
Abstract
Soybean (Glycine max (L.) Merrill) is one of the most important crop plants in the world as an important source of protein for both human consumption and livestock fodder. As flowering time contributes to yield, finding new QTLs and further identifying candidate genes associated with various flowering time are fundamental to enhancing soybean yield. In this study, a set of 120 recombinant inbred lines (RILs) which was developed from a cross of two soybean cultivars, Suinong4 (SN4) and ZK168, were genotyped by genotyping-by-sequencing (GBS) approach and phenotyped to expand the cognitive of flowering time by quantitative trait loci (QTL) analysis. Eventually, three stable QTLs related to flowering time which were detected separately located on chromosome 14, 18, and 19 under long-day (LD) conditions. We predicted candidate genes for each QTL and carried out association analyses between the putative causal alleles and flowering time. Moreover, a transient transfection assay was performed and showed that NUCLEAR FACTOR YA 1b (GmNF-YA1b) as a strong candidate for the QTL on chromosome 19 might affect flowering time by suppressing the expression of FLOWERING LOCUS T (GmFT) genes in soybean. QTLs detected in this study would provide fundamental resources for finding candidate genes and clarify the mechanisms of flowering which would be helpful for breeding novel high-yielding soybean cultivars.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean is a major legume crop used worldwide as a source of edible oil and high-quality protein for human consumption (Hoeck et al. 2003). Breeding high-yielding soybean cultivars is thus an ongoing goal of modern agriculture (Yin et al. 2018). An appropriate flowering time is important for increasing yield; thus, identifying flowering-related genes is beneficial to increase soybean yield. In addition, the timing of flowering is also a critical factor that restricts the latitudes at which plants may grow and propagate. Accordingly, breeders have exploited standing genetic variation in the photoperiod responses and flowering times of many crop species to expand their growth areas to a wide range of latitudes (Thakare et al. 2010). Therefore, the identification of alleles conferring photoperiod insensitivity and development of cultivars with flowering responses adapted to distinct geographic locations will be instrumental to improving soybean cultivars and yield (Zhao et al. 2018ab).
Soybean is a photoperiod-sensitive plant that flowers early when exposed to shorter photoperiods; daylength thus influences its flowering time and yield (Tasma et al. 2001). To date, 14 major genes or loci have been reported to be associated with flowering time and maturity in soybean: E1 (Bernard 1971; Xia et al. 2012), E2 (Bernard 1971; Watanabe et al. 2011), E3 (Buzzell 1971; Watanabe et al. 2009), E4 (Buzzell and Voldeng 1980; Saindon et al. 1989; Liu et al. 2008), E5 (McBlain and Bernard 1987), E6 (Bonato and Vello 1999; Fang et al. 2020), E7 (Cober and Voldeng 2001), E8 (Cober et al. 2010), E9 (Kong et al. 2014; Zhao et al. 2016), E10 (Samanfar et al. 2016), E11 (Wang et al. 2019), J (Ray et al. 1995; Lu et al. 2017), and TIME OF FLOWERING 11 (Tof11) and Tof12 (Lu et al. 2020). The dominant alleles of the genes or loci E1, E2, E3, E4, E8, and E10 delay flowering, while their recessive alleles promote early flowering (Zhang et al. 2007; Langewisch et al. 2014; Langewisch et al. 2017; Bernard 1971; Xia et al. 2012; Cober et al. 2010; Watanabe et al. 2011; Buzzell and Voldeng 1980; Watanabe et al. 2009; Xu et al. 2013; Samanfar et al. 2016; Lin et al. 2020).
As a typical short-day plant, soybean can be grown over a wide latitudinal range that is dictated by the complement of alleles for photoperiod-related genes. Indeed, various allelic combinations at the E1-E4 genes contribute to the adaptability of soybean to high latitude regions (Jiang et al. 2014). However, although genotypes at these genes have been widely used in breeding, they do not entirely explain adaptability to high latitudes, suggesting that additional genes remain to be identified. Two long juvenile traits, E6 and J, have been identified to extend vegetative growth under short-day environments (Hartwig and Kiihl 1979; Ray et al. 1995; Lu et al. 2017; Li et al. 2017; Fang et al. 2020). Additionally, Tof12 and Tof11 have been reported to gradually contribute to higher latitude adaptation in soybean. Tof12 and Tof11 undergone stepwise selection; the loss-of-function alleles of them were enriched in landraces, especially in the northeast regions (Lu et al. 2020).
