
Abstract
As an ancient disease, tuberculosis (TB) is a major global health threat. Therefore, there is an urgent need for an effective and safe anti-TB vaccine. In the current study, a delivery system of Fc domain of mouse IgG2a and early secreted antigenic target protein 6 (ESAT-6) was evaluated for the selective uptake of antigens by antigen-presenting cells (APCs). Thus, it was based on the immunogenicity of a fusion protein. The study was initiated by the transfer of recombinant expression vectors of pPICZαA-ESAT-6:Fcγ2a and pPICZαA-ESAT-6: His into Pichia pastoris (P. pastoris). Recombinant proteins were assessed for immunogenicity following the immunoblotting analysis. High levels of IFN-γ and IL-12 were produced to induce Th1-type cellular responses through vaccination with both recombinant proteins [ESAT-6:Fcγ2a (EF) and ESAT-6:His (EH)]. The Fc-tagged recombinant protein induced more effective Th1-type cellular responses with a low increment in IL-4 compared to PBS, BCG, and EH groups. Although in all the immunized groups, the ratio of IFN-γ/IL-4 was in favor of Th1 responses, the highest Th1/Th2 balance was observed in EF immunized group. Fc fragment of mouse IgG2a may induce a selective uptake of APCs towards the cross-presentation and formation of Th1 responses in favor of an appropriate protective anti-tuberculosis reaction. Thus, further research on Fc-fusion proteins is required to develop Fc-based TB vaccines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) has remained as a major global health threat though it is known to be an ancient disease. As reported by the World Health Organization (WHO), more than 9.6 million new cases and 1.5 million mortalities are annually caused by TB [1]. Children can be currently protected against severe TB, particularly meningitis, by BCG as the only vaccine available today. However, it has highly varied protective efficacies on pulmonary TB ranging from 0 to 80 % in adults [2]. TB appears at frequent times in most people, who are even vaccinated with BCG. Therefore, there is an urgent need of developing novel vaccines and vaccination strategies to consistently protect adults against pulmonary TB. Some vaccines, such as protein subunit and DNA vaccines, Mycobacterium tuberculosis (Mtb) attenuated mutants, and recombinant BCG over expressing certain Mtb antigens are being currently investigated [3]. So far, a number of Mtb secretory proteins as subunit vaccine candidates have been evaluated to be opposing TB. As one of the major immunodominant antigens, Early secreted antigenic target protein 6 (ESAT-6) is encoded by difference-1 region (RD1) lacking in BCG strains [4]. This antigen has been shown to induce potent immune responses against Mtb infection due to having numerous immunogenic epitopes that are recognized by T cells [5]. As a result, high protective responses against TB have been recently found to be induced by this subunit or DNA vaccine [6–8].
Yet, obtainment of an efficient uptake and processing induced by antigen-presenting cells (APCs) to provide protective immune responses against Mtb infection would remain as a major restriction to the application of protein subunit vaccines. Fusion of one or more antigenic proteins into immunoglobulin (Ig) Fc domain with the use of Fc-fusion proteins is an alternative [9]. These transgenic polypeptides have such main advantages of easily purifying Fc-containing molecules, improving the half-life of plasma, and being soluble and stable in vivo [10]. Several investigations have revealed that a critical protection against TB is induced by the Th1 immune response of an effector via Fcγ receptors (FcγRs, FcγRI, FcγRIIA, FcγRIIB, and FcγRIII) located on APCs such as dendritic cells (DCs) and macrophages for a selective antigen presentation [11–15]. In addition, a better delivery system is provided for antigens when fusing to Fc domains that bind to one or more FcγRs on APCs. This method is more effective as the half-life of the subunit vaccine and selective uptakes by DCs via FcγRs increase and consequently the killing properties of CD8+T cells and cross-presentation are potentiated to overcome intracellular pathogens [16].
Therefore, improvements of Mtb ESAT-6 immunogenicity and subsequent immune protective responses of Th1 by Fc domain fusion to ESAT-6 antigen are strongly evidenced by these data. In this study, the Fc fragment of mouse IgG2a (Fcγ2a) was fused to ESAT-6 to produce ESAT6:Fcγ2a recombinant protein and induce a Th1 immune response.
Materials and methods
Designing and synthesis of expression cassettes
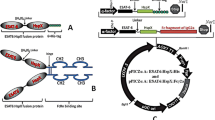
The coding sequences of ESAT-6:His (EH) [Gene Bank accession no. KT366446] and ESAT6:Fcγ2a (EF) [Gene Bank accession no. KT366445] were codon-optimized based on P. pastoris codon usage bias in order to enhance the expressions of EF and EH fusion proteins. Then, they were synthesized into pUC57 plasmid (GenScript, USA). The cutting sites of XhoI and NotI restriction enzymes and KEX2 cleavage site encoded by a nucleotide sequence were located upstream of the gene constructs. The recombinant vector pPICZαA-ESAT-6:Fcγ2a and pPICZαA-ESAT-6:His were resulted from the ligation of the synthesized gene constructs digested with XhoI and NotI (Thermo Scientific, USA) into pPICZαA (Invitrogen, USA) digested vector of the same enzyme. C-myc and His tag at C-terminal were contained by the recombinant pPICZαA-ESAT-6:His (Fig. 1). A stop codon was added at the end of the Fc fragment to prevent His tag expression in the recombinant vector pPICZαA-ESAT-6:Fcγ2a. The Luria–Bertani (LB) agar medium (Merck, Germany) containing 25 μg/mL Zeocin™ (InvivoGen, USA) was used to grow the transformed E. coli Top10 F′ cells. The recombinant plasmids were isolated from the transformant cells by a highly pure plasmid kit (Roche, Germany). Both expression plasmids were then fully sequenced over the linked sites to ensure that these constructs had appropriate and in-frame fusion products.

