
Abstract
Rice grain size and grain weight, which have a great influence on rice quality and yield, are complex quantitative traits that are mediated by grain length (GL), grain width (GW), length-to-width ratio (LWR), and grain thickness (GT). In this study, the BC1F2 and BC1F2:3 populations derived from a cross between two indica rice varieties, Guangzhan 63-4S (GZ63-4S) and Dodda, were used to locate quantitative trait loci (QTL) related to grain size. A total of 30 QTL associated with GL, GW and LWR were detected, of which six QTL were scanned repeatedly in both populations. Two QTL, qGL4 and qGL6, were selected for genetic effect validation and were subsequently fine mapped to 2.359 kb and 176 kb, respectively. LOC_Os04g52240 (known as OsKS2/OsKSL2), which encoding an ent-beyerene synthase and as the only gene found in 2.359 kb interval, was proposed to be the candidate for qGL4. Moreover, the grains of qGL4 homozygous mutant plants generated by the CRISPR-Cas9 system became shorter and wider. In addition, the qGL4 allele from GZ63-4S contributes to the increase of yield per plant. Our study not only laid the foundation for further functional study of qGL4 and map-based cloning of qGL6, but also provided genetic resources for the development of high yield and good quality rice varieties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is one of the most important staple food crops in the world and feeds more than half of the world’s population (Sasaki 2005). In the face of continuously growing population and decreasing arable land, how to further improve the grain yield of rice is a major concern of scientists and breeders. Rice yield is largely determined by three major components: grain weight, number of grains per panicle, and number of effective tillers per plant (Xing and Zhang 2010). Among them, grain weight is largely determined by grain size, which includes grain length, width, and thickness (Zuo and Li 2014). Exploring the genetic basis of grain size and grain weight will exert a crucial role in the breeding of high-yield rice varieties(Sun et al. 2024). In recent decades, many QTL associated with grain size have been cloned and shown to be involved in a variety of signaling pathways, including the guanine nucleotide-binding protein (G protein) signaling pathway, the ubiquitin–proteasome signaling pathway, the mitogen-activated protein kinase (MAPK) signaling cascade, the transcriptional regulator pathway, and the phytohormone signaling pathway (Li et al. 2018, 2019; Fan and Li 2019; Ren et al. 2023).
G proteins are guanine nucleotide-binding trimeric proteins consisting of Gα, Gβ and Gγ subunits, and are involved in transmitting signals to regulate various biological processes. GS3, encoding an atypical Gγ protein, is the first identified major QTL to negatively regulate grain size (Mao et al. 2010; Fan et al. 2006). DEP1, another atypical Gγ protein, positively regulates grain size by competitively binding to RGB1 (Gβ) with GS3 (Sun et al. 2018). In addition, OsMADS1 negatively regulates grain length through directly interacting with GS3 and DEP1 (Liu et al. 2018).
The ubiquitin–proteasome signaling pathway is involved in the regulation of rice grain size. Large-grain rice varieties represent valuable germplasm resources, harboring novel alleles that make significant contributions to grain shape(Zhao et al. 2017). The major QTL GW2, responsible for grain width and weight, was identified in WY3, a japonica variety with a KGW value of 41.9 ± 1.3 g(Song et al. 2007). GW2 encodes a RING-type E3 ubiquitin ligase and negatively regulates grain width by ubiquitinating WG1 and targeting it for degradation via the 26S proteasome pathway (Song et al. 2007; Choi et al. 2018; Hao et al. 2021). WTG1 encodes an otubain-like protease with deubiquitination activity and controls grain size and shape mainly by affecting cell expansion in the spikelet hull(Huang et al. 2017).
The OsMKKK10-OsMKK4-OsMPK6 cascade has been revealed to positively regulate grain size by promoting cell proliferation in spikelet hulls (Duan et al. 2014; Liu et al. 2015; Guo et al. 2018, 2020; Xu et al. 2018). GSN1/OsMKP1, a MAPK phosphatase, negatively regulates grain size by directly interacting with and inhibiting the dephosphorylation of OsMPK6(Guo et al. 2018). In addition, OsER1, which encodes a receptor-like protein kinase, could positively regulate grain size through the MAPK signaling cascade(Guo et al. 2020).
Many transcription factors are involved in the regulation of grain size. GLW7 encodes a plant-specific transcription factor, OSSPL13, which positively regulates glume cell size, leading to increased rice length and weight(Si et al. 2016). GLW7.1 (Grain Length, Width and Weight 7.1), which encodes the CCT motif family protein, GHD7, promotes the transcription of several cell division and expansion genes, further resulting in a larger cell size and increased cell number, and finally enhancing the grain size as well as grain weight(Liu et al. 2022). OSSPL16/GW8 is a SBP domain-containing transcription factor that regulates rice grain width and can directly bind to the promoter of GW7 and inhibit its expression (Wang et al. 2015). GS2 has transcriptional activation activity and interacts with OsGRFs, a transcriptional coactivator, which regulates cell elongation and cell division and affects grain size and grain weight of rice(Hu et al. 2015; Duan et al. 2015). GS9 encodes a protein with an unknown conserved domain and transcription factor activity, which regulates granulation by altering cell division(Zhao et al. 2018).