Additional genes also play vital roles in soybean flowering, including FT, CONSTANS (CO), APETALA 1 (AP1), and SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1). Among those, members of the FT gene family have been extensively characterized (Takeshima et al. 2016; Ogiso-Tanaka et al. 2019; Sun et al. 2019). GmFT2a and GmFT5a are the two major members of the soybean FT family, and GmFT2a was identified as the causal gene for the E9 locus (Kong et al. 20102014; Zhao et al. 2016). Another family of genes with roles in flowering time is the NUCLEAR FACTOR-Y (NF-Y) family, whose members encode highly conserved transcription factors that form complexes composed of NF-YA, NF-YB, and NF-YC subunits (Sinha et al. 19951996). Several NF-YA genes are negative regulators of flowering time, as evidenced by the late flowering of plants overexpressing some NF-YA genes (Wenkel et al. 2006; Xu et al. 2004) . Subsequently, genetic and biochemical experiments also support a positive role for NF-YA subunits in flowering time, via its direct binding to the FT promoter in a photoperiod-dependent manner (Siriwardana et al. 2016). The growth habit of soybean stems also constitutes a key adaptation and agronomic trait that directly affects flowering time, node production, and ultimately yield (Bernard 1972; Specht et al. 2001; Heatherly and Smith 2004). The causal gene mutated in the determinate stem 1 (dt1) mutant was shown to encode the soybean homolog of TERMINAL FLOWER 1 (GmTFL1) (Liu et al. 2010). More recently, Dt1 was reported to interact with the basic leucine zipper (bZIP) transcription factor GmFDc1 to repress the expression of GmAP1, thereby regulating flowering (Yue et al. 2021). Although much research has been conducted to understand the molecular basis of photoperiod-mediated regulation of soybean flowering, the regulatory pathways leading to flowering in soybean are far from completely understood, leaving many related genes related yet to be uncovered (Kong et al. 2018).
In this study, we selected two soybean cultivars (SN4 and ZK168) with the same genotype of the E1, E2, E3, E4, Dt1, and Dt2 to generate a recombinant inbred line (RIL) population, thus excluding the influence of known major genes whose variation contributes to flowering time so as to potentially unmask hidden variation at loci not previously known to regulate flowering time. We genotyped all individuals from the RIL population by genotyping-by-sequencing (GBS) and constructed a high-density genetic map. We here identified three stable quantitative trait loci (QTLs) for flowering time by two QTL mapping methods. In addition, we explored the candidate genes responsible for these QTLs and briefly analyzed the geographic distributions of their alleles. We tentatively identified the QTL mapping to chromosome 19 as GmNF-YA1b, a homolog of Arabidopsis NF-YA10, whose encoded protein may regulate flowering by binding to the promoters of GmFT2a and GmFT5a and repressing their transcription.
Materials and methods
Plant materials
We used the F6:8 RIL population S4-168, consisting of 120 lines. The S4-168 RIL population was developed by single-seed descent from a cross between the two soybean cultivars Suinong4 (SN4) (e1-as/e2/e3/E4/Dt1/dt2) and ZK168 (e1-as/e2/e3/E4/Dt1/dt2). SN4 is a major soybean variety in Northern China, and ZK168 is a e3-NIL (near-isogenic line) derived from Harosoy.
Plant cultivation
The F6:7 RILs and the two parents were sown in the experimental field in Harbin (45°43′N, 126°45′E), China, in May 2018. The F7:8 RILs and the two parents were sown in the same field in May 2019. Each row of plants was 4 m in length, with a space of 20 cm between plants and a spacing of 60 cm between rows. We sowed about 15 plants on each row and recorded progression through developmental stages, including the Ve (emergence) and R1 stages (beginning bloom) (Fehr et al. 1971).
DNA extraction
Part of a fresh trifoliate leaf was collected from each parental and RIL individual at the V2 stage and stored at − 80 °C until processing. Genomic DNA was extracted using the NuClean PlantGen DNA Kit (CWBIO, Beijing, China). DNA quality was ascertained by electrophoresis of an aliquot on a 1% agarose gel, and the DNA concentration was determined using a NanoDrop 2000 instrument (ThermoScientific, Wilmington, DE, USA).
Genotyping by high-throughput sequencing
DNA extractions from leaf samples for resequencing, and preparation of sequencing libraries, were performed according to the method reported in Cheng et al (2015). GBS technology was used to genotype the S4-168 population. The parents SN4 and ZK168 were sequenced at high coverage, while the RIL population was sequenced at low coverage to identify single-nucleotide polymorphisms corresponding to each parent (Huang et al. 2009; Davey et al. 2013).