A schematic representation of the recombinant plasmids. (A) Schematic map of pPICZα-EF. (B) Schematic map of pPICZα-EH. Digestion of the recombinant constructs was performed using XhoI and NotI restriction enzymes. They were then cloned into the pPICZαA plasmid, which was digested by XhoI and NotI, so as to provide recombinant plasmids. 5′ AOX1; alcohol oxidase 1 promoter, AOX1 TT; transcriptional terminator from P. pastoris AOX1 gene, TEF1 promoter; transcriptional elongation factor 1 promoter from Saccharomyces cerevisiae, EM7 promoter; synthetic prokaryotic promoter, Zeocin; Zeocin resistance gene, CYC1 TT; transcriptional terminator from S. cerevisiae CYC1 gene, pUCori; pUC origin of replication
Pichia pastoris transformation and transformant selection
The recombinant vectors were transformed into P. pastoris GS115 cells after being linearized through digestion with SacI. As described in P. pastoris expression manual (Invitrogen, USA), the electroporation method was employed to subsequently introduce them into the competent P. pastoris GS115 cells (Invitrogen, USA). The transformed cells were incubated at 28 °C for 3 days and then selected on YPD medium (1 % yeast extract, 2 % dextrose, 2 % peptone, and 2 % agar) plates containing 100 µg/mL Zeocin. After incubation of Zeocin-resistant colonies supplemented with 100 µg/mL Zeocin during the mentioned period, several colonies appeared on the YPD plate. Then, the largest colonies were selected for the protein expression.
Colony PCR for the selection of best transformants
Right colonies with an integrated recombinant DNA of EF and EH were selected by PCR (based on Easy Select Pichia Expression Kit, Invitrogen, USA) and were then stored for further investigations.
Small-scaled expression of recombinant proteins
To select clones with the highest levels of expression, the transformed yeast cells were corroborated by PCR and inoculated in 5 mL of BMGY in a baffled flask. They were then allowed to reach 28 °C in a shaking incubator (250 rpm) until an OD600 = 2 was achieved by the culture within 16–18 h. To induce expression of the recombinant protein, the cells were centrifuged at 5000 rpm at 4 °C for 5 min and then harvested. Afterwards, the pellet was re-suspended in buffered minimal methanol (BMMY) medium for OD600 to reach 1. The baffled flasks were then incubated at 250 rpm and 28 °C. Every 24 h, 100 % methanol was added to the final concentration of 0.5 % methanol to maintain the induction. Finally, the culture supernatants were collected after 3 days and then analyzed to see if the protein had been produced by ELISA. The recombinant proteins produced in supernatant samples were coated in ELISA plate wells at the beginning of the process. Following this, 2 % Bovine Serum Albumin (BSA) (Sigma-Aldrich, USA) was employed to block the coated plates, which were then incubated at 37 °C for 1 h. Then, goat anti-mouse IgG HRP (Santa Cruz, USA) and His probe antibody (H3) HRP (Santa Cruz, USA) were used to incubate the plates for the second time at 37 °C for 1 h following the washing steps. Using an ELISA reader (DiaMed EuroGen), the absorbance values were read at 450 nm after the substrate and stop solution were added. Finally, to perform a Medium-scale expression, the clones of highest absorbance were selected.
Medium-scale expression of recombinant proteins
One litre BMGY medium was utilized to grow the best transforming clones selected by ELISA in a shaking incubator (250 rpm) at 28 °C for the production of more proteins. This was continued until the cell density reached OD600 = 2. Then, the cells were centrifuged at 5000 rpm at 4 °C for 5 min to be harvested. Afterwards, 2 L BMMY medium was applied to re-suspend them until an OD600 of 1 was reached. The baffled flasks were incubated at 250 rpm and 28 °C for 3 days. Finally, every 24 h, 100 % methanol was added to the final concentration of 0.5 % methanol to augment the recombinant protein expression.
Purification of recombinant proteins
By centrifuging at 10,000 rpm for 10 min at the end of the expression period, the supernatant was collected after removal of the yeast cells from the culture medium and the recombinant proteins were purified. HiTrap rProtein A Sepharose Fast Flow column (GE Healthcare, USA) was used to purify EF. By adding 1 M sodium phosphate buffer and using a filter of 0.45 μm, the supernatant pH was adjusted to 7 and then filtered at the beginning of the process. Loading of the filtered supernatant onto a HiTrap column was done at a flow rate of 3 mL/min after applying binding buffer (20 mM sodium phosphate, pH 7) to wash the column. Then, a new binding buffer was utilized to wash the column again before eluting the column strip of the target proteins with an elution buffer (0.1 M sodium citrate, pH 4.6). The mentioned proteins were then collected in microtubes containing neutralization buffer (1 M Tris–HCl, pH 9).