Moreover, phytohormones play various roles in plant growth and development, stress responses, and metabolism. Some genes controlling grain size have been reported to be involved in the brassinosteroid (BR) signaling pathway and the auxin signaling pathway and the gibberellic acid (GA) signaling pathway. GS5 encodes a serine carboxypeptidase, a quantitative trait gene that controls grain width, fullness and 1000-grain weight in rice(Li et al. 2011). Enhanced GS5 expression can enhance BR signaling by occupying the extracellular leucine-rich repeat (LRR) domain of OsBAK1-7 and competitive inhibition of the interaction between OsBAK1-7 and OsMSBP1, thereby preventing endocytosis induced by OsBAK1-7 interaction with OsMSBP1(Xu et al. 2015). GW5 acts in the brassinosteroid signaling pathway to regulate grain width and weight in rice, loss of GW5 function caused wide and heavy grains, while overexpression of GW5 resulted in narrow grains (Liu et al. 2017; Duan et al. 2017). qTGW3 negatively regulates grain size and weight (Hu et al. 2018; Xia et al. 2018; Ying et al. 2018). qTGW3 encodes the GSK3/SHAGGY-like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice(Hu et al. 2018). Small Grain and Dwarf 2, encoding an HD-Zip II family transcription factor, regulates plant development by modulating gibberellin biosynthesis in rice (Chen et al. 2019). OsKS2/OsKSL2 encodes a kaurene synthase (KS), which plays a catalytic role in the early steps of GA biosynthesis(Ji et al. 2013). Ji et al. reported that a GA-responsive dwarf mutant of rice cultivar Dongjin, dwarf2, contains a mutation within the OsKS2/OsKSL2 coding region that results in silencing of OsKS2/OsKSL2 gene expression (Ji et al. 2013). However, Daisuke Tezuka et al. thought that although the study suggested the possibility that OsKS2/OsKSL2 encodes an ent-kaurene synthase that is involved in GA biosynthesis, it lacked direct biochemical analysis for its activity and genetic complementation, in which it is therefore necessary to determine the enzymatic activity of OsKS2/OsKSL2 to clarify this point(Tezuka et al. 2015). Daisuke Tezuka et al. successfully isolated a full-length cDNA for OsKS2/OsKSL2 and provided evidence that OsKS2/OsKSL2 mainly converts entCDP into ent-beyerene(Tezuka et al. 2015). Their data suggest that OsKS2/OsKSL2 is involved in phytoalexin biosynthesis rather than GA biosynthesis. However, it is still unclear whether the OsKS2/OsKSL2 gene is involved in affecting rice grain size.
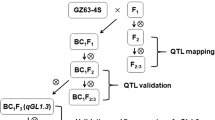
Although many QTL regulating grain size have been identified, the understanding of grain size regulation is still limited, so mining new QTL of grain size is important for better understanding the mechanism of grain size regulation and providing genetic resources for breeding applications. In this study, we mapped QTL for grain size using BC1F2 and BC1F2:3 populations derived from the cross between GZ63-4S and Dodda. The near-isogenic lines (NILs) for the two QTL, qGL4 and qGL6, were constructed to assess the genetic effects. Subsequently, the two QTL were fine mapped to intervals of 2.359 kb and 176 kb, respectively, and LOC_Os04g52240 (OsKS2/OsKSL2) was the only candidate gene in the qGL4 interval. The knockout mutants of LOC_Os04g52240 were obtained using CRISPR-Cas9 gene editing technology, and subsequent phenotypic studies verified the effect of this gene on rice grain size. In conclusion, our study laid a foundation for the final cloning and further functional studies of the two QTL qGL4 and qGL6, which may have potential value in the breeding of new high-quality and high-yield rice varieties.
Materials and methods
Population development and field experiment
The BC1F2 and BC1F2:3 were derived from a cross between two indica rice varieties, GZ 63-4S (receptor parent) and Dodda (donor parent). GZ 63-4S is an indica photo-thermo-sensitive genic male sterile line, which was developed from the indica photo-thermo-sensitive genic male sterile line Guangzhan 63S by the China North Japonica Hybrid Rice Research Center and Hefei Fengle Seed Company, carrying the infertility gene of TMS5. Dodda is an indica rice variety (belonging to the core germplasms collected by our team) (Xie et al. 2015). The BC1F2 and its derived BC1F2:3 populations used for QTL mapping were developed at the Experimental Farm of Huazhong Agricultural University (Wuhan, China) during the normal rice growing seasons in year 2016 and 2018, respectively. Twelve plants were planted in each BC1F2:3 line. To test the genetic effects of qGL4 and qGL6, we planted BC1F4 and BC1F6 populations in 2019 and 2020, respectively, at the Huazhong Agricultural University Rice Base (Wuhan, China). The progeny test was performed in the BC1F7 population. The schematic diagram of experimental design is shown in Fig. S1. The seeds were sown on May 15 every year, and the seedlings (25 or 30 days old) were transplanted onto the field with a single plant spacing of 16.5 cm and 26.4 cm between rows. The field was managed using customary agricultural practices.
Trait measurements
Harvested rice seeds were air-dried and stored at room temperature for three months prior to measurements. Seeds were imaged on a Canon LiDE110 office scanner, and images were saved at 300 dpi resolution. Image files were processed in the SmartGrain analysis software (Tanabata et al. 2012) for measurements of seed width, length, area, perimeter, and circularity. Phenotypic values of each line in BC1F2:3 population were averaged over 12 plants. GL, GW and LWR of BC1F4 population were obtained using Canon LiDE110 office scanner, while that of BC1F6 and BC1F7 populations were measured using yield trait scorer with more than 200 grains per plant(Yang et al. 2014).
Genetic map construction and QTL mapping
The parental varieties GZ63-4S and Dodda were sequenced using illumine HiSeq2000 (Illumina, San Diego, CA, USA), and the sequencing data were aligned and assembled according to the rice reference genome (Rice Genome Annotation Project, http://rice.uga.edu/, accessed on 6 February 2022) (Li and Durbin 2009). To locate QTL associated with GL, GW and LWR, 101 simple sequence repeat (SSR) or insert/deletion (InDel) polymorphic markers and 25 kompetitive allele-specific PCR (KASP) markers were used for the genotyping of 241 plants from a BC1F2 population. Rice leaf genomic DNA was extracted using the cetyltrimethylammonium bromide (CTAB) method (W.F.Thompson 1980). Genotyping was performed using 4% polyacrylamide gel (PAGE) migration as previously reported by Panaud et al.(Panaud 1996). Genotypes were obtained by silver nitrate staining and sodium hydroxide-formaldehyde solution showing DNA bands on the PAGE gel. Genetic linkage maps were constructed using the Kosambi mapping function of the MapMaker/Exp3.0 programme. QTL analysis was carried out by composite interval mapping method using Windows QTL 2.5 software (WinQTLCart 2.5) (Luciano Da Costa E. Silva 2012). Relevant primer sequences are shown in Table S2.
