established medicinal systems of the East, such as Ayurveda, Siddha and Unani [7]. Extracts of S. chirayita possess antimalarial [8], antidiabetic [9, 10], hepatoprotective [11, 12], anticarcinogenic [13], anti-inflammatory [14, 15], antioxidant [16], antiviral [17], antibacterial [16, 18] and antifungal [19] activities. These multifarious therapeutic values displayed by the plant are due to the presence of an array of pharmacologically important secondary metabolites such as xanthones, flavonoids, terpenoids, and their glycosides [20]. Increased market demand for usage in herbal drugs [21] and narrow geographic occurrence [22] have led to over exploitation of this plant species, and as a result it has been declared critically endangered by the New International Union for Conservation of Nature and Natural resources (IUCN) criteria [23].
Phytochemical profiling of S. chirayita has shown the presence of a large number of secondary metabolites including swertiamarin, mangiferin and amarogentin, which have been demonstrated to possess therapeutic values (Suppl. Table S1). Biomass is one of the significant factors that determine the final yield of desirable phytochemicals in target plant species. The endangered status has limited the accessibility of S. chirayita raw material, as well as therapeutic metabolites, thereby necessitating the development of alternate routes of biomass production. However, in vitro yield of secondary metabolites has been very low in shoot/callus/cell suspension cultures as compared to plants grown in natural habitat [24]. One of the differences between these two types of plants is their mode of nutrition. The green house grown plants exhibit photoautotrophic mode of nutrition, whilst the tissue cultured plants use photoheterotrophic mode (depending solely on the nutrient media) for their growth and development.
Various biochemical, physiological and molecular processes influence the production of biomass yield in plants [25]. The physio-biological processes, such as leaf senescence, flower/fruit size, leaf size, stem diameter and shoot biomass influence biomass yield [26, 27]. The molecular components, including key gene phosphoribosyl pyrophosphate synthetase (PRS), Dof-class transcription factor- SOB1/OBP3 and DNA-binding proteins-SOB3, ESC, SOB5, SOFL1 and SOFL2 are known to control plant growth and biomass accumulation in Arabidopsis thaliana and Nicotiana tabacum [26, 28].
Photosynthesis plays a pivotal role in maintaining the carbon pool of photosynthetic organisms. The role of carbon pool on the other hand, in determining secondary metabolite levels has been reported in Hypericum perforatum [29]. The synthesis of primary and secondary metabolites is also influenced by a multitude of intrinsic and extrinsic factors like light intensity, photoperiod, temperature, O2 and CO2 concentration, humidity, more importantly mode of nutrition (carbon sources and other elements) and others. These factors vary in photoautotrophic and photoheterotrophic modes of nutrition in different plant species [30, 31]. The influence of these factors on secondary metabolites biosynthesis has been studied extensively in plant species like Picrorhiza kurroa, Onosma paniculatum, Linum album and A. thaliana [32,33,34]. The source of nutrients in tissue cultured plants of S. chirayita (photoheterotrophs) solely depends upon the supplemented Murashige and Skoog (MS) medium, whereas in green house grown plants (photoautotrophs), the soil mixture and the photosynthesis provide the necessary nutritional requirements. We hypothesize that many of the biological, physiological, biochemical and molecular processes are altered in photoautotrophic versus photoheterotrophic modes of nutrition. These alterations should be reflected in the differences of types of biosynthetic pathways, genes, proteins and their interactions associated with those processes.
Fundamental processes like glycolysis, tricarboxylic acid cycle, pentose phosphate pathway, photosynthesis and others, are involved in the biosynthesis, degradation and interconversion of plant primary and secondary metabolites [35]. Majority of the building blocks of secondary metabolic pathways (shikimate, acetate, mevalonate, deoxyxylulose pathways and others) are derived from important intermediates such as acetyl Co-A, glucose 6-phosphate, glyceraldehyde 3-phosphate, phosphoenolpyruvate, erythrose 4-phosphate, shikimic acid, mevalonate (MVA) and 1-deoxyxylulose 5-phosphate (DXP). Therefore, it is logical to assume that the formation of these metabolites is probably influenced by the state and efficiencies of these fundamental processes which provide precursors to secondary metabolites biosynthesis. However, the contrasting levels of phytochemicals in plants adapted to differential modes of nutrition affect the status of fundamental processes, and hence the final levels of metabolites, need to be investigated. Differences in modes of nutrition also influence the types of transporters like ABC, MATE and MDR and others, which transport minerals, metabolites and photosynthetates as per the autotrophic or photoheterotrophic modes of nutrition [36].
The emergence of next generation sequencing (NGS) technologies have enabled understanding the biological processes through differential transcriptomics [37], phylogenomic analysis [38, 39], microRNA and piRNA detection [40]. In this study, we report de novo transcriptome sequencing and characterization of S. chirayita plants grown in photoautotrophic versus photoheterotrophic modes of nutrition, and also varying for the contents of three major secondary metabolites, swertiamarin, mangiferin and amarogentin using Illumina RNA-Seq method. Majority of the transcripts demonstrated considerable match with Solanum lycopersicum for both the transcriptomes during species distribution. The transcriptomes were analyzed for mevalonic acid (MVA)/methylerythritol phosphate (MEP) and phenylpropanoid pathway genes, transcription factors (TFs) and ATP-binding cassette (ABC) transporter genes, and contribution of various biological pathways implicated in photoautotrophic and photoheterotrophic modes of nutrition. The comparative transcriptomes of S. chirayita have been explored to gain insights into the underlying physio-biological processes and biochemical pathways differing in modes of nutrition. The study presented a repertoire of molecular components contributing to differences in various physio-biological processes under different modes of nutrition in S. chirayita (http://14.139.240.55/NGS/download.php).
Materials and methods
Plant material and tissue collection
Swertia chirayita plants were initially procured from Dr. Yashwant Singh Parmar University of Horticulture and Forestry, Nauni, Himachal Pradesh, India (30°86′N, 77°17′E). A voucher sample of the plant genotype having high content of secondary metabolites [41] has been submitted to the National Bureau of Plant Genetic Resources (New Delhi) with accession number IC-594053. Green house grown plants of S. chirayita (SCFG) were raised and maintained at the Jaypee University of Information technology, Waknaghat, Himachal Pradesh, India (1500 m altitude; 31°01′N, 77°04′E) with controlled conditions [light (intensity 1200–4800 W m−2), temperature (25 ± 2 °C), relative humidity (≈ 75%) and photoperiod (14 h day/10 h night). In vitro cultured S. chirayita plants (SCTC) were maintained at the tissue culture facility of the University in media supplemented with MS [42] salts, growth hormones [Kinetin (2 mg l−1), Indole-3-butyric acid (2 mg l−1) and gibberellic acid (3 mg l−1)], sucrose (30 g l−1), agar–agar (8.5 g l−1) pH 5.6–5.7 and environmentally controlled conditions [light (intensity 5800 W m−2), temperature (25 ± 2 °C), relative humidity (≈ 75%) and photoperiod (16 h day/8 h night)]. Tissue samples (roots and shoots) were procured from plants raised in the green house (1.5 years old) and in vitro (1.5 months old) conditions, frozen immediately in liquid nitrogen and stored at − 80 °C until further use.
Extraction and estimation of swertiamarin, amarogentin and mangiferin
Contents of swertiamarin, mangiferin and amarogentin were estimated using reverse phase high performance liquid chromatography on Waters HPLC system (Waters Corporation, USA) equipped with Waters 515 HPLC pumps, Waters 717 autosampler, Waters 2996 photodiode array detector and Empower software as per the method described by Kumar et al. [41]. The plant materials were homogenized in a mortar and pestle (pre-chilled) using liquid nitrogen. The powdered samples (100 mg each) were suspended in 80% methanol; vortexed and sonicated for 10 min at room temperature. The samples were then centrifuged at 10,000 rpm for 15 min. The supernatants were filtered through 0.22 μm filter, diluted fivefold with 80% methanol and quantified for swertiamarin, amarogentin and mangiferin levels. A gradient method was employed to obtain the desired separation and baseline, where the mobile phase A was composed of 0.1% TFA in water, and mobile phase B was a mixture of acetonitrile/water in a ratio of 70:30. The linear gradient at a flow rate of 1.0 ml/min was started with 15% B; 20% B in next 5 min, 70% B in next 25 min, hold for 5 min; 15% B in next 5 min, equilibrated for 5 min and detected at 240 nm UV wavelength. The phytochemicals were separated and identified on the basis of their retention time (RT) and UV-spectra comparison with the authentic standards procured from ChromaDex (Irvine, CA). The quantification was performed in triplicate and the results were recorded in terms of percentage (%) of fresh weight (fr. wt) of swertiamarin, amarogentin and mangiferin. A pictorial representation of the differences in the contents of secondary metabolites in green house grown versus tissue cultured Swertia plants has been given in Fig. 1.