Ni–NTA agarose (Qiagen, USA) was employed to purify the labeled His-tag recombinant protein (EH). Lysis buffer (0.5 % Triton X-100, 10 % glycerol, 50 mM potassium phosphate pH 7.8, 100 mM KCl, and 400 mM NaCl) was passed through Ni–NTA agarose to equilibrate the column after being washed with distilled water. Loading of the supernatant onto a Ni–NTA agarose column was done at a flow rate of 1 mL/min. Using an elution buffer (lysis buffer containing 500 mM imidazole), the protein was eluted from the band after the column was washed with lysis buffer (containing 10 and 30 mM imidazole). To concentrate and desalt the eluted fractions, an ultrafiltration of vivaspin column centrifugation 20 (Sartorius Stedim, Germany) was utilized. Finally, evaluation of the eluted fractions (containing recombinant proteins) was done by immunoblotting and using SDS-PAGE.
SDS-PAGE and western blotting
Analysis of the recombinant protein expression was performed using SDS-PAGE and Western blotting. According to Laemmli buffer system, a 12 % gel was used to visualize the recombinant protein via SDS-PAGE performance [17]. Staining of the gels was done with Coomassie Brilliant Blue G-250 following Bio-Rad Mini PROTEAN electrophoresis (Bio Rad, USA). In addition, a 12 % gel was applied to separate the proteins for Western blotting. Then, using a semi-dry approach, transferring of the proteins from the gel to polyvinylidene fluoride (PVDF) membrane (GE Healthcare, USA) was conducted. Blocking of the membranes was performed with 2 % BSA at 4 °C overnight after the electrotransferring process. Using goat anti-mouse IgG HRP to detect EF recombinant proteins at a dilution of 1/10,000, incubation of the membranes was done for probing at room temperature for 1 h. Additionally, with the help of antibody His-probe (H3) HRP at a dilution of 1/5000, incubation of the membranes was performed to detect the EH fusion protein at room temperature for 1 h. Finally, using a chemiluminescence detection system (ECL, Amersham Biotech), detection of the specific recombinant proteins was conducted.
APC-targeting of EF recombinant fusion protein
Using a direct immunofluorescence assay, confirmation of Fc-fusion protein binding to Fcγ receptor (FcγRI) on APCs was done. In brief, on slides, ESAT-6:Fcγ2a fusion was added to the immunofluorescence slides, on which mouse macrophages had been fixed and then incubated at 37 °C for 60 min. After washing the cells with 0.1 % (w/v) BSA (Sigma-Aldrich, USA) in phosphate-buffered saline (PBS), they were fixed with methanol/acetone (v/v) at −20 °C and incubated with fluorescent antibodies consisting of PE anti-mouse CD64 (FcγRI) (BioLegend, USA) and goat anti-mouse IgG2a-FITC (Santa Cruz, USA) in 3 % (w/v) BSA in a humidified chamber for 120 min. Ultimately, a fluorescence microscope (Nikon Eclipse E200, Japan) was employed to view the slides and obtain the images.
Subunit vaccine preparation
50 µg of each fusion protein was diluted in sterile phosphate-buffered saline (PBS) to prepare an appropriate concentration. Using a lipid film method, preparation of an adjuvant formulation of trehalose-6,6-dibehenate (TDB) (IvivoGen, USA) and dimethyl dioctadecyl ammonium bromide (DDA) (Sigma-Aldrich, USA) was done [18]. Before injecting the vaccine, a mixture of CAF01 adjuvant (DDA/TDB; 250/50 µg) and the recombinant proteins was prepared.
Immunization process of mice
6–8 week aged female C57BL/6 mice were purchased from the Pasteur Institute of Iran and maintained at the animal house of Bu-Ali Research Institute (Mashhad, Iran) under a condition of special pathogen void. The study protocols were approved by the Animal Institutional Committee of MUMS (Mashhad University of Medical Sciences) (permit no: 910072). The mice were randomly divided into six groups (5 mice per group): (1) the group receiving 200 μL PBS (negative control), (2) the group receiving BCG 5 × 105 colony forming units (CFU), (3) the group receiving 50 µg of EF in 100 μL of PBS plus 100 μL of DDA/TDB adjuvant, (4) the group receiving 50 µg of EH in 100 μL of PBS plus 100 μL of DDA/TDB adjuvant, (5) the group primed with BCG and stimulated by ESAT-6: Fcγ2a subunit vaccine (50 µg of EF in 100 μL of PBS plus 100 μL of DDA/TDB adjuvant), and (6) the group primed with BCG and stimulated by EH subunit vaccine (50 µg of EH in 100 μL of PBS plus 100 μL of DDA/TDB adjuvant). Immunization of the mice was done through a subcutaneous injection four times at 2 week intervals. The PBS or BCG primed groups received one-time injection at 0 week. Other groups received three injections at 0, 4, and 6 week. Two groups were immunized with BCG plus each subunit vaccine and then boosted by each recombinant protein mixed with DDA/TDB twice at a 14-d interval.
Evaluation of cytokines production
Two suspensions of the spleen cells of the mice groups were prepared following aseptical removal of the spleens after 2 weeks of final immunization. Lymphocytes cells were seeded in 24 well plate (4 × 106 cells/well) and cultured in 1 mL of RPMI 1640 medium (Biosera, UK) and stimulated with EH (5 µg/mL) and EF (5 µg/mL) for 72 h (37 °C, 5 % CO2). Phytohemagglutinin (PHA, Gibco,USA) (5 µg/mL) was applied as a positive control. The supernatant was collected after incubation and IFN-γ, IL-12, IL-4, and IL-17 levels were determined by commercial ELISA kits (eBioscience, USA) based on manufacturer’s instructions.