Effect validation and fine mapping
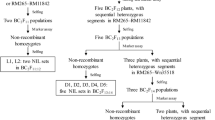
Disregarding the genes that had been cloned, we selected qGL4, which has a large LOD and Add, and qGL6, which was localized in two-year repeats, for the main study. Based on the two flanking markers within the localization intervals of qGL4 and qGL6, we selected lines that were heterozygous for target region in the BC1F2:3 population and developed the BC1F4 population for effect validation. To fine map these two QTL, we developed two BC1F6 populations consisting of 1200 and 1100 individuals respectively to screen recombinants, and conduct effect validation. Furthermore, specific markers were developed for genotyping and composite interval mapping (Luciano Da Costa E. Silva 2012). The BC1F7 population was then used for progeny testing within the small intervals.
RNA extraction, reverse transcription, and quantitative RT-PCR
Total RNA was extracted from 8 cm, 12 cm, 14 cm and 16 cm young panicles using RNA extraction kit (TRIzol, Invitrogen, Carlsbad, CA, USA). Approximately 2 μg of total RNA was used as a template for reverse transcription to generate cDNA, total RNA was pretreated with DNase I (Invitrogen), and first-strand cDNA was synthesized using oligo (dT)18 as primer (Promega, Madison, WI, USA). Real-time PCR was carried out using Bio-Rad T100™ real-time PCR system (Bio-Rad, Hercules, CA, USA) with the SYBR Green I mix (TaKaRa, Shiga, Japan) on the QuantStudio6Flex instrument (Applied Biosystems, Carlsbad, CA, USA). All tests were performed at least three biological repeats. OsActin1 was used as an internal control, and relative expression level was calculated by 2−ΔΔCt. The relevant primers for this analysis are listed in Table S2.
Design of the OsKS2/OsKSL2 target sites and construction of the CRISPR/Cas9 targeting vector
We designed target sites at bases 57–76 of the first exon of the OsKS2/OsKSL2 gene to produce transgenic lines. The online tool CRISPR GE was used to design the target sites(Xie et al. 2017). The target sequences for the OsKS2/OsKSL2 gene were GTGGATCCAGCGGAACCAGC. The U6-sgRNA expression cassette encoding the 20 nucleotides (nt) OsKS2/OsKSL2 target sequence was amplified by polymerase chain reaction (PCR) and ligated into the pCXUN vector, which Kpn I digested to create the pCXUN-U6-sgRNA intermediate vector. The constructed CRISPR/Cas9 final vector was introduced into the NIL-GZ63-4S receptor using Agrobacterium tumfaciens-mediated genetic transformation(Hiei et al. 1997). The primer sequences used to construct the vector are listed in Table S2.
Statistical analysis
Correlation analysis was plotted using OmicStudioKits (v. 1.8.1) online tool at https://www.omicstudio.cn/tool. Significant differences between two sets of data were presented as the mean ± standard error and performed using the two-tailed student’s t-test. Microsoft Excel 2016 was used to perform the preliminary processing and analysis of phenotypic data.
Results
Phenotypic variation and correlation of the B1F and BC1F2:3 populations
The GZ63-4S is a classic example of a photoperiod- and thermo-sensitive genic male sterile line, exhibiting male sterility during the regular growing seasons in Wuhan. Consequently, seed harvesting was not feasible, precluding any comparative analysis of grain size between the two parent lines. All the traits in BC1F2 and its derived BC1F2:3 populations were continuously variable and normally distributed in 2016 and 2018, respectively, showing typical quantitative trait characteristics (Fig. 1).

Frequency distribution of GL (A), GW (B) and LWR (C) in the BC1F2 and BC1F2:3 populations in year 2016 and 2018. The vertical axis represents the number of BC1F2 and BC1F2:3 lines, with white and black bars, respectively. Arrow indicates the value of Dodda
Correlation analysis showed that GL was positively correlated with LWR (the correlation coefficient between GL16 and LWR16 was 0.42, and the correlation coefficient between GL18 and LWR18 was 0.41), while GW was negatively correlated with LWR (the correlation coefficient between GW16 and LWR16 was -0.58, and the correlation coefficient between GW18 and LWR18 was -0.68) (Fig. 2).