Construction of the genetic map
SNPs were assigned to their chromosomes by alignment against the soybean reference genome (Glycine max Wm82.a2.v1) downloaded from Phytozome (http://phytozome.jgi.doe.gov). Segregation distortion was tested using a chi-square test. Markers were assigned to 20 linkage groups (or chromosomes) based on their physical position, and the genetic distances were estimated when generating the genetic map.
QTL analysis
The inclusive composite interval mapping (ICIM) method was used to detect QTLs with the QTL IciMapping software (Meng et al. 2015), treating data for each year as a separate phenotype. Permutation tests (PT) were performed to calculate logarithm of the odds (LOD) thresholds to a significance level of P < 0.05 (N = 1,000). A QTL was considered significant when its LOD score was higher than the threshold across both years. In addition, QTLs with LOD scores higher than 2.5 but lower than the LOD thresholds were retained. To validate the ICIM results, composite interval mapping (CIM) was also performed using Windows QTL Cartographer V2.5 (WinQTL, Wang et al. 2012). The LOD threshold was set empirically to 2.5.
Identification of candidate genes
The soybean reference genome and annotation files (Schmutz et al. 2010) were downloaded from Phytozome (http://phytozome.jgi.doe.gov). The candidate genes within the QTL intervals were categorized through gene ontology analysis with the online tools Gene Ontology (GO; http://geneontology.org/) and WEGO (http://wego.genomics.org.cn) to obtain detailed pathway, gene ontology, and annotation information. We organized the functional annotation information for all genes within the QTL interval which had been identified in both 2 years and selected the most likely candidate genes within variations that exist in two parents of the GBS analysis consequence.
Haplotype calling and association analysis
Resequencing data of the 1295-accession panel, VCF files, and the flowering time data from the 424-accession panel used in this study were obtained from Lu et al (2020). The 1295-accession panel consisted of 146 wild soybeans, 575 landraces, and 574 improved cultivars. The 424-accession panel comprised 78 wild soybeans, 132 landraces, and 214 improved cultivars. VCF files were processed using the VCFtools software (v.0.1.16). Field evaluations were performed in six locations spanning latitudes 23°N (Guangzhou) to 45°N (Harbin); the significance of association was calculated with IBM SPSS software 20 (https://www.ibm.com/analytics/spss-statistics-software) with Student’s t-tests, P < 0.05.
RNA extraction, reverse transcription, and quantitative PCR (RT-qPCR)
Total RNA was extracted from different SN4 and ZK168 tissues (stem, ternate leaf, euphylla, shoot apical meristem, apical meristem, flower) with the Ultrapure RNA Kit (CWBIO, China). First-strand cDNAs were synthesized with the PrimeScript RT Reagent Kit with genomic DNA Eraser (Takara, Japan) and their concentrations were determined with a NanoDrop 2000 instrument (ThermoScientific, Wilmington, DE, USA). Quantitative PCR (RT-qPCR) was performed using a LightCycler 480 instrument (Roche). The expression levels of GmTubulin (Glyma.05G157300) were used as reference for normalization. Three biological replicates were used in all assays, and each sample was analyzed in technical triplicate. Primer sequences used for RT-qPCR are listed in Supplementary Table 1.
Transient transfection assays
Around 3 kb of promoter sequences from the FT2a and FT5a were PCR-amplified from genomic DNA extracted from leaves of Williams82 and cloned into pGreen0800-LUC/REN to generate the pFT2a-LUC/REN and pFT5a-LUC/REN reporters. The GmNF-YA1b coding regions were amplified from cDNAs reverse-transcribed from total RNA extracted from SN4 and ZK168 leaves and cloned into the p35S: Flag vector, yielding the effector constructs p35S:NFYA1b-Flag (GmNFYA1b-SN4-type and GmNFYA1b-ZK168-type). All reporter and effector constructs were introduced into Agrobacterium (Agrobacterium tumefaciens) strain GV3101. Agrobacteria were grown overnight and resuspended in infiltration buffer to a final OD600nm of 0.6 ~ 0.8. Agrobacterium suspensions harboring each reporter and effector construct were mixed and co-infiltrated into Nicotiana benthamiana leaves. Each combination of constructs was infiltrated in at least three leaves from individual N. benthamiana plants. The Renilla Luciferase Assay System (Promega) was used to measure the quantitative values of LUC and REN activities. Three biological replicates were used in all assays. Primer sequences used for cloning are listed in Supplementary Table 2.