Statistical analysis
The data were expressed as mean ± standard deviation (SD) of the duplicate samples. Tukey’s multiple comparison tests of ONE-WAY ANOVA were used to determine the statistical significance of the differences at p < 0.05.
Results
Designing and cloning of the expression cassettes
The schematic Fig. 1 shows the designed recombinant fusion proteins. After these gene constructs were synthesized into pUC57 plasmid, they were sub-cloned into pPICZαA vector containing α-factor signal sequence for the recombinant proteins to be additionally secreted by the cells, as well as AOX1 promoter.
Transformation and screening of transformants
Positive clones were selected on YPDS, which contained 100 μg/mL Zeocin™ after transformation. Then, the recombinant pPICZαA vectors were observed to have integrated into P. pastoris cells after the genomic DNA was isolated from the transfected clones. This was corroborated by colony PCR by using α-factor specific primers and AOX1 (data not shown).
Expression of recombinant proteins
Based on the ELISA assay, the clones with the highest absorbance were selected for a Medium-scale expression following the screening of small-scale expression. Then, HiTrap rProtein A Sepharose Fast Flow Column (for EF) and Ni–NTA Agarose (for EH) was utilized to purify the recombinant proteins collected from the supernatant. The protein bands of nearly 35 and 12 kDa represented Western blot and SDS-PAGE analyses of the eluted fractions for EF (Fig. 2a, b) and EH (Fig. 2c, d) recombinant proteins, respectively. Also, 35 and 12 kDa were calculated for the molecular weights of EF and EH proteins based on amino acid sequences, respectively (ExPASy, PeptideMass).

Analysis of the Recombinant Proteins by SDS-PAGE and Western blots a EF recombinant protein (12 % SDS-PAGE, Coomassie blue staining). Lane 1: protein marker; Lane 2 and 3 fractions of the eluted recombinant protein (~35 kDa). b EF recombinant protein (Western blot). c EH recombinant protein (12 % SDS-PAGE, Coomassie blue staining); Lane 1: protein marker; Lane 2 and 3 fractions of the eluted recombinant protein (~12 kDa). d EH recombinant protein (Western blot)
APC-targeting of EF recombinant fusion protein
Direct immunofluorescence assay was employed to demonstrate Fc-fusion recombinant protein (ESAT-6:Fcγ2a) binding to Fcγ receptor (FcγRI) on APCs. Thus, it demonstrates the binding of EF to the FcγRI on mouse macrophage cells (Fig. 3).

Co-localization of FcγRI on macrophages and EF recombinant fusion protein a Immunofluorescence staining of macrophages fixed with methanol/acetone associated with goat anti-mouse IgG2a-FITC and PE anti-mouse CD64 (FcγRI) as a negative control (red signal indicating macrophages stained with PE anti-mouse CD64 (FcγRI) antibody). b Light microscopy image obtained from the same field. c Immunofluorescence staining of macrophages with PE anti-mouse CD64 (FcγRI) and goat anti-mouse IgG2a-FITC representing EF fusion binding to FcγRI. Red signal displaying macrophages stained with PE anti-mouse CD64 (FcγRI) antibody; Green signal (green ring around the cells) exhibiting EF recombinant fusion stained with goat anti-mouse IgG2a-FITC antibody. PE phycoerythrin , FITC fluorescein isothiocyanate
Assessment of immune responses to recombinant proteins
After grouping of C57BL/6 mice, they were subcutaneously immunized with BCG, PBS, or recombinant proteins, i.e. emulsified EH or EF in DDA-TDB. Afterwards, the levels of IL-12, IFN-γ, IL-4, and IL-17 produced by the stimulated spleen cells with EH (5 µg/mL), EF (5 µg/mL), and PHA (5 µg/mL) were evaluated to determine the cellular immune responses of Th1, Th2, and Th17 models in the different groups of the immunized mice.
IL-12 and IFN-γ (Th1 response)
A significantly higher concentration of IFN-γ were found in the mice immunized with EH compared to those of PBS and BCG groups (p < 0.05) (Fig. 4a). Also, a significantly higher level of IFN-γ in mice vaccinated with EF was discovered in comparison to those of BCG, PBS, and EH groups (p < 0.05). In addition, a significantly higher concentration of IFN-γ was observed in the mice immunized with EH compared to the groups immunized with PBS and BCG (p < 0.05) (Fig. 4b). Moreover, a significantly higher level of IL-12 was produced in mice vaccinated with EF in comparison to those of EH, PBS, and BCG groups (p < 0.05). The results revealed that the specific CMI potentiated with EF was to a higher degree than those amplified by EH, PBS, and BCG groups.

Cytokine level released from the splenocytes of the immunized mice. The splenocytes were prepared from individual mice (5 in each group) and cultured in the presence of EF and EH recombinant proteins 2 weeks after the last immunization. Then, ELISA assay of cytokine was employed to determine the levels of a IFN-γ, b IL-12, and c IL-4 after 72 h. three independent experiments were used to express the data as the mean value ±SD of each group (n = 5). PHA; phytohemagglutinin. The significant differences among groups are represented by different letters
IL-4 (Th2 response)
No significant correlation was seen between the group immunized with recombinant proteins (EH or EF) and the control groups (PBS and BCG) in the case of IL-4 level (p > 0.05). Furthermore, the higher levels of IL-4 in the mice vaccinated with EF was found compared to those of EH, PBS, and BCG groups, but it was not a significant difference (p > 0.05) (Fig. 4c). A comparison between IFN-γ and IL-4 productions was revealed that immunization with EF could stimulate Th1 immune response (Fig. 5a).