Correlation coefficients among grain size-related traits in the BC1F2 and BC1F2:3 populations in year 2016 and 2018
QTL mapping and validation
QTL analysis revealed that 30 QTL related to GL, GW and LWR were detected, with 8 QTL for GL, 12 QTL for GW and 10 QTL for LWR (Fig. 3, Table 1). Eight QTL for GL were detected in two populations and distributed on chromosomes 2, 3, 4, 5, 6, 7 and 11 respectively, with phenotypic variation explained by each QTL ranging from 0.01% to 22.41% (Fig. 3, Table 1). For qGL2, qGL3, qGL4, qGL7, and qGL11.2, the alleles from GZ63-4S had positive effects, and the alleles showing positive effects for the remaining QTL were from Dodda. qGL3 and qGL6 were repeatedly detected in 2016 and 2018. In 2016, qGL3 and qGL6 accounted for 8.56% and 22.41% of the phenotypic variation, respectively, while in 2018, they explained 11.64% and 13.80% of the phenotypic variation, respectively. The remaining GL QTL were detected only in a single year, for example, qGL5 and qGL11.2 could only be detected in 2016, while the rest could only be found in 2018. Twelve QTL for GW were detected in two populations and distributed on chromosomes 2, 5, 6, 7, 8, 9, 10, 11 and 12, with phenotypic variation explained by each QTL ranging from 0.06% to 21.66% (Fig. 3, Table 1). Of these, the beneficial alleles of qGW2.1, qGW2.2, qGW5.1, qGW5.2, qGW6, qGW7, qGW8, and qGW9 were from GZ63-4S, while that of others were from Dodda. Ten LWR-related QTL were identified in both populations, distributed on chromosomes 2, 3, 4, 5, 6, 9 and 11 (Fig. 2, Table 1). Among them, four QTL, including qLWR2.1, qLWR3.1, qLWR3.2 and qLWR9.2, were repeatedly detected in both populations, while the rest LWR QTL were only detected in one population. For qLWR2.1 and qLWR9.2, the alleles from Dodda had positive effects, while the alleles from GZ63-4S conferred positive effects for qLWR3.1 and qLWR3.2.

Genetic linkage map of grain-size-related QTLs detected in the BC1F2 and BC1F2:3 populations
To verify the authenticity of the QTL we identified, qGL4 with a large LOD value and qGL6, which was repeatedly detected in two years, were selected to perform the genetic effect validation and fine mapping. The development of near-isogenic lines is common practice. Thus, plants carrying heterozygous target QTL regions were screened from the BC1F2:3 population and were subjected to several rounds of self-crosses for reducing the heterozygosity of non-target regions. Our strategy not only constructed near-isogenic lines for the target QTL, but also save tedious hybridisation work.
Validation and fine mapping of qGL4
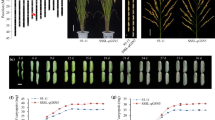
Effect on grain length was evaluated in the BC1F4 and BC1F6 NIL populations of qGL4 in Wuhan in 2019 and 2020, respectively, and the results showed that NIL-GZ63-4SqGL4 had significantly longer grains than NIL-DoddaqGL4 in both years (Fig. 4A-B). For the fine mapping of qGL4, a BC1F6 population consisting of 1200 individuals was developed, and 133 recombinants between GZY4-28.27 and GZY4-32.64 were identified. Subsequently, 14 InDel markers were developed for genotyping these recombinants according to the sequence variation of the two parents. Combining the contemporary phenotypes of these recombinants, qGL4 was further mapped to a 361-kb genomic interval with flanking markers GZY4-30.968 and GZY4-31.329 (Fig. S4). There were also 40 recombinants between markers GZY4-30.968 and GZY4-31.329, and among them, 9 key recombinants were screened by using 4 additional polymorphic molecular markers. The progeny tests of 9 recombinants showed that qGL4 locus was finally narrowed down to a 2.359-kb genomic interval with flanking markers CX4-31.033787 and cx4-31.036146 (Fig. 4C).

Effect validation and fine mapping of qGL4. A Grain length difference of qGL4 among near-isogenic lines in Wuhan in 2019 and 2020. B Seed grain morphology of NIL-DoddaqGL4 and NIL-GZ63-4SqGL4. Scale bar:1 cm. C Fine mapping of qGL4. Data are represented as mean ± s.e.m. (n ≥ 10). The P values were obtained by a two-tailed T-test. *and ** mean significant differences at P < 0.05 and P < 0.01 level, respectively
According to the Rice Annotation Project annotation, (http://rapdb.dna.afrc.go.jp/), this region contains only one annotated gene, LOC_Os04g52240, encoding an ent-beyerene synthase and known as OsKS2/OsKSL2. Previous research reported that OsKS2/OsKSL2 gene may be involved in GA biosynthesis (Ji et al. 2013) or phytoalexin biosynthesis (Tezuka et al. 2015). However, the effect of this gene on grain shape and its regulatory mechanism are still unclear.
Comparative biparental sequencing of qGL4 candidate interval and genetic transformation and functional validation of qGL4
Comparative biparental sequencing revealed two SNP variants in the OsKS2/OsKSL2 promoter region, one SNP variant in the first exon and nine SNP variants in the first intron (Fig. 5A). However, the SNP on the first exon was a nonsense mutation that did not result in an amino acid change. Considering that the variants in the promoter might cause differences in the relative expression levels among the near-isogenic lines, so we examined the relative expression levels of OsKS2/OsKSL2 in 8 cm, 12 cm, 14 cm and 16 cm young spikes of the near-isogenic lines. RT-qPCR results showed that OsKS2/OsKSL2 differed significantly between 12 and 14 cm young spikes, indicating that the differences in grain length among the near-isogenic lines caused by OsKS2/OsKSL2 might be due to differences in its relative expression (Fig. 5B).

Parental sequencing comparison and genetic transformation analysis of qGL4. A Localization of biparental variant sites between intervals. Red markers are sgRNA target site in the CRISPR-Cas9 system. bar:100 bp. B Relative expression levels of qGL4 between near-isogenic lines. C Sequences of sgRNA target sites and allelic mutant sequences in the CRISPR-Cas9 system. Black bold letters for sgRNA target, red letters for base insertion, “-” for base deletion. D Grain length and grain width of knockout mutants and wild type. E Seed morphology of wild type and knockout mutants. Scale bar: 1 cm. Data are represented as mean ± s.e.m. The P values were obtained by a two-tailed T-test. * and **, indicate significant differences at P < 0.05 and P < 0.01, respectively. Different lowercase letters mean significant differences at P < 0.05 level in Duncan’s multiple range test
In order to further clarify the effect of gene OsKS2/OsKSL2 on grain size, we performed gene editing of this gene using the CRISPR-Cas9 system in the NIL-GZ63-4SqGL4 genetic background for the construction of OsKS2/OsKSL2 knockout materials. We designed the sequence in the first exon of the OsKS2/OsKSL2 gene (GTGGATCCAGCGGAACCAGC) as a target site for sgRNA with the aim of obtaining amino acid mutations adjacent to the GGG structural domain (Fig. 5C). The gene editing experiments yielded three allelic mutant phenotypes, named KO-1, KO-2, and KO-3, respectively. We examined the grain size of the mutants, and in agreement with the expected results, the grain length of all three allelic mutant phenotypes became shorter than that of the wild type in the NIL-GZ63-4SqGL4 background. In contrast, it was also found that the grain width of the KO-1 and KO-2 allelic mutants became wider than that of the wild type (Fig. 5D-E).