Contents of major secondary metabolites in Swertia chirayita plants grown in green house conditions (photoautotrophic, SCFG) and tissue culture (photoheterotrophic, SCTC)
Illumina HiSeq 2000-2 × 100 PE library preparation
RaFlex™ total RNA isolation kit (GeNei™, Bangalore, India) was used to isolate total RNA from both tissue samples following manufacturer’s instructions. The quality of RNA was assessed on 1% denaturing agarose gel and quantity was determined using NanoDrop 8000 spectrophotometer (Thermo Scientific, USA). Illumina TruSeq RNA Library Preparation Kit was used to prepare pair-end cDNA sequencing libraries for both the samples. The steps included for library preparation were mRNA fragmentation, reverse transcription, second strand synthesis, pair-end adapter ligation and concluded with the index PCR amplification of adaptor-ligated library. The quantity and quality of the library was assessed using HT DNA High Sensitivity LabChip® Kit on a Caliper LabChip GX alalyzer (PerkinElmer Inc., Santa Clara, CA).
Cluster generation and de novo sequence assembly
A workflow diagram depicting de novo transcriptome assembly, annotation and computational analysis of S. chirayita transcriptomes is given in Fig. 2. The template fragment was sequenced in both forward and reverse directions using paired-end sequencing technology. For cluster generation, template DNA molecules were hybridized onto the flow cell surface coated with oligonucleotides. Bridge amplification process was used to amplify immobilized DNA template so as to generate clonal DNA clusters. TruSeq PE Cluster kit v3-cBot-HS (Illumina, USA) was used in this process of cluster generation. The binding of the sample to complementary adapter oligos on paired-end flow cells was carried out through kit reagents. The adapters were designed in a manner to permit selective cleavage of the forward DNA strand soon after the reverse strand gets resynthesized. In order to sequence each cluster DNA on a flow cell, TruSeq SBS v3-HS kit (Illumina, USA) was used employing sequencing by synthesis technology on the HiSeq 2000. After cluster generation, adaptor trimming was performed followed by quality filteration (mean quality score ≥ 20) using Trimmomatic tool to remove low quality/trash reads. The repeated reads were removed and the high quality sequences were also processed for rRNA sequences. After quality filtration (mean quality score ≥ 20) and adaptor trimming, the high quality (HQ) reads for both samples were assembled with Velvet pipeline for different k-mer length (k-mer 43, 45, 47, 49, 51, 53, 55). At last, based on the N50 and covered transcriptome length, one best k-mer assembly was chosen for all sample and its respective transcript contigs were used for further analysis.

Work flow for de novo whole transcriptome sequencing, assembly, annotation and analyses in SCFG and SCTC transcriptomes of S. chirayita
Functional annotation, GO mapping and COG analysis
For functional annotation analysis, similarity search was performed using BLASTx program against the non-redundant (nr) NCBI nucleotide database (E-value of 10−5). These assembled transcripts were also checked against UniProtKB/Swiss-Prot database for sequence annotation [43]. Blast2GO program was used for identification and high-throughput functional annotation of assembled transcripts [44]. It classified transcripts using BLAST program and represented them into cellular component, molecular function and biological processes ontologies (http://www.geneontology.org/). Furthermore, the transcripts were ligned up to clusters of orthologous groups (COGs) database to predict and classify putative functions according to COG functional annotation. Top BLAST hits for the transcript sequences (E-value < 10−5) against COGs database were selected and mapped onto their respective COG IDs.
Read mapping and transcript abundance prediction
In silico expression level of all the assembled transcripts was measured using RSEMv1.2.5 software package. RSEM estimates transcript abundance based on the mapping of RNA-Seq reads to the assembled transcriptome. RSEM calculates maximum likelihood abundance estimates, posterior mean estimates and 95% credibility intervals for genes/isoforms [45]. RSEM generated FPKM (Fragments per Kilobase per Million) values measure expression of even poorly expressed transcripts. This output was automated by running two programs of RSEM (rsem-prepare-reference and rsem-calculate-expression) together with bowtie read aligner.
Domain identification using Pfam database
Domains are basic building blocks of proteins which can be used for functional class prediction [46]. Conserved protein domains were identified using Hmmer tool for searching protein sequences against Pfam domain database. Pfam library of HMMs for protein families were obtained from Pfam website http://pfam.janelia.org/), and pfam_scan.pl, a perl program was used for domain identification.
Mining secondary metabolic pathway genes in differential transcriptomes of S. chirayita
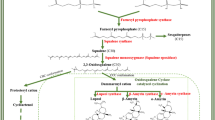
The biosynthesis of swertiamarin/amarogentin and mangiferin in S. chirayita follows the common MVA/MEP/phenylpropanoid route (Fig. 3). S. chirayita transcriptomes were mined for potential MVA/MEP/phenylpropanoid pathway genes using in-house developed perl scripts. The assembled transcripts from both the transcriptomes (SCFG and SCTC) were scanned against NR protein database available at NCBI by using BLASTx algorithm (E-value of 10−5). Fifteen genes, coding for AACT, HMGS, HMGR, MVK, PMK, MVDD, DXS, DXR, ISPD, ISPE, ISPF, ISPG, ISPH, IPPI and GDPS from MVA/MEP/pathways, and 13 genes, encoding DAHPS, DHQS, DHQD, SAK, EPSPS, CS, CM, PAT, ADH, ADT, PHAT, PAL and C4H from phenylpropanoid pathway were identified. Further, in silico transcript abundance was quantified for these genes for both the photoautotrophic and photoheterotrophic modes of nutrition to assess the contribution and relevance of MVA/MEP/phenylpropanoid pathways in the biosynthesis of secondary metabolites in S. chirayita.

Schematic representation of swertiamarin, amarogentin a and mangiferin b biosynthesis pathways in S. chirayita. The picture is adapted with modification from [24]. The differentially expressed transcripts in SCFG versus SCTC conditions are given in a tabular form
Expression analysis of fundamental pathway genes through RT-qPCR
Verso cDNA synthesis kit (Thermo Scientific, Wilmington, DE) containing anchored oligo(dT) primers and Verso RT Enhancer was used to synthesize first strand cDNA from total RNA (1 µg) following the manufacturer’s protocol. BLAST similarity search program (http://www.ncbi.nlm.nih.gov/BLAST) was employed to retrieve gene sequences of the fundamental processes such as glycolysis, TCA cycle, oxidative pentose phosphate pathway and Calvin cycle of photosynthesis from the whole transcriptome data of S. chirayita. Primer3 [47] was used to design gene specific primers from the retrieved gene sequences. The expression status of genes was analysed through quantitative RT-PCR (RT-qPCR). RT-qPCR reactions were run on a CFX96 system (Bio-Rad; Hercules, CA) with the iScript one step RT PCR kit (Bio-Rad; Hercules, CA). The cycling conditions comprised denaturation (5 min, 94 °C), followed by 40 cycles each of denaturation (20 s, 94 °C), annealing (30 s, at different temperatures), followed by elongation (20 s, 72 °C). All reactions were run in triplicate. The expression data were normalized using multiple internal control genes as described previously [48]. To start with, a set of six candidate reference genes encoding actin, eEF1a, β-tubulin, histone H3, 26s rRNA and GAPDH were taken into consideration for their expression across different tissues under study and their suitability as reference genes were analyzed using BestKeeper [49]. BestKeeper analysis identified eEF1a, β-tubulin and Actin genes to be considered further for our analysis based on their score (data not provided). Finally, for normalisation and relative quantification, geometric average of all the three reference genes was done as per the method described previously [48]. Comparative CT (ΔΔCt) method [50] was used to determine relative fold changes from the Quantification cycle (Cq) values. Additionally, a melt curve analysis, comprising melting curve ramping from 65 to 95 °C with an increasing temperature of 0.5 °C for 5 s (1 cycle) was performed in order to evaluate the specificity of RT-qPCR assay. A list of primer sequences used in RT-qPCR analysis is given in Suppl. Table S2.
Pathway mapping using KEGG
The pathway annotation was performed using KEGG Automatic Annotation Server (KAAS) ([51]; http://www.genome.jp/tools/kaas/). KAAS assigned “KEGG orthology (KO) identifiers” to transcripts, which were mapped onto enzyme commission (EC) number with default bit score threshold of 60. The BRITE functional hierarchies categorized transcripts into genetic and environmental information processing, and cellular processes.
Functional classification by NCBI biosystems
NCBI Biosystems database provides access and updated records from KEGG, Pathway Interaction Database, Gene Ontology, BioCyc and Wikipathways [52]. To functionally classify assembled transcripts, these were mapped onto known molecules that interact in a biological system using in-house developed scripts. Computational mining was performed on the annotated file for the transcripts and their respective gene IDs. These mined gene IDs were mapped onto their respective biological processes using an unique bsid identifier. Duplicate entries of similar biological process for different gene Ids were clubbed together.