Releases of cytokine from the splenocytes of the immunized mice. a Comparison of IL-12, IFN-γ, and IL-4 secretions in the mice vaccinated. b IL-17 cytokine production in the immunized group. No statistically significant differences were found between the groups based on IL-17 production. c The ratio of IFNγ/IL-4 in the vaccinated mice. The significantly higher ratio of IFN-γ/IL-4 in the stimulated splenic cells of EF obtained from the mice immunized with EF compared to the other groups. three independent experiments were applied to express the data as the mean value ±SD of each group (n = 5). PHA phytohemagglutinin. The significant differences among groups are represented by different letters
IL-17 (Th17 response)
Less than 1 pg/mL of IL-17 as Th17 marker was seen to have been produced in all the groups, which was lower than the limit of detection level (Fig. 5b).
IFN-γ/IL-4 ratio (Th1/Th2 balance)
The cytokine pattern was indicative of a significantly higher IFN-γ/IL-4 ratio in the group immunized with EF compared to the other groups (p < 0.05) (Fig. 5c). Although, the immunized groups had strong Th1 responses, the highest Th1/Th2 ratio was observed in the EF immunized group.
BCG priming and subunit boosting
Two study groups were primed with BCG and then, the primed animals were boosted with the subunit fused proteins in DDA/TDB at a certain time. The secretions of IFN-γ, IL-12, IL-4, and IL-17 were assessed in the activated splenic lymphocytes with EF, EH (5 μg/mL), or PHA (5 μg/mL). A significantly higher concentration of IFN-γ was discovered in the group primed with BCG and boosted with EF compared to the BCG-primed groups boosted by EH, BCG, and PBS (p < 0.05). In addition, a higher IFN-γ concentration was observed in the BCG-immunized animals boosted with EH in comparison to those of BCG and PBS groups (p < 0.05) (Fig. 4a). A higher IL-12 level was found in the group primed with BCG and boosted with EF compared to those of PBS and BCG groups (p < 0.05) (Fig. 4b).
Although IL-4 secretion level was higher than BCG-primed group boosted with EF, there was no statistically significant difference between the treated groups (Fig. 4c). The level of IL-17 was lower than the minimum detection limit of ELISA kit (Fig. 5b).
Discussion
Protection of BCG vaccine against TB has been evidenced in children, but it has been shown to have a highly variable efficacy in the prophylaxis of adult pulmonary TB. Therefore, it is urgently needed to develop and test new strategies for vaccination against TB. In the current research, it was attempted to produce improved responses to ESAT-6 antigen by adding Fcγ2a as a new delivery system. Previous studies have shown that targeting of immunodominantn peptides to FcγRs on antigen-presenting cells can potentiate its selectively uptake and increase CMI and antibody responses both in vitro and in vivo [11–15]. These reports demonstrate that the immunogenicity of Fc-fused proteins could be enhanced by immunoglobulin Fc domain (especially IgG) so that fusion with antigens, such as ESAT-6 from Mtb, can be formed, which can be used as a delivery system. In this study, to boost specific immune responses against Mtb, ESAT-6 antigen was fused to mouse Fcγ2a to construct ESAT6:Fcγ2a so as to facilitate the selective antigen intake of ESAT6 protein by FcγRs on APCs and its immunogenicity was then compared to a similar His-tagged construct (ESAT6:His).
Traditionally, cellular immune responses have been generally deemed to mediate the protection against intracellular microbes. Generally, Th1 cells producing IFN-γ play a major role in protection against TB by enhancing macrophage ability to engulf and kill Mtb [19]. Undoubtedly, Th1 cell activity, which leads to IFN-γ, IL-2, and TNF-α production, provides the immunity protection against Mtb. Yet, the question is raised as to whether host protection against Mtb infection or the disease development is only provided by Th1. The studies conducted in the previous decade have shown that host defense during Mtb infection is mainly provided by Th17 cells that recruit the inflammatory cells, especially neutrophils, in the acute local sites. Furthermore, during the Mtb infection, IFN-γ protective effects are augmented by IL-17 [20]. Another dominant cytokine is interleukin-12 (IL-12), by which Th1 immunity induction and maintenance are enhanced. The shift from naïve T-cells towards Th1-type cells is more critically influenced by IL-12 production by APCs [21]. M. tuberculosis infection dissemination and the disease development are further promoted by Th2 anti-inflammatory cytokines like IL-4 in contrast to Th1 and Th17 cytokines [22]. The microenvironment at the inflammation site determines the reaction outcomes by activating appropriate transcription factors to induce protective responses. In the presence of such suitable cytokines as IL-12 and IL-4, the activated T-cells are differentiated towards Th1 and Th2 cells, respectively. These changes are accrued in the presence of antigen [23]. Therefore, the efficacy of a new vaccine candidate against TB highly depends on an effective inducibility of T cell response, especially Th1 response.