qGL4 allele from GZ63-4S can improve rice yield
To assess the effect of qGL4 on rice yield, we examined yield-related traits in NILs. Compared to NIL-DoddaqGL4, NIL-GZ63-4SqGL4 exhibited the substantial enhancement in both grain length and grain width, as well as the thousand-grain weight (Fig. 6A-C). Although NIL-GZ63-4SqGL4 exhibited a reduction in panicle length and the number of primary branches than NIL-DoddaqGL4, the filled grain number per panicle of NIL-GZ63-4SqGL4 also increased, which resulted in the improvement of yield per plant of NIL-GZ63-4SqGL4 (Fig. 6D-H). These results indicate that the qGL4 allele from GZ63-4S can improve rice yield.

The yield-related traits in the NILs of qGL4. A–H Grain length (mm), grain width (mm), thousand‐grain weight (g), number of tillers per plant, panicle length (cm), primary branch number, filled grains number per panicle, and grain yield per plant in the NILs. All phenotypic data in (A–H) were measured from paddy‐grown NIL plants grown under normal cultivation conditions. Data are represented as mean ± s.e.m. (n = 12). The student’s t‐test was used to produce P values (*, ** indicate significance at P < 0.05 and P < 0.01 level, respectively)
Validation and fine mapping of qGL6
To verify the genetic effect of qGL6, grain size traits were evaluated in the BC1F4 and BC1F6 NIL populations in Wuhan in 2019 and 2020, respectively. The results showed that significant differences in GL, GW and thousand-grain weight were observed between NIL-GZ63-4SqGL6 and NIL-Dodda qGL6 in both years (Fig. 7A-B). To fine map qGL6, we constructed a BC1F6 population of 1100 plants and screened to 81 recombinants with two markers, RM20048 and GZY6-21.34. Subsequently, 13 markers were developed to genotype the recombinants and the grain size traits were examined. qGL6 was further mapped to a 475-kb genomic interval with flanking markers GZY6-19.791 and GZY20.266 by composite interval mapping (Fig. S7). There are also 20 recombinants of qGL6 between markers GZY6-19.791 and GZY20.266, and among them, 10 key recombinants were screened by using 3 additional polymorphic molecular markers. The progeny tests of 10 recombinants showed that qGL6 was narrowed down to a 176-kb interval marked by GZY6-19.871 and GZY6-20.047 (Fig. 7C).

Effect validation and fine mapping of qGL6. A Grain length of near-isogenic lines in Wuhan in 2019 and 2020. B Seed morphology of NIL-DoddaqGL6 and NIL-GZ63-4SqGL6. Scale bar: 1 cm. C Fine mapping of qGL6. Data are represented as mean ± s.e.m. (n ≥ 13). The P valueS were obtained by a two-tailed T-test. *and ** mean significant differences at P < 0.05 and P < 0.01 level, respectively
According to the annotation information of Nipponbare (http://rice.uga.edu/cgi-bin/gbrowse/rice/#search), the target region of qGL6 contains six annotated genes (LOC_Os06g34360, LOC_Os06g34390, LOC_Os06g34400, LOC_Os06g34420, LOC_Os06g34430, LOC_Os06g34440). Among these genes, LOC_Os06g34360, LOC_Os06g34390 and LOC_Os06g34400 encode a C3HC4 zinc finger protein. LOC_Os06g34420 encodes a DEAD/DEAH box helicase domain protein. LOC_Os06g34430 encodes zinc finger protein. LOC_Os06g34440 encodes dnaJ domain containing protein. We further compared the sequence variations of these candidate genes including the 2 kb promoter and the coding sequence between GZ63-4S and Dodda (Table S3). The result showed that the variations occurred in the promoter, intron, and 5′ and 3′ untranslated regions (UTR).
Discussion
The QTL detected in the original population is sometimes unstable and therefore requires further effect validation. The most common and effective strategy is to use near-isogenic lines. In this study, we selected lines from the BC1F2:3 population that were heterozygous for each QTL in the locus interval to generate high-generation populations in case the target regions were lost in subsequent selfing. In the experimental context, the parent line GZ63-4S, being a sterile strain, necessitated a strategy to reduce heterozygosity in non-target regions. To achieve this, we employed a successive selfing process across generations with the selected germplasm. Concurrently, we utilized molecular marker-assisted screening to guarantee the maintenance of heterozygosity within the target locus intervals. This approach not only preserved the required heterozygosity but also alleviated the labor-intensive burden of re-screening for fertility in each subsequent generation following backcrossing. Zhou et al. also employed this method to precisely identify 5 QTLs influencing rice yield and quality, and accumulated the genetic basis for the cloning of these 5 QTLs, providing valuable resources for the breeding of high-yield and high-quality rice varieties(Zhou et al. 2024).