Mining for common and unique genes in S. chirayita transcriptomes
The comparative analysis between SCTC and SCFG transcriptomes was performed to identify common and unique genes in green house versus tissue cultured plants of S. chirayita through BLAST similarity search approach (E-value of 10−5). For mining of common and unique genes, all the assembeled transcripts from one transcriptome were searched for sequence simalarity against the other (i.e. SCFG vs. SCTC). If the significant hit from the first (SCFG) is observed for at least a transcript in the second transcriptome (SCTC), it is refered to as a common transcript/gene. On the contrary, in case of “no hit” scenario, the transcript is referred being unique.
Identification of transcription factors families and ABC transporters involved in the biosynthesis and transport of metabolites
The final level of phytochemicals in plant cells is controled by the coordinated transcriptional regulation of the biosynthetic pathway genes [53]. For the annotation of plant transcription factors (TFs), transcriptional regulators (TRs) and chromatin regulators (CRs) in SCFG and SCTC transcriptomes, PlantTFcat [54], a high-performance web-based tool was used. A comparative analysis of TFs obtained from both the data sets (SCFG and SCTC) was done in order to reveal unique and common TFs for further assessment. A systematic literature search for transcription factor families identified eight classes (AP2-EREBP, bHLH, bZIP, DOF, MYB, NAC, WRKY and Zinc-finger) of TF families associated with the biosynthesis of secondary metabolites [55]. A master list was prepared containing transcripts belonging to these eight classes of transcription factor families. Pfam keyword search with ‘ABC transporters’ was performed in the Pfam database [56] to mine all possible candidate ABC transporter genes in the transcriptomes of S. chirayita. The resulting hits were also checked manually for their role as transporters in plants. Functionally characterized, ABC transporters in Arabidopsis [57] were retrieved from literature and mined in SCFG and SCTC transcriptomes to dissect their possible role in S. chirayita.
Website development and platform used
Related material and detailed results for S. chirayita transcriptome datasets have been uploaded on our previously developed next generation sequencing and analysis website [58]. The website is hosted on a DELL PowerEdge™ T410 server with 16 cores 2.67 GHz Intel R Xenon processors and loaded with 64-bit CentOS linux distribution operating system.
Results and discussion
The transcriptome data provide a valuable resource for rapid elucidation of pathways and their mapping through graphical connectivity diagrams along with the identification and subsequent characterization of biosynthesis pathway genes, transcription factors (TFs), microRNAs (miRNAs), transporters and others [59, 60]. In the current study, we have made an attempt to assemble and characterize transcriptomes of S. chirayita using paired-end sequencing method (Illumina), an initiative taken towards exploring molecular biology of this plant species.
HPLC quantification
Swertiamarin content in the green house grown Swertia plant (SCFG) was observed to be ~ 8.2 folds higher than that of the tissue cultured plants, whereas amarogentin and mangiferin contents were found to be 5 and 2.5 folds higher, respectively in the former (Fig. 1). The difference in levels of these secondary metabolits may be attributed to the differential nutitional modes (photoautotrophic vs. photoheterotrophic) of the plants.
Paired-end sequencing and de novo sequence assembly
Despite recent development of many bioinformatics tools for NGS data assembly and analysis, a challenge in de novo assembly of short reads in the absence of a reference genome still remains [59, 61]. Paired-end (PE) Illumina sequencing generated a total of 43,306,144 and 23,075,416 raw reads of 8661 and 4615 MB data for green house and tissue cultured S. chirayita samples, respectively. Subsequent filtering of the raw PE-reads produced a total of 41,031,326 and 21,859,688 high quality (HQ) reads which were further assembled using Velvet pipeline. k-mer 51 and k-mer 47 emerged as best k-mers for performing assembly of the HQ reads for green house and tissue cultured tissue samples, respectively. A total of 57,460 assembled transcripts with an average length of 1143.47 bp and average GC content of 41% were obtained for green house S. chirayita sample (SCFG) (Table 1). The maximum size of transcript amongst the assembled transcript was 10,838 bp while the minimum transcript size obtained was 200 bp long. Similarly, for tissue cultured S. chirayita sample (SCTC), a total of 43,702 transcripts with an average length of 1085.47 bp and average GC content of 41% were obtained. The maximum and minimum transcript sizes for the assembled transcripts for the tissue cultured sample were 7803 and 200 bp, respectively. Differential growth conditions may have accounted for the difference in the number of transcripts generated for green house and tissue cultured S. chirayita samples.
Functional annotation of transcripts
The functional annotation of S. chirayita transcriptomes was carried out using non-redundant (nr) database and UniProtKB/Swiss-Prot protein database at NCBI by BLASTx algorithm (E-value of 10−5). BLASTx resulted in the annotation of 50,795 transcripts with significant BLAST hits, while no hits were observed for 6665 transcripts out of total 57,460 assembled high quality transcripts for SCFG. Significant BLAST hits with nr database were found for 39,150 sequences of SCTC, whereas no hits were found for 4552 transcripts out of 43,702 assembled high quality sequences. For both the samples, maximum number of transcripts showed significant similarity with Solanum lycopersicum, followed by Vitis vinifera, and so forth (Figs. 4, 5). GenScan prediction tool, based on the Arabidopsis model matrix parameter, yielded 35,493 CDS (33,024 with significant BLAST hits and 2469 without BLAST hits), 44,277 exons and 34,493 peptides for SCFG, and 26,349 CDS (25,634 with significant BLAST hits and 715 without BLAST hits), 31,757 exons and 31,757 peptides for SCTC (Table 2). Annotation against UniProtKB/Swiss-Prot database yielded significant annotation of 30,903 from 57,460 assembled sequences in SCFG and 23,563 from 43,702 assembled sequences in SCTC transcriptomes, respectively, representing best possible hits (Suppl. Table S3).

Species distribution of the top Blastx hits of SCFG transcripts. (Color figure online)

Species distribution of the top Blastx hits of SCTC transcripts. (Color figure online)
Functional classification by GO and COG
GO classification is based on orthology and direct experimental evidence with more detailed functional annotation and analysis of gene products (42,988 terms). On the other hand, COGs classification is based on orthology only, where genes are assigned to broad categories (24) corresponding to their ancient conserved domain. Therefore, to unify gene and gene product the annotated transcripts were mapped onto GO database. The assembled transcripts, 57,460 for SCFG and 43,702 for SCTC were subjected to Blast2GO program [44], which yielded 18,090 and 2102 functional terms for SCFG and SCTC, respectively. These transcripts were further classified into three major categories, the majority of the assignments belonged to molecular function (8277 and 45.75% for SCFG; 1105 and 52.56% for SCTC), followed by the biological process (6221 and 34.38% for SCFG; 559 and 26.59% for SCTC) and cellular component (3589 and 19.83% for SCFG; 435 and 20.69% for SCTC) (Figs. 6, 7; Suppl. Table S4). Among all categories, cell, catalytic and metabolic process were the most abundant classes in cellular component, molecular function and biological processes, respectively. These observations were in agreement with previously assigned GO terms in A. thaliana, P. kurroa, Medicago truncatula and Cicer arietinum [59, 62].

Distribution of GO annotated transcripts in SCFG transcriptome of S. chirayita

Distribution of GO annotated transcripts in SCTC transcriptome of S. chirayita
To categorize and predict potential function, the transcripts were were ligned up against COG database using the BLASTx program. The transcripts showing significant similarity (E-value of 10−5) with those in the database were classified into the respective functional classes. COG assignments were given to 12,826 and 9565 transcripts of SCFG and SCTC, respectively. COG analysis revealed that SCFG contained higher number of transcripts representing different functional classes compared to the SCTC sample (Figs. 8, 9). The higher number of transcripts in SCFG compared to SCTC might be due to differential environmental conditions encountered by S. chirayita plants.

Distribution of COG classified transcripts of SCFG transcriptome of S. chirayita

Distribution of COG classified transcripts of SCTC transcriptome of S. chirayita
On further analysis, we categorized transcripts into 25 functional classes, where the most frequent functional category was observed to be “general function prediction (symbol R)” for both tissue samples (2405 in SCFG and 1820 in SCTC) followed by “post-translational modification, protein turnover, chaperones (symbol O)” (1624 in SCFG and 1164 in SCTC), “translation, ribosomal structure and biogenesis (symbol J)” (1184 in SCFG and 907 in SCTC), “carbohydrate transport and metabolism (symbol G)” (783 in SCFG and 622 in SCTC) and others, which was similar to the study done on Taxodium [63]. Furthermore, 528 and 313 transcripts of SCFG and SCTC, respectively were classified into “secondary metabolites biosynthesis, transport and catabolism (category symbol Q)” in S. chirayita.