Results
Phenotypic variation in the parents and RILs
We recorded various traits associated with flowering time for all RILs and the two parental cultivars over two consecutive years (2018 and 2019) in Harbin, China (Supplementary Fig. 1 and Supplementary Table 3). We observed transgressive segregation in the RIL population, indicating that flowering time in soybean is under polygenic control. Correlation analysis was conducted on the 2-year data of all traits recorded of the RIL population; the result shows that R1 (flowering time trait that means days from emergence to first open flower appeared on 50% of the plants in one line) were significantly correlated in 2 years. The absolute value of the skewness of the mean R1 values in the RIL population followed an approximately normal distribution, as evidence by the absolute skewness values below 1. We therefore used the R1 data for both years to detect QTLs.
Construction of a high-density genetic map
Both of the parental cultivars SN4 and ZK168 were resequenced at a higher coverage level of 14.7X and 8.1X, individually, to detect SNP markers and call variations between parents. For SN4 and ZK168, a total of 16,182,987,000 and 8,878,524,900 bases were identified, respectively. The Q30 ratio of SN4 and ZK168 was 94.66% and 91.35% while the GC content of SN4 and ZK168 was 40.41% and 35.85%, respectively (Supplementary Table 4). Finally, a total of 2942 polymorphic SNP markers were obtained and used in linkage map construction of 20 chromosomes (Supplementary Fig. 2 and Supplementary Table 5). The genetic distance between the markers was estimated in cM using the QTL IciMapping software.
QTL mapping for flowering time
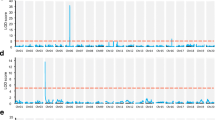
With the linkage map described above, we then turned to the identification of QTLs for flowering time using the inclusive composite interval mapping (ICIM) method. The threshold of the LOD scores for evaluating the statistical significance of QTL effects is 3.43 and 3.56 of 2 years, respectively. Three QTLs were detected by the ICIM method named as qR1-L, qR1-G, and qR1-B2 (R1: flowering time trait that means days from emergence to first open flower appeared on 50% of the plants in one line; L: Soybean Linkage Group L; G: Soybean Linkage Group G; B2: Soybean Linkage Group B2) (Fig. 1 and Supplementary Table 6). qR1-L mapped to chromosome 19, with a confidence interval spanning the region from bp 45,564,991 to bp 48,271,467 (using the genomic coordinates for the Wm82.a2.v1reference genome) with a LOD score of 6.72 in 2018 and 4.10 in 2019. The qR1-L QTL explained 12.1–17.4% of the observed phenotypic variation, depending on the year. qR1-G mapped to chromosome 18 in the region from bp 56,210,047 to bp 57,696,740. qR1-G reached significance only in 2019, with a LOD score of 3.65, but was close to significant in 2018, with a LOD score of 2.78. This QTL explained 4.6–8.5% of the observed phenotypic variation. Finally, qR1-B2 mapped to chromosome 14, within an interval from bp 34,291,536 to bp 39,844,217 and LOD scores of 3.57 and 4.39. The LOD of qR1-L and qR1-B2 was higher than the LOD threshold in both 2 years whereas the LOD of qR1-G was only greater than the LOD threshold in 2019. The LOD value of qR1-G in 2018 was not exceeding the threshold, but because the LOD value was higher than 2.5, we also considered it as a credible QTL.

Quantitative trait locus (QTL) mapping by ICIM. a QTLs detected for flowering time in 2018. b QTLs for flowering time in 2019. Chr, chromosome. The dashed red lines indicate the logarithm of the odds (LOD) threshold for each year
In addition, we independently detected QTLs with the WinQTL software to validate the ICIM results. Indeed, WinQTL detected three QTLs that overlap with the qR1-L, qR1-G, and qR1-B2 which were detected by ICIM (Supplementary Fig. 3 and Supplementary Table 7). As the mapping interval for qR1-B2 from WinQTL was larger than that obtained from ICIM, we used the WinQTL confidence interval when searching for candidate genes of qR1-B2. We also identified several year-specific QTLs: for instance, we noticed two strong QTLs mapping to chromosome 4 with high LOD scores of 6.12 and 8.57, respectively, when using the ICIM method. However, these two QTL peaks rose above significance only in 2019 and showed LOD scores of at most 2.0 in 2018. Similarly, WinQTL identified three QTLs mapping to chromosomes 8, 9, and 20 either in 2018 or 2019. Since these QTLs were detected only in 1 year, they were not considered further (Supplementary Table 8). The instability of these QTLs may be caused by different meteorological conditions between 2018 and 2019, including temperatures and rainfalls (Supplementary Table 9).