ESAT-6 is a major immunodominant antigen among the proteins secreted from Mtb capable of inducing a protective immunity against TB [5]. ESAT-6 has been shown by several studies to be the only virulent factor with a potentiality of modulating immune responses though Th1 cells can recognize the majority of epitopes identified in ESAT-6. The mentioned studies showed that IFN-γ and IL-12 secretions were significantly inhibited by ESAT-6 by binding to TLR2 (Toll-like receptor-2) and making an interference with TLR signaling [24–26]. In the current research, ESAT-6 protein was fused with Fcγ2a to prevent binding to TLR2 and targeting at FcγR on APCs. Similar to some previous studies, significantly higher levels of IFN-γ and IL-12 inducing Th1 immune responses were found in the mice only vaccinated with ESAT-6 (EH) compared to the groups immunized with BCG and PBS (Fig. 4a, b) [27–29]. The results obtained might be dependent on ESAT-6 purity or concentration, method of administration, selection of host or vector for expression, and selection of the adjuvant. Sereinig et al. demonstrated that Th1 immune response could be induced by the intranasal immunization of mice and guinea pigs with influenza virus NS vectors that are capable of expressing ESAT-6 and thus, the animals were protected against tuberculosis [30].
In this study, EH and EF recombinant proteins as highly effective factors on promoting Th1 persistent immune responses were formulated by choosing DDA/TDB adjuvant [31]. Nevertheless, investigators have sought to utilize some other expression systems to overcome such difficulties as insolubility, poor yield, and gene instability for the production of mycobacterial antigens in E. coli [32, 33].
In the present research, P. pastoris expression system was employed to produce recombinant proteins with several advantages such as post-translational modifications (disulfide bond formation, proteolytic processing, glycosylation, and protein folding), high levels of productivity in cheap media, and fast growth rate associated with easy manipulation and culture [34].
As expected, EH recombinant protein induced the immune cells to produce high levels of IL-12 and IFN-γ, but to a significantly less degree than EF. Although Mtb ESAT-6 is an immunodominant antigen, the main difference between the mentioned fused immunogenes for the secretion of IL-12 and IFN-γ is tagging. Fc had a powerful impact on the immunogenicity of Fc fusion recombinant protein (EF), most likely by affecting the APCs (Fig. 4a, b5a). The results obtained were indicative of significantly higher levels of IFN-γ and IL-12 in the mice vaccinated with EF compared to the groups immunized with BCG and PBS (Fig. 4a and b). In addition, as the other studies exhibited a mixed immune response of augmented Th1 and declined Th2 responses, the elevated IL-4 level in the mice vaccinated only with EF recombinant protein or primed with BCG provided a protection against TB (Fig. 4c) and thus this study was consistent with the mentioned researches [35, 36].
In the present study, the concentration of IL-17 as a marker of Th17 in all groups was less than the lower limit of detection (Fig. 5b). Thus, these results confirmed that both recombinant proteins pushed the immune cells to produce strong antigen-specific CMI response in mice with a more effective response for Fc fusion protein.
Th2 cytokines, IL-4, IL-5, and IL-10 inhibited the excessive response of Th1. There happens an interaction between Th1 and Th2 cells to inhibit each other’s activities by blocking either the functions of the opposite cell type receptor or its maturation [37]. Generally, an imbalance is observed between TB development and pathogenesis of Th1/Th2 cytokines [38]. In this investigation, EF immunized group represented the highest Th1/Th2 balance (IFN-γ/IL-4 ratio) (Fig. 5c). This result was indicative of the stronger action of EF than EH to shift Th1/Th2 balance towards Th1 responses.
Several investigations revealed that the immunity induced by BCG can be boosted by the prime-boost vaccination strategy [39]. The findings of this study represented that an interesting vaccine strategy against TB can be obtained by producing a high level of Th1 immune response in the mice immunized with EF fusion protein of DDA/TDB adjuvant (Fig. 4a, b).
According to the results achieved, a combination of EF recombinant protein and DDA/TDB adjuvant can induce more strenuous cellular immune responses than EH. The previous studies demonstrated that antigen targeting to FcγRs on APCs not only changes its processing and trafficking to T cells, but also increases its binding and uptake in comparison to a soluble antigen [15, 40]. Thus, it seems that inducing protective cellular immunity against intracellular pathogens is mainly caused by CD4+ and CD8+ Th1 cells, which can be stimulated by the application of Fc-fusion proteins. A cross-presentation leading to CD8+ T cell responses of an effector can be induced by an antigen presentation and cross-linking mediated by FcγRs on DCs [41]. Moreover, due to an avidity effect, a high-affinity binding is provided by the homo-dimeric nature of Fc-fusion proteins [42]. Utilization of FcγR antigen targeting may be involved in a main restriction of its potential for the interaction with the inhibitory FcγR (FcγR IIB) on APCs resulting in a tolerogenic immune response. Nonetheless, one of the reasons for its binding to the stimulatory Fc receptor can be the co-localization of Fc-fusion protein produced from yeast and FcγRI (CD 64) as the mentioned receptor is engaged in a cross-presentation and cell-mediated immunity induction (Fig. 3). Besides, the results of fluorescence immunoassay (FIA) indicated that binding to its main receptor (FcγRI or CD64) would adequately occur due to the protein folding.