Pathway mapping using KEGG
KEGG automatic annotation server (KAAS) was employed to map transcripts onto their biological pathways. Bi-directional best hit scheme was employed for KEGG orthology (KO) assignments with defalult BHR (> 0.95). EC number was assigned to 4510 and 3541 transcripts from SCFG and SCTC sample, using KAAS (Suppl. Table S5). From a total of 57,460 transcripts in SCFG, 8690 KO assigned transcripts were mapped onto 342 KEGG pathways. Out of 8690 transcripts, 830 (12.65%) were related to metabolic pathways, 351 (5.35%) to the biosynthesis of secondary metabolites, 20 (0.30%) to citrate cycle (TCA cycle), 20 (0.30%) to pentose phosphate pathway and 35 (0.53%) to glycolysis/gluconeogenesis. Similarly, in SCTC, out of 43,702 assembled transcripts, 6991 transcripts were assigned KO, which were mapped to 341 KEGG pathways. Out of 6991 transcripts, 796 (12.86%) were related to metabolic pathways, 341 (5.50%) to the biosynthesis of secondary metabolites, 22 (0.355%) to tricarboxylic acid (TCA) cycle, 17 (0.274%) to pentose phosphate pathway and 32 (0.51%) to glycolysis/gluconeogenesis (Suppl. Table S6). The BRITE functional hierarchy categorized the transcripts hierarchically thereby linking them with biological systems such as metabolism, genetic information processing and cellular processes in S. chirayita. KEGG analysis results will immensely help to explore the biological information (metabolic pathways, biochemical reactions, cellular processes, pathway interactions and others) hidden in the mass of data and will assist in gaining insight into the biology of this species. Moreover, the comparative KEGG analysis for both the transcriptome datasets will help to unravel the major enzymes and pathways encoding for important secondary metabolites in S. chirayita.
Functional classification using NCBI biosystems
All the transcripts from both the transcriptomes were further analyzed using NCBI BioSystems database (Suppl. Table S7). These transcripts were categorised into five major classes, namely; genetic information processing, metabolism, cellular processes, organismal systems and environmental information processing. Highest number of transcripts belonged to the metabolism category (349, 46.65%), followed by genetic information processing (341, 45.58%), cellular processes (33, 4.41%), organismal systems (15, 2.00%) and environmental information processing (10, 1.33%) in SCFG transcriptome. On the other hand for SCTC, genetic information processing formed the biggest category SCTC (280, 47.61%), followed by metabolism (268, 45.57%), cellular processes (20, 3.40%), organismal systems (13, 2.21%) and environmental information processing (7, 1.25%).
These major five categories were further subcategorized to gain insight into their biological roles. For SCFG sample, metabolism category included 99 transcripts in carbohydrate metabolism, 64 transcripts in energy metabolism, 42 transcripts in lipid metabolism, 29 transcripts in nucleotide metabolism, 48 transcripts in amino acid metabolism, 25 transcripts in metabolism of other amino acids, 10 transcripts in glycan biosynthesis and metabolism, 22 transcripts in metabolism of cofactors and vitamins, 10 transcripts in metabolism of terpenoids and polyketides, 7 transcripts in biosynthesis of other secondary metabolites. Genetic information processing included 71 transcripts in transcription, 190 transcripts in translation, 80 transcripts in folding, sorting and degradation, 28 transcripts in replication and repair. Further, environmental information processing included 4 transcripts in membrane transport and 6 transcripts in signal transduction, whereas cellular processes included 33 transcripts in transport and catabolism. Organismal systems included 4 transcripts in immune system and 11 transcripts in environmental adaptation. For SCTC sample, metabolism category included 61 transcripts in carbohydrate metabolism, 58 transcripts energy metabolism, 27 transcripts lipid metabolism, 26 transcripts in nucleotide metabolism, 36 transcripts in amino acid metabolism, 10 transcripts in metabolism of other amino acids, 8 transcripts in glycan biosynthesis and metabolism, 26 transcripts in metabolism of cofactors and vitamins, 7 transcripts in metabolism of terpenoids and polyketides and 9 transcripts in biosynthesis of other secondary metabolites. Genetic information processing included 61 transcripts in transcription, 137 transcripts in translation, 57 transcripts in folding, sorting and degradation, and 25 transcripts in replication and repair. Again, environmental information processing included 4 transcripts in membrane transport, 3 transcripts in signal transduction, whereas cellular processes included 20 transcripts in transport and catabolism. Organismal systems included 3 transcripts in immune system and 10 transcripts in environmental adaptation. Functional classification using biosystems yielded significant results depicting ongoing important processes such as genetic information processing and various metabolic processes implicated in SCFG versus SCTC transcriptomes.
Due to non-availability of reference genome, estimation of the number of genes and the level of transcript coverage is difficult. However, BLAST similarity search provides a platform to functionally annotate the transcripts obtained from lesser explored species by comparing them against unique known proteins available in different public databases. All the above ways of annotating unknown transcripts assigned functions to major fraction of the transcripts in S. chirayita.
Comparative analysis between SCTC and SCFG transcriptomes to unravel unique/common genes
Comparative analysis of SCTC and SCFG transcriptomes was performed to find out the common and unique genes in tissue cultured versus green house grown plants of S. chirayita. This analysis was performed in order to pull out information related to genes those are specifically correlated with secondary metabolites biosynthesis in S. chirayita influenced by differential mode of nutrition. Out of the total transcripts (57,460), 47,464 transcripts (82.60%) in SCFG were common to SCTC transcriptome, whereas 9996 transcripts were uniquely present in SCFG transcriptome (Suppl. Table S8). Further analysis evaluated the transcriptomes abundance for common and unique genes across the SCFG and SCTC transcriptomes. Transcript abundance, ranged from 0 to 2244.45 for common genes and 0 to 322.47 for uniquely present genes in SCFG transcriptomes. In case of SCTC transcriptome, transcript_1599 showed the highest transcript abundance (2244.45 FPKM), whereas its corresponding transcript_956 in SCTC showed 582.38 FPKM value. The presence of common genes in SCTC and SCFG transcriptomes infers the common function of genes in various physio-biological processes in S. chirayita. The unique genes present in SCTC (and/or SCFG) transcriptomes might be because of differential modes of nutrition between the two sample types. A graphical representation of the differentially expressed genes in various processes in SCFG and SCTC transcriptomes has been given in Fig. 10. The figure illustrates the distribution of genes of various processes as analysed through GO annotation (Fig. 10a–c), KEGG annotation (Fig. 10d–f) and heatmap of the differentially expressed transcripts of primary and secondary metabolic pathways in SCFG and SCTC transcriptomes (Fig. 10g). The heatmap was generated using using Multiple Experiment Viewer (MEV v4.9.0). In the common tranceipts region of the Venn diagram, a transcript of SCFG may have one or more than one common transcripts in SCTC and vice versa. The common identified genes from SCTC and SCFG transcriptomes may be responsible for fundamental processes as well as conserved defense responses against environmental regimes.

Differentially expressed genes of various processes between SCFG and SCTC transcriptomes: a–c Venn diagram of shared GO terms; d–f Venn diagram of shared KEGG processes; g heatmap of the differentially expressed genes (log normalised FPKM values) of primary and secondary metabolic pathway genes in SCFG and SCTC. Common transcripts of SCFG and SCTC represented in Venn diagrams have one or more than one transcripts in common and vice versa. (Color figure online)
Unique and common genes have been identified in plant species such as A. thaliana, Jatropha curcas, Ricinus communis and others, for their role in plant defense mechanisms [64, 65]. The common genes present in SCTC and SCFG transcriptomes also support the fact of duplication events occurring in plant species [66].
Mining secondary metabolite pathway genes in S. chirayita transcriptomes
The swertiamarin, mangiferin and amarogentin are the major secondary metabolites in S. chirayita biosynthesized through MVA/MEP and the phenylpropanoid route of terpenoids biosynthesis (Fig. 3; [24]). The SCFG and SCTC transcriptomes were mined for genes involved in the secondary metabolites biosynthesis pathways in S. chirayita using in-house perl scripts. Fifteen genes of MVA/MEP pathways, coding for AACT, HMGS, HMGR, MVK, PMK, MVDD, DXS, DXR, ISPD, ISPE, ISPF, ISPG, ISPH, IPPI and GDPS and 13 genes, encoding DAHPS, DHQS, DHQD, SAK, EPSPS, CS, CM, PAT, ADH, ADT, PHAT, PAL and C4H of phenylpropanoid pathway were identified in SCFG and SCTC transcriptomes. The transcriptomes of several plant species, such as P. kurroa, Aconitum heterophyllum, Podophyllum hexandrum and others, have been mined to decipher metabolic pathway genes involved in the biosynthesis of secondary metabolites [59, 67,68,69]. The transcript abundance was also checked for these pathways genes by FPKM method using RSEM. The transcript abundance analysis of MVA/MEP/phenylpropanoid pathways revealed that most of the genes (17 genes) showed higher transcript abundance in SCFG compared to SCTC transcriptomes, thereby suggesting their significant contribution in secondary metabolites production in S. chirayita (Suppl. Table S9). Moreover, genes showing higher transcript abundance could be suitable targets for implementation of genetic intervention strategies aimed towards enhancement of metabolite contents in S. chirayita. The FPKM based transcript abundance method has already been used in many plant species, including P. hexandrum, P. kurroa, Malus domestica, Camellia sinensis and others, for determining the relative contribution of pathways genes in secondary metabolite production [59, 67, 70, 71].