Candidate gene prediction of qR1-G
The mapping interval for the qR1-G included 83 genes, which we characterized by Gene Ontology (GO) analysis (Supplementary Table 10). Among them, 49, 18, and 79 were functionally annotated to the categories of cellular components, molecular functions, and biological processes, individually (Supplementary Fig. 4). A single gene, Glyma.18G281400, was related to transcriptional regulation. Of the 83 genes within the mapping interval, 15 were polymorphic, with nonsynonymous mutations between the two parents (Supplementary Table 11). All the 15 genes’ functional annotations are shown in Supplementary Table 12. Notably, Glyma.18G281400 was among these polymorphic genes. Glyma.18G281400 encoded an APETALA2 (AP2)-EREBP (ETHYLENE-RESPONSIVE ELEMENT-BINDING PROTEIN)-type transcription factor. EREBPs with AP2 domains play a role in regulating plant development (Zhao et al. 2006; Wang et al. 2014; Kuluev et al. 2015). A previous research has reported that two AP2 domain-encoding genes, SCHLAFMÜTZE and SCHNARCHZAPFEN, repressed flowering in Arabidopsis thaliana (Schmid et al. 2003). Therefore, Glyma.18G281400 was considered as a candidate gene for qR1-G.
To test the association between the genotype of Glyma.18G281400 and flowering time, we exploited the genotype collected from the 1295-accession panel, from which we defined seven haplotypes producing three distinct predicted alleles. However, these nonreference alleles were rare across all accessions (Fig. 2a and Supplementary Table 11). However, the haplotype 2 (H2), which carried a 9-bp insertion after 665th and a single-nucleotide polymorphism (SNP) at 430th bp, appeared to be associated with flowering time. We then turned to the 424-accession panel and compared flowering time for accessions with the H2 and H3-H7 (reference allele); these showed no significant differences in the flowering time data collected at Guangzhou in 2019, but did display contrasting flowering times at other locations (Harbin in 2019, Wuhan in 2019, Zhengzhou in 2018 and 2019, and Hefei in 2018) (Fig. 2b–g). These results suggest that Glyma.18G281400 may be associated with flowering time at medium and high latitudes but not at low latitudes in China. Therefore, we consider Glyma.18G281400 to be a likely candidate gene for qR1-G, but do not exclude the remaining 14 genes within the interval, listed in Supplementary Table 12.

Haplotypes and correlation analysis with flowering time for the candidate gene Glyma.18G281400. a Haplotypes of Glyma.18G281400 in a panel of 1295 soybean varieties. Blue indicates the reference allele from W82; green indicates the polymorphic allele. b–g Flowering times for landraces and improved cultivars from 424-accession panel carrying the Glyma.18G281400 haplotypes H2 or H3-H7, in different regions: b Harbin in 2019; c Zhengzhou in 2019; d Wuhan in 2019; e Guangzhou in 2019; f Zhengzhou in 2018; g Hefei in 2018. The numbers at the bottom represent numbers of individuals. Haplotypes were extracted from the 1295-accession panel, which comprises 146 wild soybeans, 575 landraces, and 574 improved cultivars. The horizontal lines indicate the median values. Statistical significance was determined by Student’s t-test
Candidate gene prediction of qR1-B2
As with qR1-G, we compared the coding sequences of genes within the mapping interval between the two parental cultivars and identified 49 genes with polymorphisms in their exons (Supplementary Table 13). Among these, annotation information was available for 35 genes, prompting us to carry out a GO analysis (Supplementary Table 14). Of the 35 genes, we obtained functional annotations for nine genes in terms of cellular components, 31 genes for molecular functions, and 25 genes for biological processes (Supplementary Fig. 5). We retained all 35 genes as candidates for qR1-B2 and their functional annotations are listed in Supplementary Table 15. Three genes were related to transcriptional regulation, namely Glyma.14G159400, Glyma.14G160600, and Glyma.14G161900. Of these genes, Glyma.14G159400 encoded a transcription factor with a jumonji (jmjC) domain. The rice (Oryza sativa) gene photoperiod sensitivity-14 (Se14) was previously shown to delay flowering under long-day conditions and encoded a jmjc domain–containing protein (Takayuki et al. 2014), pointing to Glyma.14G159400 as a potential candidate gene for qR1-B2.