Due to comparative biparental sequencing of the localization intervals revealed two SNPs in the qGL4 promoter region, one SNP in the first exon and nine SNPs in the first intron (Fig. 5A). However, the SNP variants in the first exon was a nonsense mutation that did not result in an amino acid change. Difference in the relative expression levels of qGL4 in 12 cm and 14 cm young spikes among the NILs indicated that two SNPs in the promoter region of qGL4 may be responsible for variation in grain size among near-isogenic lines. According to the cis-acting element prediction website (https://www.dna.affrc.go.jp/PLACE/?action=newplace), there is a TACT cis-element in the Dodda promoter region of qGL4 due to the SNP (-70 bp), but not in the GZ63-4S promoter region. Subsequently, we will focus on this SNP locus and design relevant experiments to verify whether this SNP causes differences in relative expression levels between qGL4 near-isogenic lines.
The excellent allele of qGL4 derived from GZ63-4S can not only increase grain length but also increase single plant yield, which can be used for selection and improvement of rice grain size and yield. The genetic effect of qGL6 could be detected in the NIL population planted for two consecutive years, which proved the stability and reliability of the QTL and was suitable for subsequent gene cloning. In short, our work laid the foundation for the functional study of qGL4 and subsequent gene cloning of qGL6, and the excellent allele of qGL4 from GZ63-4S provided genetic resources for the cultivation of high quality and high yield rice varieties.
Material and data availability
The authors declare that all relevant data supporting the findings of this study are included in the main manuscript file or Supplementary Information or are available from the corresponding author upon reasonable request.
References
Chen W, Cheng Z, Liu L, Wang M, You X, Wang J, Zhang F, Zhou C, Zhang Z, Zhang H, You S, Wang Y, Luo S, Zhang J, Wang J, Wang J, Zhao Z, Guo X, Lei C, Zhang X, Lin Q, Ren Y, Zhu S, Wan J (2019) Small Grain and Dwarf 2, encoding an HD-Zip II family transcription factor, regulates plant development by modulating gibberellin biosynthesis in rice. Plant Science 288. https://doi.org/10.1016/j.plantsci.2019.110208
Choi B, Kim Y, Markkandan K, Koo Y, Song J, Seo H (2018) GW2 Functions as an E3 Ubiquitin Ligase for Rice Expansin-Like 1. International Journal of Molecular Sciences 19 (7). https://doi.org/10.3390/ijms19071904
Duan P, Rao Y, Zeng D, Yang Y, Xu R, Zhang B, Dong G, Qian Q, Li Y (2014) SMALL GRAIN 1, which encodes a mitogen-activated protein kinase kinase 4, influences grain size in rice. Plant J 77(4):547–557. https://doi.org/10.1111/tpj.12405
Duan P, Xu J, Zeng D, Zhang B, Geng M, Zhang G, Huang K, Huang L, Xu R, Ge S, Qian Q, Li Y (2017) Natural Variation in the Promoter of GSE5 Contributes to Grain Size Diversity in Rice. Mol Plant 10(5):685–694. https://doi.org/10.1016/j.molp.2017.03.009
Duan P, Ni S, Wang J, Zhang B, Xu R, Wang Y, Chen H, Zhu X, Li Y (2015) Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nature Plants 2 (1). https://doi.org/10.1038/nplants.2015.203
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112(6):1164–1171. https://doi.org/10.1007/s00122-006-0218-1
Fan Y, Li Y (2019) Molecular, cellular and Yin-Yang regulation of grain size and number in rice. Molecular Breeding 39 (12). https://doi.org/10.1007/s11032-019-1078-0
Guo T, Chen K, Dong N-Q, Shi C-L, Ye W-W, Gao J-P, Shan J-X, Lin H-X (2018) GRAIN SIZE AND NUMBER1 Negatively Regulates the OsMKKK10-OsMKK4-OsMPK6 Cascade to Coordinate the Trade-off between Grain Number per Panicle and Grain Size in Rice. Plant Cell 30(4):871–888. https://doi.org/10.1105/tpc.17.00959
Guo T, Lu Z-Q, Shan J-X, Ye W-W, Dong N-Q, Lin H-X (2020) ERECTA1 Acts Upstream of the OsMKKK10-OsMKK4-OsMPK6 Cascade to Control Spikelet Number by Regulating Cytokinin Metabolism in Rice. Plant Cell 32(9):2763–2779. https://doi.org/10.1105/tpc.20.00351
Hao J, Wang D, Wu Y, Huang K, Duan P, Li N, Xu R, Zeng D, Dong G, Zhang B, Zhang L, Inzé D, Qian Q, Li Y (2021) The GW2-WG1-OsbZIP47 pathway controls grain size and weight in rice. Mol Plant 14(8):1266–1280. https://doi.org/10.1016/j.molp.2021.04.011
Hiei Y, Komari T, Kubo T (1997) Transformation of rice mediated by Agrobacterium tumefaciens. Plant Mol Biol 35(1–2):205–218. https://doi.org/10.1023/A:1005847615493
Hu J, Wang Y, Fang Y, Zeng L, Xu J, Yu H, Shi Z, Pan J, Zhang D, Kang S, Zhu L, Dong G, Guo L, Zeng D, Zhang G, Xie L, Xiong G, Li J, Qian Q (2015) A Rare Allele of GS2 Enhances Grain Size and Grain Yield in Rice. Mol Plant 8(10):1455–1465. https://doi.org/10.1016/j.molp.2015.07.002