Experimental validation of FPKM data for genes of fundamental processes using RT-qPCR
Expression analysis of primary metabolic pathway genes belonging to glycolysis, TCA cycle, oxidative pentose phosphate pathway and Calvin cycle were performed by RT-qPCR to further validate the expression pattern observed through in silico transcript abundance analysis (FPKM). Sixteen genes (coding for IDH, G6PD, PRK, OGDH, DLD, DLST, HXK, PFK1, PGK, FBP, RUBISCO, ENO, FUM, GPI, SUCLG and RPE) involved in various fundamental processes were selected for experimental validation on the basis of higher in silico transcript abundance values (Suppl. Table S10). Their relative expression across the SCFG and SCTC samples were assessed through RT-qPCR. Quantitative real-time PCR analysis revealed that 10 genes (encoding G6PD, OGDH, DLD, HXK, PFK1, FBP, RUBISCO, FUM, SUCLG and RPE) out of 16 showed elevated expression levels (≥ twofold) in SCFG as compared to SCTC (Fig. 11), which is in agreement with the RNA-seq data. Thus, gene expression observed through different platforms i.e. FPKM and qRT-PCR based was found in tune with each other. Previously, these two methods have been used to validate the expression pattern of genes in plant species such as P. hexandrum, C. sinensis, Solanum tubersoum, M. domestica and others [67, 70,71,72]. Our results are in agreement with the previous studies, where the expression pattern of biosynthesis pathway genes varied in tune with metabolite contents such as shikonin in Arnebia euchroma, picrosides in P. kurroa, and catechins in C. sinensis, podophyllotoxin in P. hexandrum, and aconites in A. heterophyllum [59, 66,67,68,69].

Relative fold changes in the expression pattern of fundamental pathway genes in SCFG with respect to SCTC. Error bars correspond to percentage error (5%)
Multiple pathways contribute to autophototrophic and photoheterotrophic modes of nutrition in relation to secondary metabolite biosynthesis in S. chirayita
The contribution of various metabolic pathways pertaining to metabolites biosynthesis can be better understood through mining of transcriptomes. In S. chirayita, the differentially regulated multiple pathways encountered in differential growth regimes (SCFG and SCTC) are suggested to be contributing towards growth and biosynthesis of secondary metabolites. The primary metabolic pathways such as glycolysis, TCA cycle, oxidative pentose phosphate pathway and the light independent Calvin cycle of photosynthesis provide necessary precursors for the biosynthesis of building blocks of secondary metabolism in plant systems. As a matter of fact, if the biosynthesis of secondary metabolites is directly influenced by the levels of precursors and intermediates of primary metabolism, it should get reflected in the abundance of transcripts encoding those pathway enzymatic steps. In our study, the contents of major secondary metabolites were higher in SCFG samples compared to SCTC, which was in tune with the expression patterns of primary metabolic pathways.
In silico transcript abundance revealed that nineteen genes from primary metabolic pathways (Fig. 12), including 7 genes of glycolysis (encoding HXK, GPI, PFK1, GAPDH, PGK, ENO and PVK), 6 genes of TCA cycle (coding for IDH, OGDH, DLD, DLST, FUM and SUCLG), 3 genes of pentose phosphate pathway (encoding G6PD, 6PGL and RPE) and 3 genes of Calvin cycle (coding for RuBisCO, FBP and PRK) showed elevated levels of transcripts abundance in SCFG. Similarly, 17 genes from secondary metabolic pathways (Fig. 13), that include 9 genes (coding for HMGS, MVK, PMK, ISPD, ISPE, ISPF, IPPI, GDPS and MVDD) from MVA/MEP pathways and 8 genes (encoding DAHPS, DHQS, EPSPS, SAK, CS, ADH, PAL and C4H) from phenylpropanoid pathway showed higher transcript abundance in SCFG compared to SCTC transcriptomes, thereby suggesting their major contribution in the biosynthesis of metabolites (Suppl. Tables S9 and S10). The contribution of primary and secondary metabolic pathway genes was further validated through RT-qPCR to infer their role in the biosynthesis of metabolites in S. chirayita. Expression patterns of most of the genes of the metabolic pathways, observed through two different approaches (FPKM based and RT-qPCR), showed higher gene expression levels in tissues with higher secondary metabolites content and vice versa. Our results are in agreement with previous reports which highlighted the contribution of MVA, MEP and phenylpropanoid pathways genes, where genes coding for HMGR, MVK, PMK, ISPD, ISPE, IPPI, GDPS, CS, ADH, PAL and C4H showed enhanced expression in relation to the biosynthesis and accumulation of target metabolites such as podophyllotoxin, aconites, picrosides, withanolides, phytosterols and terpenoids in P. hexandrum, A. heterophyllum, P. kurroa, Salvia miltiorrhiza, Croton sulyratus and Catharanthus roseus, respectively [59, 67,68,69, 73,74,75,76] (Figs. 11, 12, 13). However, the outcomes of current study need functional validation to correlate the final enzyme levels in relation to transcripts amounts with the contents of target metabolites.

Transcript abundance of primary metabolic pathways genes: a glycolysis pathway b TCA pathway c Pentose phosphate pathway d Calvin cycle in SCFG and SCTC transcriptomes. (Color figure online)

Transcript abundance of secondary metabolic pathways genes: a MVA/MEP pathway b phenylpropanoid/shikimic acid pahtway in SCFG and SCTC transcriptomes. (Color figure online)
Identification of transcription factors families and ABC transporters in transcriptomes
Transcription factors (TFs) and transporters regulate various functions of cells, such as growth, development, transportation, nutrition, response to external and internal stimuli, and various primary and secondary metabolism acting in spatiotemporal manner. In this study, PlantTFcat identified several TF/TR/CR genes (4100 in SCFG and 3210 in SCTC) belonging to 108 TF families. In SCFG, out of the 872 TF-coding transcripts, 108 were unique to SCFG. Similarly, BLAST hits for TFs involved in secondary metabolites in SCTC were 614, out of which 28 transcripts were found to be unique to SCTC. Overall 21.26% transcripts contributed to the biosynthesis of secondary metabolites in SCFG compared to only 19.12% transcripts in SCTC. All these TFs associated with secondary metabolites biosynthesis belong to eight classes (AP2-EREBP, bHLH, bZIP, DOF, MYB, NAC, WRKY and Zinc-finger) of transcription factor families. Regulatory surveillance by these transcription factors can be assessed for their role in the biosynthesis of major phytochemicals in S. chirayita as demonstrated earlier in terpene biosynthesis in S. lycopersicum [77] and artemisinin biosynthesis in Artemisia annua [78].
ATP-binding cassette (ABC) transporters are one of the largest families in plants known for their role in transporting various macro- and micromolecules across biological membranes [79,80,81]. ABC transporters in plant cells act as importers of sugars and other carbohydrates, amino acids, peptides, polyamines, metal ions and others, whereas exporters of polysaccharides, proteins, lipids and hydrophobic drugs including toxins such as hemolysin [82, 83]. Pfam domain database identified a total of 39,376 domain/family in SCFG transcriptome, whereas 28,261 in SCTC transctiptome. A total of 3166 and 3075 unique domain/family classes were observed in SCFG and SCTC transcriptomes, respectively. The diffrerential statistics revealed 158 domain/family unique to SCFG transcriptome, 67 domain /family unique to SCTC transcriptome and 3008 doamin/family common to both the transcriptomes (Suppl. Table S11). Pfam domain database revelaed 299 and 207 ABC-type transporters domains (including PDR_associated, ABC_membrane_2 domains and Cytochrom_C_asm family) from 144 SCFG and 129 SCTC transcripts, respectively. A list of ABC transporter families identified in S. chirayita transcriptomes is given in Table 3. On further analysis, 125 transcripts were identified as common transcripts in both the transcriptomes, 19 transcripts were uniquely present in SCFG transcriptome, whereas 4 transcripts were unique to SCTC transcriptome. No clan was found from a total of 39 and 25 peptide sequences in SCFG and SCTC, respectively (Suppl. Table S12).