To test this hypothesis, we conducted a correlation analysis between the genotype of Glyma.14G159400 and flowering time. We distinguished nine haplotypes of Glyma.14G159400 across the 1295-accession panel and discovered that the allele of Glyma.14G159400 from SN4 (3206th-T) also consists of generally natural varieties (Fig. 3a). We also determined evolutionary relationships between haplotypes, which led us to conclude that the most common haplotype, H9, may have originated from haplotype H6, with a concomitant marked reduction in the proportion of wild varieties (Fig. 3b). We then compared the flowering time of accessions carrying either the C allele or the T allele at nucleotide 3206 (3206-C or 3206-T): this analysis revealed that the 3206-T allele flowers significantly later than the 3206-C allele at all six locations tested (Fig. 3c–h). This observation indicated that the polymorphism at nucleotide 3206 in Glyma.14G159400 may lead to variation in flowering time, supporting a role for Glyma.14G159400 in the control of flowering time across diverse genetic soybean backgrounds and environmental conditions. Therefore, we consider Glyma.14G159400 as a more likely candidate gene for qR1-B2, but do not exclude the remaining genes in the mapping interval, listed in Supplementary Table 15.

Haplotypes, origins, and correlation analysis with flowering time of Glyma.14G159400. a Haplotypes of Glyma.14G159400. b Haplotype origins of Glyma.14G159400. c–h Flowering time of Glyma.14G159400 3206th-C type and 3206th-T type in landraces and improved cultivars in 424 sub-accessions. Flowering time in different regions: c Harbin, 2019; d Zhengzhou, 2019; e Wuhan, 2019; f Guangzhou, 2019; g Zhengzhou, 2018; h Hefei, 2018. Haplotypes were extracted from the 1295 panel of 146 wild soybeans, 575 landraces, and 574 improved cultivars. Red color represented the wild soybeans, green color represented the landraces, and blue color represented the improved cultivars in haplotype origins analysis. The horizontal line indicates the median value. Statistical significance was determined by Student’s t-test
Candidate gene prediction of qR1-L
qR1-L mapped to chromosome 19, within an interval that overlaps with the well-known flowering-related gene E3, but sequence comparison showed no differences at E3 between the two parents. Likewise, the growth habit gene Dt1 recently reported to regulate flowering time in soybean (Yue et al. 2021), also mapped in close proximity to the qR1-L interval, but we detected no polymorphisms between the two parents, indicating the existence of a new gene modulating flowering time defined by qR1-L. We determined that 131 genes within the mapping interval have nonsynonymous or frameshift mutations between the two parental cultivars (Supplementary Table 16). Among them, 87 genes have been annotated by GO (Supplementary Table 17). GO analysis showed that 24 genes have functional annotation related to their cellular components, while 78 genes were functionally annotated for putative molecular functions and 47 genes were functionally annotated for biological processes (Supplementary Fig. 6). Importantly, Glyma.19G200800, referred to as GmNF-YA1b, was the only gene related to transcriptional regulation, as determined by GO analysis. GmNF-YA1b is homologous to Arabidopsis NF-YA10, which belongs to the NF-YA family and was previously shown to regulate flowering time in Arabidopsis (Wenkel et al. 2006; Xu et al. 2004; Siriwardana et al. 2016). Consequently, although a series of genes which were listed in Supplementary Table 18 are all candidate genes of qR1-L, we consider the GmNF-YA1b as a more likely candidate gene of the locus.
GmNF-YA1b is involved in flowering time control
As a first step in characterizing GmNY-A1b and its role in flowering time, we PCR-amplified the coding regions (CDS) from first-strand cDNAs prepared from SN4 and ZK168 total RNA and sequenced the PCR products. We note here that the current primary transcript for Glyma.19G200800, as defined in Phytozome v12.1, contains 48 additional nucleotides at the 5′ end of the CDS that are not included here and may reflect alternative splicing, as previously reported (Schaarschmidt et al. 2013). In addition, we observed a T-G polymorphism at nucleotide 154 (Supplementary Fig. 7), resulting in a nonsynonymous change of an Alanine to Serine (A52S).
NF-YA were previously shown to directly bind the distal CCAAT box in the FT promoter and activate its transcription, thus acting as positive regulators of flowering in the long-day plant species Arabidopsis (Siriwardana et al. 2016). To assess the function of GmNF-YA1b in soybean, we performed transient infiltration assays in Nicotiana benthamiana leaves with luciferase (LUC) reporters, whereby LUC transcription was driven by the GmFT2a or GmFT5a promoters, either alone or co-infiltrated with the SN4 or ZK168 alleles of GmNF-YA1b as effectors. Expression of both GmNF-YA1b-SN4 and GmNF-YA1b-ZK168 repressed LUC transcription, as demonstrated by a reduction in LUC activity, indicating that GmNF-YA1b can repress GmFT2a and GmFT5a transcription (Fig. 4). However, we did notice significant differences in the extent of transcriptional repression imparted by the two alleles, as the SN4 allele was more effective at repressing GmFT2a and GmFT5a transcription than the ZK168 allele. Therefore, although fine-mapping is critical to identify the causal locus behind qR1-L, we concluded that GmNF-YA1b is the most likely candidate for this QTL, as the protein it encodes affects flowering by repressing the expression of two florigen genes, GmFT2a and GmFT5a.