Hu Z, Lu SJ, Wang MJ, He H, Sun L, Wang H, Liu XH, Jiang L, Sun JL, Xin X, Kong W, Chu C, Xue H-W, Yang J, Luo X, Liu JX (2018) A Novel QTL qTGW3 Encodes the GSK3/SHAGGY-Like Kinase OsGSK5/OsSK41 that Interacts with OsARF4 to Negatively Regulate Grain Size and Weight in Rice. Mol Plant 11(5):736–749. https://doi.org/10.1016/j.molp.2018.03.005
Huang K, Wang D, Duan P, Zhang B, Xu R, Li N, Li Y (2017) WIDE AND THICK GRAIN 1, which encodes an otubain-like protease with deubiquitination activity, influences grain size and shape in rice. Plant J 91(5):849–860. https://doi.org/10.1111/tpj.13613
Ji SH, Gururani MA, Lee JW, Ahn BO, Chun SC, Peeters T (2013) Isolation and characterisation of a dwarf rice mutant exhibiting defective gibberellins biosynthesis. Plant Biol 16(2):428–439. https://doi.org/10.1111/plb.12069
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li Y, Fan C, Xing Y, Jiang Y, Luo L, Sun L, Shao D, Xu C, Li X, Xiao J, He Y, Zhang Q (2011) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43(12):1266–1269. https://doi.org/10.1038/ng.977
Li N, Xu R, Duan P, Li Y (2018) Control of grain size in rice. Plant Reproduction 31(3):237–251. https://doi.org/10.1007/s00497-018-0333-6
Li N, Xu R, Li Y (2019) Molecular Networks of Seed Size Control in Plants. Annu Rev Plant Biol 70(1):435–463. https://doi.org/10.1146/annurev-arplant-050718-095851
Liu S, Hua L, Dong S, Chen H, Zhu X, Je J, Zhang F, Li Y, Fang X, Chen F (2015) OsMAPK6, a mitogen-activated protein kinase, influences rice grain size and biomass production. Plant J 84(4):672–681. https://doi.org/10.1111/tpj.13025
Liu Q, Han R, Wu K, Zhang J, Ye Y, Wang S, Chen J, Pan Y, Li Q, Xu X, Zhou J, Tao D, Wu Y, Fu X (2018) G-protein βγ subunits determine grain size through interaction with MADS-domain transcription factors in rice. Nat Commun 9:852. https://doi.org/10.1038/s41467-018-03047-9
Liu J, Chen J, Zheng X, Wu F, Lin Q, Heng Y, Tian P, Cheng Z, Yu X, Zhou K, Zhang X, Guo X, Wang J, Wang H, Wan J (2017) GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nature Plants 3 (5). https://doi.org/10.1038/nplants.2017.43
Liu R, Feng Q, Li P, Lou G, Chen G, Jiang H, Gao G, Zhang Q, Xiao J, Li X, Xiong L, He Y (2022) GLW7.1, a Strong Functional Allele of Ghd7, Enhances Grain Size in Rice. International Journal of Molecular Sciences 23 (15). https://doi.org/10.3390/ijms23158715
Luciano Da Costa E, Silva SW, Zeng Z-B (2012) Composite Interval Mapping and Multiple Interval Mapping_ Procedures and Guidelines for Using Windows QTL Cartographer. Methods Mol Biol 871:75–119. https://doi.org/10.1007/978-1-61779-785-9_6
Mao H, Sun S, Yao J, Wang C, Yu S, Xu C, Li X, Zhang Q (2010) Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci 107(45):19579–19584. https://doi.org/10.1073/pnas.1014419107
Panaud OC, X., McCouch, S, (1996) Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice (Oryza sativa L.). Mol Gen Genet 252:597–607. https://doi.org/10.1007/BF02172406
Ren D, Ding C, Qian Q (2023) Molecular bases of rice grain size and quality for optimized productivity. Science Bulletin 68(3):314–350. https://doi.org/10.1016/j.scib.2023.01.026
Sasaki T (2005) The map-based sequence of the rice genome. Nature 436(7052):793–800. https://doi.org/10.1038/nature03895
Si L, Chen J, Xuehui H, Gong H, Luo J, Hou Q, Zhou T, Lu T, Zhu J, Shangguan Y, Chen E, Gong C, Zhao Q, Jing Y, Zhao Y, Cui L, Fan D, Lu Y, Han B (2016) OsSPL13 controls grain size in cultivated rice. Nature genetics 48. https://doi.org/10.1038/ng.3518
Song X-J, Huang W, Shi M, Zhu M-Z, Lin H-X (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39(5):623–630. https://doi.org/10.1038/ng2014
Sun S, Wang L, Mao H, Shao L, Li X, Xiao J, Ouyang Y, Zhang Q (2018) A G-protein pathway determines grain size in rice. Nature Communications 9 (1). https://doi.org/10.1038/s41467-018-03141-y
Sun P, Zheng Y, Li P, Ye H, Zhou H, Gao G, Zhang Q, He Y (2024) Dissection and validation of quantitative trait loci (QTLs) conferring grain size and grain weight in rice. Euphytica 220 (3). https://doi.org/10.1007/s10681-024-03310-9
Tanabata T, Shibaya T, Hori K, Ebana K, Yano M (2012) SmartGrain: High-Throughput Phenotyping Software for Measuring Seed Shape through Image Analysis. Plant Physiol 160(4):1871–1880. https://doi.org/10.1104/pp.112.205120
Tezuka D, Ito A, Mitsuhashi W, Toyomasu T, Imai R (2015) The rice ent-KAURENE SYNTHASE LIKE 2 encodes a functional ent-beyerene synthase. Biochem Biophys Res Commun 460(3):766–771. https://doi.org/10.1016/j.bbrc.2015.03.104
W.F. Thompson MGMa, (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Wang S, Li S, Liu Q, Wu K, Zhang J, Shuansuo W, Wang Y, Chen X, Zhang y, Gao C, Wang F, Huang H, Fu X (2015) The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nature genetics 47. https://doi.org/10.1038/ng.3352