Additionally, we also checked the status of 22 functionally characterized ABC transporters of A. thaliana against SCFG and SCTC transcriptomes to decipher their roles in S. chirayita. The in silico transcript abundance analysis revealed that out of 22, 13 ABC transporters genes showed higher transcript abundance in SCTC compared to SCFG suggesting their major contribution in different physio-biological processes in SCTC. AtABCG11, AtABCG13, AtABCG32 and AtABCG26 have been characterized in flower for their role in cutin formation and pollen exine formation. AtABCC5 and AtABCI13/14/15 were found in seeds playing a role in insP6 loading to the seeds in addition to cutin and plastid lipid formation. AtABCB1/19, AtABCD1, AtABCG11/12/32 and AtABCI13/14/15 were characterized in shoot for their role in fatty acyl-CoA import to peroxisome, auxin transport, cuticle and plastid lipid formation. AtABCB14, AtABCC5, AtABCG11/12/32, AtABCG40 in leaf were assessed for stomatal regulation, insP6 transport, cuticle formation and ABA import. AtABCB1/4/19, AtABCG11, AtABCG25 and AtABCI16/17 in the root have been found to be associated with auxin transport, suberin formation, ABA import and aluminum tolerance, whereas AtABCC1, AtABCC2, AtABCG19 and AtABCG36 genes were validated for their ubiquitous role in folate and chlorophyll catabolite transport, kanamycin tolerance, metal/metalloid tolerance, biotic and abiotic stress tolerance (Table 4).
The identified common transcripts between SCFG and SCTC transcriptomes suggested their role in transportation of various essential macro- and micromolecules under differential modes of nutrition in S. chirayita. Uniquely present transcripts in SCFG and SCTC transcriptomes could be associated with the transport of specific nutrients implicated in the biosynthesis and accumulation of primary and secondary metabolites in S. chirayita. To further filter out the important ABC transporters in SCFG and SCTC transcriptomes, the transcript abundance based on FPKM value was calculated using RSEM. Results revealed that in silico transcript abundance of ABC transporters genes was higher in SCTC compared to SCFG which suggest their major contribution in the transport of nutrients in SCTC. The transcript abundance ranged from 0.04 to 156.03 FPKM value in SCFG and ranged from 0.05 to 147.92 in SCTC transcriptome (Suppl. Table S13). Previously, the possible roles of molecular components (pathway genes, TFs and ABC transporters) implicated in various physio-biological processes such as growth and development, seed germination, secondary metabolism, responses to stress and others, has been reported on the basis of in silico transcript abundance in plant species like A. heterophyllum [58], A. euchroma [84] and P. hexandrum [67].
Several studies have demonstrated photoautotrophy as a physiological adaptation that is achieved through a change in environment such as depletion of sucrose and controlled condition of light and CO2 [85]. These factors are highly compromised in tissue cultured plants of S. chirayita accounting for its photoheterotrophic behaviour. A decline in the photosynthetic activity is seen when sucrose is present in the medium as the plants preferentially utilize this carbon source. Depletion of sucrose results in reestablishment of photosynthetic carbon fixation along with increase in chlorophyll content of the system [85]. CO2 concentration is an essential component required for photosynthesis and a transition from photomixotrophy to photoautotrophy has been established by feeding CO2 as illustrated in hairy root cultures of Acmella oppositifolia [86]. CO2 fixation in plants operates through two carboxylating enzyme systems. Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) enzyme involved in carbon dioxide fixation in plants, that adds CO2 to ribulose-1,5-bisphosphate to form two molecules of 3-phosphoglycerate. In the other system, phosphoenolpyruvate carboxylase (PEPCase) plays a key role in binding CO2 to phosphoenolpyruvate, a glycolytic intermediate, to create oxaloacetate. This oxaloacetate is then converted back to puruvate with the release of CO2 which is ultimately utilised in carbon fixation by RuBisCO and the Calvin cycle. A lower RuBisCO:PEPCase transcript abundance ratio was observed in tissue cultured S. chirayita compared to the photoautotrophic field grown plants possibly accounting for the difference in their net photosynthetic rate (data not shown). However, what needs to be investigated is this difference also being reflected at enzyme level.
Concluding remarks
The detailed analysis of two transcriptomes of S. chirayita grown under differential mode of nutrition revealed crucial molecular components differing in various physio-biological processes. The study provided deep refinements into the existing knowledge of biosynthesis of major phytochemicals in S. chirayita, and the molecular dynamics associated with it through identification and comparative analysis of MVA/MEP and phenylpropanoid pathways genes, transcription factors (TFs) and genes of ABC transporters. Through this study, we have made an attempt to link the contribution of important, yet much less explored fundamental processes to secondary metabolites biosynthesis in plant systems by comparing transcriptomes from differentially grown plant samples. De novo transcriptomes characterization identified 30 genes belonging to various fundamental processes like glycolysis, TCA cycle, oxidative pentose phosphate pathway and light independent Calvin cycle of photosynthesis and 28 genes of secondary metabolic pathways (MVA/MEP/phenylpropanoid pathways). A similar expression pattern observed through FPKM and RT-qPCR data in relation to metabolites content validated the sequencing and assembly procedures employed. This in turn supports our idea of the roles played by differentially regulated pathways (primary and secondary) in plants differing in their modes of nutrition (photoautotrophic vs. photoheterotrophic) those ultimately decide the final metabolite levels. The transcriptomics data generated in the present work has immense value and opens new window for exploring and designing genetic intervention strategies such as marker discovery for conservation as well as improving the metabolites contents in an endangered medicinal herb, S. chirayita. Further experimental validation needs to be done in order to assess the exact biological functions for the data sets generated through computational annotations procedures. Our study provides a new bench mark for future endeavors by providing the transcriptome resource to scientific community in the open website http://14.139.240.55/NGS/download.php.
Abbreviations
- 6PGD:
-
6-Phosphogluconate dehydrogenase
- 6PGL:
-
6-Phosphogluconolactonase
- 7-DLGT:
-
7-Deoxyloganetic acid glucosyl transferase
- 8-HGO:
-
8-Hydroxygeraniol oxidoreductase
- AAC:
-
Acetyl-CoA carboxylase
- AACT:
-
Acetoacetyl-CoA thiolase
- ADH:
-
Arogenate dehydrogenase
- ADT:
-
Prephenate dehydratase
- AH:
-
Aconitate hydratase
- C3H:
-
p-Coumarate 3-hydroxylase
- C4H:
-
Trans-cinnamate 4-hydroxylase
- CM:
-
Chorismate mutase
- COG:
-
Clusters of orthologous groups
- CS:
-
Chorismate synthase
- CRS:
-
Citrate (si)-synthase
- DAHPS:
-
3-Deoxy-D-arabinoheptulosonate-7-phosphate synthase
- DHQD:
-
3-Dehydroquinate dehydratase
- DHQS:
-
3-Dehydroquinate synthase
- DL7H:
-
7-Deoxyloganic acid hydroxylase
- DLD:
-
Dihydrolipoamide dehydrogenase (part of oxoglutarate dehydrogenase complex)
- DLST:
-
Dihydrolipoamide S-succinyl transferase (part of oxoglutarate dehydrogenase complex)
- DXR:
-
1-Deoxy-D-xylulose 5-phosphate reductoisomerase
- DXS:
-
1-Deoxy-D-xylulose 5-phosphate synthase
- ENO:
-
Enolase
- EPSPS:
-
5-Enolpyruvylshikimate-3-phosphate synthase
- FBA:
-
Fructose bisphosphate aldolase
- FBP:
-
Fructose bisphosphatase
- FUM:
-
Fumarate hydratase
- G10H:
-
Geraniol 10-hydroxylase/8-oxidase
- G6PD:
-
Glucose 6-phosphate dehydrogenase
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GDPS:
-
Geranyl diphosphate synthase
- GES:
-
Geraniol synthase
- GO:
-
Gene ontology
- GPI:
-
Phosphoglucose isomerise
- HMGR:
-
3-Hydroxy-3-methylglutaryl-CoA reductase
- HMGS:
-
3-Hydroxy-3-methylglutaryl-CoA synthase
- HXK:
-
Hexokinase
- IDH:
-
Isocitrate dehydrogenase
- IO:
-
Iridoid oxidase
- IPPI:
-
Isopentenyl diphosphate isomerase
- IS:
-
Iridoid synthase
- ISPD:
-
2-C-Methyl-D-erythritol 4-phosphate cytidylyltransferase
- ISPE:
-
4-(Cytidine-5′-diphospho)-2-C-methyl-D-erythritol kinase
- ISPF:
-
2-C-Methyl-D-erythritol 2,4-cyclodiphosphate synthase
- ISPG:
-
(E)-4-Hydroxy-3-methylbut-2-enyl diphosphate synthase
- ISPH:
-
(E)-4-Hydroxy-3-methylbut-2-enyl diphosphate reductase
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LMT:
-
Loganic acid O-methyltransferase
- MEP:
-
2-C-Methyl-D-erythritol 4-phosphate
- MQO:
-
Malate:quinone oxidoreductase
- MVA:
-
Mevalonate
- MVDD:
-
Mevalonate diphosphate decarboxylase
- MVK:
-
Mevalonate kinase
- OGDH:
-
Oxoglutarate dehydrogenase E1 subunit (part of oxoglutarate dehydrogenase complex)
- PAL:
-
Phenylalanine ammonia lyase
- PAT:
-
Aspartate–prephenate aminotransferase
- PFK1:
-
Phosphofructokinase
- PGAM:
-
Phosphoglycerate mutase
- PGK:
-
Phosphoglycerate kinase
- PHAT:
-
Phenylalanine (histidine) aminotransferase
- PMK:
-
Phosphomevalonate kinase
- PRK:
-
3-Epimerase phosphoribulokinase
- PVK:
-
Pyruvate kinase
- RPE:
-
Ribulose 5-phosphate 3-epimerase
- RPI:
-
Ribose 5-phosphate isomerase
- RuBisCO:
-
Ribulose-1,5-bisphosphate carboxylase/oxygenase
- SAK:
-
Shikimate kinase
- SCFG:
-
Swertia chirayita green house grown plant
- SCTC:
-
Swertia chirayita tissue cultured plants
- SDH:
-
Shikimate dehydrogenase
- SLS:
-
Secologanin synthase
- SUCLG:
-
Succinate-CoA ligase
- TAL:
-
Tyrosine ammonia-lyase
- TALD:
-
Transaldolase
- TKT:
-
Transketolase
- TPI:
-
Triosephosphate isomerise
References
Rao MR, Palada MC, Becker BN (2004) Medicinal and aromatic plants in agroforestry systems. Agrofor Syst 61–62(1–3):107–122
Kala CP, Dhyani PP, Sajwan BS (2006) Developing the medicinal plants sector in northern India: challenges and opportunities. J Ethnobiol Ethnomed 2:32
Hasan SZ, Misra V, Singh S et al (2009) Current status of herbal drugs and their future perspectives. Biol Forum Int J 1:12–17
Sharma A (2004) Global medicinal plants demand may touch $5 trillion by 2050. Indian Express
Joshi K, Chavan P, Warude D, Patwardhan B (2004) Molecular markers in herbal drug technology. Curr Sci 87:159–165
Joshi P, Dhawan V (2005) Swertia chirayita : an overview. Curr Sci 89:635–640
Williamson EM (2002) Major herbs of Ayurveda. Churchill Livingstone, New York
Bhat GP, Surolia N (2001) In vitro antimalarial activity of extracts of three plants used in the traditional medicine of India. Am J Trop Med Hyg 65:304–308
Alam KD, Ali MS, Mahjabeen S, Hassan MR, Rahman MF, Chowdhury RMAA. (2011) Potential hypoglycemic effect of Swertia chirata: an Indian subcontinent herb with important medicinal value. Pharmacol Online 2:642–647
Arya R, Sharma S, Singh S (2011) Antidiabetic effect of whole plant extract and fractions of Swertia chirayita Buch.-Ham. Planta Med 77:138
Nagalekshmi R, Menon A, Chandrasekharan DK, Nair CKK (2011) Hepatoprotective activity of Andrographis paniculata and Swertia chirayita. Food Chem Toxicol 49:3367–3373
Verma VK, Sarwa KK, Kumar A, Zaman MK (2013) Comparison of hepatoprotective activity of Swertia chirayita and Andrographis paniculata plant of north-east India against CCl4 induced hepatotoxic rats. J Pharm Res 7(7):647–653
Saha P, Mandal S, Das A, Das PC, Das S (2004) Evaluation of the anticarcinogenic activity of Swertia chirata Buch.Ham, an Indian medicinal plant, on DMBA-induced mouse skin carcinogenesis model. Phytother Res 18:373–378
Banerjee S, Sur TK, Mandal S, Das PC, Sikdar S (2010) Assessment of the anti-inflammatory effects of Swertia chirata in acute and chronic experimental models in male albino rats. Indian J Pharmacol 32:21–24
Das SC, Bhadra S, Roy S, Saha SK, Islam MS, Bachar SC (2012) Analgesic and anti-inflammatory activities of ethanolic root extract of Swertia chirata (Gentianaceae). Jordan J Biol 5:31–36
Roy P, Abdulsalam FI, Pandey DK, Bhattacharjee A, Eruvaram NR, Malik T (2015) Evaluation of antioxidant, antibacterial, and antidiabetic potential of two traditional medicinal plants of India: Swertia cordata and Swertia chirayita. Pharmacognosy Res 7(Suppl 1):S57
Verma H, Patil PR, Kolhapure RM, Gopalkrishna V (2008) Antiviral activity of the Indian medicinal plant extract, Swertia chirata against herpes simplex viruses: a study by in-vitro and molecular approach. Indian J Med Microbiol 26:322
Rehman S, Latif A, Ahmad S, Khan AU (2011) In-vitro antibacterial screening of Swertia chirayita Linn. against some gram negative pathogenic strains. Int J Pharm Res Dev 4:188–194
Laxmi A, Siddhartha S, Archana M (2011) Antimicrobial screening of methanol and aqueous extracts of Swertia chirata. Int J Pharm Pharm Sci 3(Suppl 4):142–146
Patil K, Dhande S, Kadam V (2013) Therapeutic Swertia chirata: an overview. Res J Pharmacogn Phytochem 5:199–207
Badola HK, Pal M (2002) Endangered medicinal plant species in Himachal Pradesh. Curr Sci 83:797–798
Bhat JA, Kumar M, Negi A, Todaria N (2013) Informants’ consensus on ethnomedicinal plants in Kedarnath Wildlife Sanctuary of Indian Himalayas. J Med Plant Res 7:148–154
IUCN S (2008) The IUCN Red List of Threatened Species, 1994–2007 version. IUCN, Gland
Padhan JK, Kumar V, Sood H, Singh TR, Chauhan RS (2015) Contents of therapeutic metabolites in Swertia chirayita correlate with the expression profiles of multiple genes in corresponding biosynthesis pathways. Phytochemistry 116:38–47
Kovarik B (2013) Biofuels in history, in: Singh BP (ed) Biofuel crops: production, physiology and genetics. CABI Publishing, Wallingford, pp 1–22
Zhang J, Wrage EL, Vankova R, Malbeck J, Neff MM (2006) Over-expression of SOB5 suggests the involvement of a novel plant protein in cytokinin-mediated development. Plant J 46:834–848
Parkash J, Vaidya T, Kirti S, Dutt S (2014) Translation initiation factor 5A in Picrorhiza is up-regulated during leaf senescence and in response to abscisic acid. Gene 542:1–7
Koslowsky S, Riegler H, Bergmüller E, Zrenner R (2008) Higher biomass accumulation by increasing phosphoribosylpyrophosphate synthetase activity in Arabidopsis thaliana and Nicotiana tabacum. Plant Biotechnol J 6:281–294
Mosaleeyanon K, Zobayed SMA, Afreen F, Kozai T (2005) Relationships between net photosynthetic rate and secondary metabolite contents in St. John’s wort. Plant Sci 169(3):523–531
Fine PVA, Miller ZJ, Mesones I et al (2006) The growth-defense trade-off and habitat specialization by plants in Amazonian forests. Ecology 87:S150–S162
Vogt T (2010) Phenylpropanoid biosynthesis. Mol Plant 3:2–20
Casal JJ, Yanovsky MJ (2005) Regulation of gene expression by light. Int J Dev Biol 49:501
Kawoosa T, Singh H, Kumar A et al (2010) Light and temperature regulated terpene biosynthesis: hepatoprotective monoterpene picroside accumulation in Picrorhiza kurrooa. Funct Integr Genom 10:393–404
Paulilo MTS, Lapa FS, Falkenberg MdB (2010) Effect of light intensity and growth substratum on plant development and production of secondary metabolites in Cordia curassavica (Jacq.) Roem Schult. Rev Árvore 34:417–423
Dewick PM (2001) Secondary metabolism: the building blocks and construction mechanisms. In: Medicinal natural products: a biosynthetic approach. Wiley, Chichester, pp 7–38
Dreyer I, Horeau C, Lemaillet G et al (1999) Identification and characterization of plant transporters. J Exp Bot 50:1073–1087
Jiménez-Guri E, Huerta-Cepas J, Cozzuto L et al (2013) Comparative transcriptomics of early dipteran development. BMC Genom 14:123
Smith SA, Wilson NG, Goetz FE et al (2011) Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature 480:364–367
Hartmann S, Helm C, Nickel B et al (2012) Exploiting gene families for phylogenomic analysis of Myzostomid transcriptome data. PLoS ONE 7:e29843
Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nat Rev Genet 12:671–682
Kumar V, Chauhan R, Sood H (2013) In vitro production and efficient quantification of major phytopharmaceuticals in an endangered medicinal herb, Swertia chirata. Int J Biotechnol Bioeng Res 4:495–506
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Magrane M, Consortium U (2011) UniProt knowledgebase: a hub of integrated protein data. Database 2011:bar009
Conesa A, Götz S, García-Gómez JM et al (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323
Jaiswal V, Chanumolu SK, Gupta A, Chauhan RS, Rout C (2013) Jenner-predict server: prediction of protein vaccine candidates (PVCs) in bacteria based on host-pathogen interactions. BMC Bioinform 14:211
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Vandesompele J, De Preter K, Pattyn F et al (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C T method. Nat Protoc 3:1101–1108
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182–W185
Geer LY, Marchler-Bauer A, Geer RC et al (2010) The NCBI BioSystems database. Nucleic Acids Res 38:D492–D496
Vom Endt D, Kijne JW, Memelink J (2002) Transcription factors controlling plant secondary metabolism: what regulates the regulators? Phytochemistry 61:107–114
Dai X, Sinharoy S, Udvardi M, Zhao PX (2013) PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinform 14:321
Yang CQ, Fang X, Wu XM et al (2012) Transcriptional regulation of plant secondary metabolism. J Integr Plant Biol 54:703–712
Goossens A, Häkkinen ST, Laakso I, Oksman-Caldentey KM, Inzé D (2003) Secretion of secondary metabolites by ATP-binding cassette transporters in plant cell suspension cultures. Plant Physiol 131:1161–1164
Kang J, Park J, Choi H et al (2011) Plant ABC transporters. Arabidopsis Book 9:e0153
Pal T, Malhotra N, Chanumolu SK, Chauhan RS (2015) Next-generation sequencing (NGS) transcriptomes reveal association of multiple genes and pathways contributing to secondary metabolites accumulation in tuberous roots of Aconitum heterophyllum Wall. Planta 242(1):239–258
Gahlan P, Singh HR, Shankar R et al (2012) De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments. BMC Genom 13:126
Bhattacharyya D, Sinha R, Hazra S, Datta R, Chattopadhyay S (2013) De novo transcriptome analysis using 454 pyrosequencing of the Himalayan Mayapple, Podophyllum hexandrum. BMC Genom 14:748
Simpson JT, Wong K, Jackman SD et al (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123
Pradhan S, Bandhiwal N, Shah N et al (2014) Global transcriptome analysis of developing chickpea (Cicer arietinum L.) seeds. Front Plant Sci 5:698
Qi B, Yang Y, Yin Y, Xu M, Li H (2014) De novo sequencing, assembly, and analysis of the Taxodium ‘Zhongshansa’ roots and shoots transcriptome in response to short-term waterlogging. BMC Plant Biol 14:201
Armisén D, Lecharny A, Aubourg S (2008) Unique genes in plants: specificities and conserved features throughout evolution. BMC Evol Biol 8:280
Sood A, Jaiswal V, Chanumolu SK et al (2014) Mining whole genomes and transcriptomes of Jatropha (Jatropha curcas) and Castor bean (Ricinus communis) for NBS-LRR genes and defense response associated transcription factors. Mol Biol Rep 41:7683–7695
Haake V, Cook D, Riechmann JL et al (2002) Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis. Plant Physiol 130:639–648
Kumar P, Pal T, Sharma N et al. (2015) Expression analysis of biosynthetic pathway genes vis-à-vis podophyllotoxin content in Podophyllum hexandrum Royle. Protoplasma 252:1–10
Pandit S, Shitiz K, Sood H, Naik PK, Chauhan RS (2013) Expression pattern of fifteen genes of non-mevalonate (MEP) and mevalonate (MVA) pathways in different tissues of endangered medicinal herb Picrorhiza kurroa with respect to picrosides content. Mol Biol Rep 40:1053–1063
Malhotra N, Kumar V, Sood H, Singh TR, Chauhan RS (2014) Multiple genes of mevalonate and non-mevalonate pathways contribute to high aconites content in an endangered medicinal herb, Aconitum heterophyllum Wall. Phytochemistry 108:26–34
Singh AK, Sharma V, Pal AK, Acharyam V, Ahuja PS (2013) Genome-wide organization and expression profiling of the NAC transcription factor family in potato (Solanum tuberosum L.). DNA Res 20:403–423
Paul A, Jha A, Bhardwaj S et al (2014) RNA-seq-mediated transcriptome analysis of actively growing and winter dormant shoots identifies non-deciduous habit of evergreen tree tea during winters. Sci Rep 4:5932
Arya P, Kumar G, Acharya V, Singh AK (2014) Genome-wide identification and expression analysis of NBS-encoding genes in Malus × domestica and expansion of NBS genes family in Rosaceae. PLoS ONE 9:e107987
Veau B, Courtois M, Oudin A et al (2000) Cloning and expression of cDNAs encoding two enzymes of the MEP pathway in Catharanthus roseus. BBA-Gene Struct Expr 1517:159–163
De-Eknamkul W, Potduang B (2003) Biosynthesis of β-sitosterol and stigmasterol in Croton sublyratus proceeds via a mixed origin of isoprene units. Phytochemistry 62:389–398
Chaurasiya ND, Sangwan NS, Sabir F, Misra L, Sangwan RS (2012) Withanolide biosynthesis recruits both mevalonate and DOXP pathways of isoprenogenesis in Ashwagandha Withania somnifera L. (Dunal). Plant Cell Rep 31:1889–1897
Yang L, Ding G, Lin H et al (2013) Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis. PLoS ONE 8:e80464
Spyropoulou EA, Haring MA, Schuurink RC (2014) Expression of Terpenoids 1, a glandular trichome-specific transcription factor from tomato that activates the terpene synthase 5 promoter. Plant Mol Biol 84:345–357
Yu ZX, Li JX, Yang CQ et al (2012) The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol Plant 5:353–365
Theodoulou FL (2000) Plant ABC transporter. BBA-Biomembr 1465:79–103
Holland IB, Cole SPC, Kuchler K, Higgins CF (2003) ABC proteins: from bacteria to man. Academic Press, Amsterdam
Jeong CB, Kim BM, Lee JS, Rhee JS (2014) Genome-wide identification of whole ATP-binding cassette (ABC) transporters in the intertidal copepod Tigriopus japonicas. BMC Genom 15:651
Garmory HS, Titball RW (2004) ATP-binding cassette transporters are targets for the development of antibacterial vaccines and therapies. Infect Immun 72:6757–6763
Davidson AL, Dassa E, Orelle C, Chen J (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72:317–364
Singh RS, Gara RK, Bhardwaj PK et al (2010) Expression of 3-hydroxy-3-methylglutaryl-CoA reductase, p-hydroxybenzoate-m-geranyltransferase and genes of phenylpropanoid pathway exhibits positive correlation with shikonins content in arnebia [Arnebia euchroma (Royle) Johnston]. BMC Mol Biol 11:88
Hardy T, Chaumont D, Wessinger ME, Bournat P (1987) Photoautotrophic suspension cultures II: transition from photoheterotrophic to photoautotrophic growth. J Plant Physiol 130(4):351–361
Flores HE, Dai YR, Cuello JL, Maldonado-Mendoza IE, Loyola-Vargas VM (1993) Green roots: photosynthesis and photoautotrophy in an underground plant organ. Plant Physiol 101(2):363–371
Acknowledgements
The authors are thankful to the Department of Biotechnology, Ministry of Science and Technology, Government of India for providing financial support to RSC in the form of a programme support on high-value medicinal plants.
Author information
Authors and Affiliations
Contributions
RSC conceptualized and hypothesized the biological questions. HS and JKP generated the study materials. TP and JKP did the computational analyses, JKP and PK analyzed literature into experiments, results and interpretations. All authors have contributed to, seen, and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Data accessibility All scripts and sequence data used in this study are accessible via http://14.139.240.55/NGS/download.php.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pal, T., Padhan, J.K., Kumar, P. et al. Comparative transcriptomics uncovers differences in photoautotrophic versus photoheterotrophic modes of nutrition in relation to secondary metabolites biosynthesis in Swertia chirayita. Mol Biol Rep 45, 77–98 (2018). https://doi.org/10.1007/s11033-017-4135-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-017-4135-y