GmNF-YA1b inhibit the expression of GmFT2a/GmFT5a. a Schematic representation of the constructs used for transient infiltration assay of GmNF-YA1b and GmFT2a. b Luciferase activity under control of GmFT2a promoter regulated by different GmNF-YA1b alleles. c Schematic representation of the constructs used for transient infiltration assay of GmNF-YA1b and GmFT5a. d Luciferase activity under control of GmFT5a promoter regulated by different GmNF-YA1b alleles. Data are shown as means ± SD from three independent replicates (indicated as individual dots). Duncan’s test was used for test of significance. Different letters indicate significant differences between means
We next investigated the expression levels of GmNF-YA1b in the parental cultivars SN4 and ZK168 by RT-qPCR in plants grown under long-day (LD) conditions (16-h light/8-h dark) in a growth chamber. GmNF-YA1b was expressed in all tissues examined, especially in leaves and flowers (Supplementary Fig. 8a). GmNF-YA1b also exhibited a diurnal pattern in leaves, with a minor peak 8 h after lights on (Zeitgeber time 8, ZT8) and a major peak in the dark part of the diurnal cycle, 4 h after lights off at ZT20 (Supplementary Fig. 9b). We also determined the expression patterns of GmFT genes grown in the same conditions: both GmFT2a and GmFT5a were more highly expressed in ZK168 than in SN4 plants (Supplementary Fig. 8c and 8d), meaning that GmFT2a and GmFT5a expression differs between the two parental cultivars in LD conditions.
Haplotype analysis and geographical latitudinal distribution of GmNF-YA1b
We defined eight GmNF-YA1b haplotypes in the 1295-accession panel, with seven polymorphisms whose combinations generate five distinct functional alleles (Fig. 5a). We compared the GmNF-YA1b protein sequences to find its homologous genes in soybean, Arabidopsis thaliana (https://phytozome.jgi.doe.gov/pz/portal.html), and several legumes (https://www.legumeinfo.org), and further reproduced the phylogenetic tree (Supplementary Fig. 9). Four NF-YA10 homologous genes in soybean were clustered into the clade, which was further subdivided into two groups (Supplementary Fig. 9a). We also investigated the frequency of the nonsynonymous variant S52A across GmNF-YA1b homologs and established that the serine residue is common to all legumes, soybean, and Arabidopsis, while alanine is specific to GmNF-YA1b from SN4 (Supplementary Fig. 9b). Combined with the results of protein homologous alignment and haplotypes origin analysis, we found that all the haplotypes originated from the H2 by a SNP and the common H8 was differentiated from H7 (Fig. 5b). Variant S52A also existed in a large number of natural accessions. We therefore examined the association between the genotype at GmNF-YA1b (defined by the S52 [bp 154-G] or A52 [bp 154-T] alleles) and flowering time in the 424-accession panel measured at six field sites with different latitudes in China. We observed significant associations between the genotype at this position and flowering time (Fig. 5e–j).

Haplotypes, origins, latitude distribution, and correlation analysis of GmNF-YA1b. a Haplotypes of GmNF-YA1b. b Haplotype origins of GmNF-YA1b. c Distribution of GmNF-YA1b alleles 154-G and 154-T in landraces and improved cultivars according to their region of origin. d Distribution of GmNF-YA1b alleles across wild varieties, landraces, and cultivars. e–j Flowering times for landraces and improved cultivars from the 424-accession panel carrying the 154-G or 154-T allele at GmNF-YA1b, in different regions: e Harbin in 2019; f Zhengzhou in 2019; g Wuhan in 2019; h Guangzhou in 2019; i Zhengzhou in 2018; j Hefei in 2018. Haplotypes were extracted from the 1295-accession panel. Red indicates wild soybeans, green indicates landraces, and blue indicates improved cultivars. Geographic distributions were extracted from the 1295-accession panel. The horizontal line indicates the median value. Statistical significance was determined by Student’s t-test. LL, low latitude; ML, middle latitude; HL, high latitude. W, wild varieties; L, landrace accessions; C, improved cultivars; W, L, and C were from the 1295-accession panel
The 424 soybean accessions were mainly collected from across Southern and Northern China, as well as the Huanghuai region, and comprised 78 wild soybeans, 132 landraces, and 214 improved cultivars. Since both parents of the RIL population employed here were collected at high latitude, we selected only improved cultivars from the Northeast region (with high latitude) to further explore the correlation between the two alleles at GmNF-YA1b and flowering time. Indeed, the flowering times of cultivars carrying either the 154-G or the 154-T allele of GmNF-YA1b differed significantly for those collected from middle- and high-latitude regions (Supplementary Fig. 10a–10f). These results confirmed that GmNF-YA1b affects flowering and the 154-G allele leads to early flowering, while the 154-T allele confers late flowering. These results further confirm that GmNF-YA1b is related to flowering time and the 154th-G allele leads to early flowering while the 154th-T allele contributes to late-flowering phenotype.