Xia D, Zhou H, Liu R, Dan W, Li P, Wu B, Chen J, Wang L, Gao G, Zhang Q, He Y (2018) GL3.3, a Novel QTL Encoding a GSK3/SHAGGY-like Kinase, Epistatically Interacts with GS3 to Produce Extra-long Grains in Rice. Mol Plant 11(5):754–756. https://doi.org/10.1016/j.molp.2018.03.006
Xie X, Ma X, Zhu Q, Zeng D, Li G, Liu Y-G (2017) CRISPR-GE: A Convenient Software Toolkit for CRISPR-Based Genome Editing. Mol Plant 10(9):1246–1249. https://doi.org/10.1016/j.molp.2017.06.004
Xie W, Wang G, Yuan M, Yao W, Lyu K, Zhao H, Yang M, Li P, Zhang X, Yuan J, Wang Q, Liu F, Dong H, Zhang L, Li X, Meng X, Zhang W, Xiong L, He Y, Wang S, Yu S, Xu C, Luo J, Li X, Xiao J, Lian X, Zhang Q (2015) Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proceedings of the National Academy of Sciences 112 (39). https://doi.org/10.1073/pnas.1515919112
Xing Y, Zhang Q (2010) Genetic and Molecular Bases of Rice Yield. Annu Rev Plant Biol 61(1):421–442. https://doi.org/10.1146/annurev-arplant-042809-112209
Xu R, Duan P, Yu H, Zhou Z, Zhang B, Wang R, Li J, Zhang G, Zhuang S, Lyu J, Li N, Chai T, Tian Z, Yao S, Li Y (2018) Control of Grain Size and Weight by the OsMKKK10-OsMKK4-OsMAPK6 Signaling Pathway in Rice. Mol Plant 11(6):860–873. https://doi.org/10.1016/j.molp.2018.04.004
Xu C, Liu Y, Li Y, Xu X, Xu C, Li X, Xiao J, Zhang Q (2015) Differential expression of GS5 regulates grain size in rice. Journal of experimental botany 66. https://doi.org/10.1093/jxb/erv058
Yang W, Guo Z, Huang C, Duan L, Chen G, Jiang N, Fang W, Feng H, Xie W, Lian X, Wang G, Luo Q, Zhang Q, Liu Q, Xiong L (2014) Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nature Communications 5 (1). https://doi.org/10.1038/ncomms6087
Ying J-Z, Ma M, Bai C, Huang X-H, Liu J-L, Fan Y-Y, Song X-J (2018) TGW3, a Major QTL that Negatively Modulates Grain Length and Weight in Rice. Mol Plant 11(5):750–753. https://doi.org/10.1016/j.molp.2018.03.007
Zhao D, Li P, Wang L, Sun L, Xia D, Luo L, Gao G, Zhang Q, He Y (2017) Genetic dissection of large grain shape in rice cultivar ‘Nanyangzhan’ and validation of a grain thickness QTL (qGT3.1) and a grain length QTL (qGL3.4). Molecular Breeding 37 (3). https://doi.org/10.1007/s11032-017-0638-4
Zhao D-S, Li Q-F, Zhang C-Q, Zhang C, Yang Q-Q, Pan L-X, Ren X-Y, Lu J, Gu M-H, Liu Q-Q (2018) GS9 acts as a transcriptional activator to regulate rice grain shape and appearance quality. Nature Communications 9 (1). https://doi.org/10.1038/s41467-018-03616-y
Zhou Y, Yang H, Liu E, Liu R, Alam M, Gao H, Gao G, Zhang Q, Li Y, Xiong L, He Y (2024) Fine Mapping of Five Grain Size QTLs Which Affect Grain Yield and Quality in Rice. International Journal of Molecular Sciences 25 (8). https://doi.org/10.3390/ijms25084149
Zuo J, Li J (2014) Molecular Genetic Dissection of Quantitative Trait Loci Regulating Rice Grain Size. Annu Rev Genet 48(1):99–118. https://doi.org/10.1146/annurev-genet-120213-092138
Funding
This work was supported by grants from STI 2030-Major Project (2023ZD04069), Natural Science Foundation of Hubei Province of China (2022CFB835), Natural Science Foundation of China (32201879), and Earmarked fund for Agriculture Research System in China (CARS-01–01).
Author information
Authors and Affiliations
Contributions
Yuanyuan Zheng performed most of the experiments; Minqi Li, Ping Sun and Yanhua Li participated in part of phenotyping and genotyping; Yuanyuan Zheng and Guangming Lou performed various data analysis; Guanjun Gao and Qinglu Zhang participated in partial field experiments; Yuanyuan Zheng, Bian Wu and Yuqing He designed experiments; Yuanyuan Zheng wrote the manuscript and Guangming Lou improved it; All authors discussed and commented on the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
The experiments comply with the ethical standards in the country in which they were performed.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
11032_2024_1502_MOESM1_ESM.docx
Supplementary Materials 1: Fig. S1 Seed grain morphology of Dodda and GZ63-4S. Scale bar:1 cm; Fig. S2 The schematic diagram of experimental design; Fig. S3 the plant photo of NIL-DoddaqGL4 and NIL-GZ63-4SqGL4. Scale bar:10 cm; Fig. S4 Mapping of the qGL4 locus using 133 recombinants; Fig. S5 the plant photo of the wild type and transgenic plants. Scale bar:10 cm; Fig. S6 the plant photo of NIL-DoddaqGL6 and NIL-GZ63-4SqGL6. Scale bar:10 cm; Fig. S7 Mapping of the qGL6 locus using 81 recombinants; Table S1 The phenotypic performance of four grain size traits in Dodda and GZ63-4S; Table S2 Primers used in this study; Table S3 The sequence variations of the candidate genes between GZ63-4S and Dodda.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zheng, Y., Li, M., Sun, P. et al. QTL detection for grain shape and fine mapping of two novel locus qGL4 and qGL6 in rice. Mol Breeding 44, 62 (2024). https://doi.org/10.1007/s11032-024-01502-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-024-01502-8