Fc fragment of IgG supported the idea of the antigen selective uptake inducing cross-presentation and forming an appropriate anti-TB immune response in the context of Th1/Th2 and Th17/Treg balances. This is very pivotal for protection and particularly prevention from damage. However, comparing the findings of the present study with our previous results obtained from the studies of Soleimanpour et al. and Farsiani et al. [15, 43], in which subunit vaccine candidates consisting of multi-stage Mtb antigens were used, demonstrated that ESAT-6 fused to another Mtb antigen is in favor of a stronger Th1 immune response. There are two different interpretations. Firstly, in those studies, ESAT has been put in the Nterminal region of the fusion proteins, i.e. the inhibitory effect of C-terminus of ESAT has been prevented. Secondly, those have been multi-stage Fc-fused vaccines and have had more immunodominant epitops, which have been able to efficiently potentiate CMI immune responses [15, 43, 44]. Also, Fc-tag in EF has conferred other advantages on the proposed vaccine strategy, including ease of purification by using protein A column under mild conditions and increased protein stability conferred by the Fc fragment. Fc-tag may also have an adjuvant property, which usually occurs in the interaction between Fc in the recombinant protein and Fcγ receptors on APCs and activates immune reactions [15, 44].
The results obtained from cytokine production revealed that the generation of Th1 immune responses of both CD4+ and CD8+ can be induced by EF recombinant proteins. Therefore, the protective immune response against TB can be evaluated by a suitable animal model, such as a non-human primate of Mtb. Thus, the current study was confined by this type of assessment.
It can be concluded that EF recombinant protein is an efficient subunit vaccine candidate, which can be used for the prevention of Mtb infection. However, further research must be conducted to determine whether EF immunization induces suitable protection in more appropriate animals such as guinea pigs and monkey. This is the beginning stage of exploring Fc domain capacity of antibodies for the achievement of efficient immune responses against Mtb infection. Still, novel anti-TB vaccines should be designed for new antigens and delivery systems in the future.
References
World Health Organization (2015) Global tuberculosis report 2015 (WHO). www.who.int/about/licensing/copyright_form/en/index.html
Nuttall JJ, Davies MA, Hussey GD, Eley BS (2008) Bacillus Calmette-Guerin (BCG) vaccine-induced complications in children treated with highly active antiretroviral therapy. Int J Infect Dis 12:e99–e105
Logan KE, Chambers MA, Hewinson RG, Hogarth PJ (2005) Frequency of IFN-gamma producing cells correlates with adjuvant enhancement of bacille Calmette-Guerin induced protection against Mycobacterium bovis. Vaccine 23:5526–5532
Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S et al (1999) Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284:1520–1523
Brodin P, Rosenkrands I, Andersen P, Cole ST, Brosch R (2004) ESAT-6 proteins: protective antigens and virulence factors? Trends Microbiol 12:500–508
Jiang Q, Zhang J, Chen X, Xia M, Lu Y, Qiu W et al (2013) A novel recombinant DNA vaccine encoding Mycobacterium tuberculosis ESAT-6 and FL protects against Mycobacterium tuberculosis challenge in mice. J Biomed Res 27:406–420
Brandt L, Elhay M, Rosenkrands I, Lindblad EB, Andersen P (2000) ESAT-6 subunit vaccination against Mycobacterium tuberculosis. Infect Immun 68:791–795
Weinrich Olsen A, van Pinxteren LA, Meng Okkels L, Birk Rasmussen P, Andersen P (2001) Protection of mice with a tuberculosis subunit vaccine based on a fusion protein of antigen 85b and esat-6. Infect Immun 69:2773–2778
Zaghouani H, Steinman R, Nonacs R, Shah H, Gerhard W, Bona C (1993) Presentation of a viral T cell epitope expressed in the CDR3 region of a self immunoglobulin molecule. Science 259:224–227
Carter PJ (2011) Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res 317:1261–1269
Adamova E, Walsh MC, Gosselin DR, Hale K, Preissler MT, Graziano RF et al (2005) Enhanced antigen-specific antibody and cytokine responses when targeting antigen to human Fc-gamma receptor type I using an anti-human Fc-gamma receptor type I-streptavidin fusion protein in an adjuvant-free system. Immunol Invest 34:417–429
Keler T, Guyre PM, Vitale LA, Sundarapandiyan K, van De Winkel JG, Deo YM et al (2000) Targeting weak antigens to CD64 elicits potent humoral responses in human CD64 transgenic mice. J Immunol 165:6738–6742
Rawool DB, Bitsaktsis C, Li Y, Gosselin DR, Lin Y, Kurkure NV et al (2008) Utilization of Fc receptors as a mucosal vaccine strategy against an intracellular bacterium, Francisella tularensis. J Immunol 180:5548–5557
Walsh MC, Banas JA, Mudzinski SP, Preissler MT, Graziano RF, Gosselin EJ (2003) A two-component modular approach for enhancing T-cell activation utilizing a unique anti-FcgammaRI-streptavidin construct and microspheres coated with biotinylated-antigen. Biomol Eng 20:21–33
Soleimanpour S, Farsiani F, Mosavat A, Ghazvini K, Eydgahi MR, Sankian M et al (2015) APC targeting enhances immunigenicity of a novel multistage Fc-fusion tuberculosis vaccine in mice. Appl Microbiol Biotechnol 99:10467–10480
den Haan JM, Lehar SM, Bevan MJ (2000) CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med 192:1685–1696
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Davidsen J, Rosenkrands I, Christensen D, Vangala A, Kirby D, Perrie Y et al (2005) Characterization of cationic liposomes based on dimethyldioctadecylammonium and synthetic cord factor from M. tuberculosis (trehalose 6,6′-dibehenate)-a novel adjuvant inducing both strong CMI and antibody responses. Biochim Biophys Acta 1718:22–31
Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR (1993) An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med 178:2249–2254
Lockhart E, Green AM, Flynn JL (2006) IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol 177:4662–4669
Cooper AM (2009) Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27:393–422
Mosmann TR, Coffman RL (1989) TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7:145–173
Ansel KM, Lee DU, Rao A (2003) An epigenetic view of helper T cell differentiation. Nat Immunol 4:616–623
Wang X, Barnes PF, Huang F, Alvarez IB, Neuenschwander PF, Sherman DR et al (2012) Early secreted antigenic target of 6-kDa protein of Mycobacterium tuberculosis primes dendritic cells to stimulate Th17 and inhibit Th1 immune responses. J Immunol 189:3092–3103
Wang X, Barnes PF, Dobos-Elder KM, Townsend JC, Chung YT, Shams H et al (2009) ESAT-6 inhibits production of IFN-gamma by Mycobacterium tuberculosis-responsive human T cells. J Immunol 182:3668–3677
Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A et al (2007) Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol 8:610–618
Langermans JA, Doherty TM, Vervenne RA, van der Laan T, Lyashchenko K, Greenwald R et al (2005) Protection of macaques against Mycobacterium tuberculosis infection by a subunit vaccine based on a fusion protein of antigen 85B and ESAT-6. Vaccine 23:2740–2750
Pym AS, Brodin P, Majlessi L, Brosch R, Demangel C, Williams A et al (2003) Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat Med 9:533–539
Olsen AW, Williams A, Okkels LM, Hatch G, Andersen P (2004) Protective effect of a tuberculosis subunit vaccine based on a fusion of antigen 85B and ESAT-6 in the aerosol guinea pig model. Infect Immun 72:6148–6150
Sereinig S, Stukova M, Zabolotnyh N, Ferko B, Kittel C, Romanova J et al (2006) Influenza virus NS vectors expressing the mycobacterium tuberculosis ESAT-6 protein induce CD4 + Th1 immune response and protect animals against tuberculosis challenge. Clin Vaccine Immunol 13:898–904
Agger EM, Rosenkrands I, Hansen J, Brahimi K, Vandahl BS, Aagaard C et al (2008) Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): a versatile adjuvant for vaccines with different immunological requirements. PLoS One 3:e3116
Korepanova A, Gao FP, Hua Y, Qin H, Nakamoto RK, Cross TA (2005) Cloning and expression of multiple integral membrane proteins from Mycobacterium tuberculosis in Escherichia coli. Protein Sci 14:148–158
Andersson GE, Sharp PM (1996) Codon usage in the Mycobacterium tuberculosis complex. Microbiology 142:915–925
Li P, Anumanthan A, Gao XG, Ilangovan K, Suzara VV, Duzgunes N et al (2007) Expression of recombinant proteins in Pichia pastoris. Appl Biochem Biotechnol 142:105–124
Ordway DJ, Costa L, Martins M, Silveira H, Amaral L, Arroz MJ et al (2004) Increased Interleukin-4 production by CD8 and gammadelta T cells in health-care workers is associated with the subsequent development of active tuberculosis. J Infect Dis 190:756–766
Demissie A, Abebe M, Aseffa A, Rook G, Fletcher H, Zumla A et al (2004) Healthy individuals that control a latent infection with Mycobacterium tuberculosis express high levels of Th1 cytokines and the IL-4 antagonist IL-4delta2. J Immunol 172:6938–6943
Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M et al (2007) T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27:505–517
Reljic R, Paul MJ, Arias MA (2009) Cytokine therapy of tuberculosis at the crossroads. Expert Rev Respir Med 3:53–66
Goonetilleke NP, McShane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV (2003) Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J Immunol 171:1602–1609
Heyman B (2000) Regulation of antibody responses via antibodies, complement, and Fc receptors. Annu Rev Immunol 18:709–737
Probst HC, Lagnel J, Kollias G, van den Broek M (2003) Inducible transgenic mice reveal resting dendritic cells as potent inducers of CD8 + T cell tolerance. Immunity 18:713–720
Goldenberg MM (1999) Etanercept, a novel drug for the treatment of patients with severe, active rheumatoid arthritis. Clin Ther 21:75–87
Farsiani H, Mosavat A, Soleimanpour S, Sadeghian H, Akbar Eydgahi MR, Ghazvini K, Sankian M et al (2016) Fc-based delivery system enhances immunogenicity of a tuberculosis subunit vaccine candidate consisting of the ESAT-6:CFP-10 complex. Mole Biosyst. doi:10.1039/c6mb00174b
Soleimanpour S, Hassannia T, Motiee M, Amini AA, Rezaee SAR (2016) Fcγ1 fragment of IgG1 as a powerful affinity tag in recombinant Fc-fusion proteins: immunological, biochemical and therapeutic properties. Crit Rev Biotechnol 6:1–22
Acknowledgments
This study has been supported by vice chancellor for research of Mashhad University of Medical Sciences (Grant no. 910072). This study was a part of a PhD dissertation by Abdollah Kebriaei.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Kebriaei, A., Derakhshan, M., Meshkat, Z. et al. Construction and immunogenicity of a new Fc-based subunit vaccine candidate against Mycobacterium tuberculosis . Mol Biol Rep 43, 911–922 (2016). https://doi.org/10.1007/s11033-016-4024-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-016-4024-9