We next examined the distribution of the two major GmNF-YA1b alleles within the Chinese accessions of the 1295-accession panel as a function of their latitude of origin. For both landraces and improved cultivars, the relative representation of the early flowering allele 154-G gradually increased from low to high latitude (Fig. 5c). We then determined the relative representation of the early flowering allele within wild accessions, landraces, and improved cultivars: the early flowering allele 154-G was only present in 12% of wild varieties, but gradually increased to 71% in improved cultivars, providing a strong indication that it has undergone selection in landraces and has been widely utilized in modern breeding (Fig. 5d). Taken together, these results support the notion that GmNF-YA1b is a latitudinal adaptation gene and that its early flowering allele 154-G has been widely incorporated in past breeding efforts in Northern China.
Discussion
Soybean plays key roles in global food security and agronomic sustainability. The discovery of new genes controlling agronomic traits will help increase yield (Zhang et al. 2004). Among these valuable traits, flowering time dictates the length of the vegetative growth period and defines the geographical range of soybean. Therefore, deciphering the molecular basis of variation in flowering time not only will reveal signatures of artificial selection as a consequence of breeding, but also holds great significance for the domestication, diversity, and improvement of soybean (Zhao et al. 2018).
Molecular markers are widely used in plant research, including for the identification of QTLs (Sonah et al. 2013; Semagn and Bjrnstad 2006). Among the various markers available, single-nucleotide polymorphisms (SNPs) are highly abundant, easier to identify than structural variants, evenly distributed over the genome, and are hence especially appropriate for molecular applications (Rafalski 2002). However, high-throughput genotyping at a handful of markers is relatively time-consuming (Sonah et al. 2013). Accordingly, next-generation sequencing (NGS) approaches have emerged to expedite analysis and increase throughput (Pareek et al. 2011). Among the many NGS technologies available, genotyping by sequencing (GBS) boasts a genome reduction step via digestion with a restriction enzyme, followed by a simplified library production procedure, which enhances its suitability for sequencing many individuals (Elshire et al. 2011). Due to its low cost and fast turnaround times, GBS is now widely used to sequence multiple genomes, including those of soybean. In this study, we applied the GBS approach to genotype an RIL population and detect QTLs for flowering time. We detected three QTLs for the R1 trait, which we named qR1-L, qR1-G, and qR1-B2, that mapped to chromosomes 19, 18, and 14, respectively. While other QTLs have been reported to map to the same intervals (Supplementary Table 19), none of the underlying causal genes has been identified. Here, we explored the list of candidate genes included in the mapping intervals for these QTLs, selected high-confidence candidates for all three QTLs, and performed a functional validation to one candidate gene for qR1-L.
Within the mapping interval of qR1-G, we identified 15 genes with nonsynonymous mutations between the parents (Supplementary Table 12). One gene, Glyma.18G281400, is related to transcriptional regulation and was polymorphic between the parents, making it a more likely candidate for qR1-G. Glyma.18G281400 encodes a member of the AP2-EREBP-type superfamily of transcription factors, several of which affect flowering in Arabidopsis (Weigel 1995; Okamuro et al. 1997; Schmid et al. 2003). We further explored the potential of Glyma.18G281400 as a candidate gene for the underlying QTL by analyzing the distribution of its haplotypes across a soybean panel (Fig. 2). We followed the same methods for qR1-B2 and qR1-L, and identified one probable candidate gene for each QTL: Glyma.14G159400 (for qR1-B2) and Glyma.19G200800, which we named GmNF-YA1b (for qR1-L).
Nuclear factor Y (NF-Y), also known as CCAAT-binding factor (CBF), is a complex composed of three subunits, NF-YA, NF-YB, and NF-YC, that specifically binds to the evolutionarily conserved CCAAT motif (Hou et al. 2014; Mantovani 1999; Kusnetsov et al. 1999). The florigen FT, encoded by a gene whose promoter includes a CCAAT box, is the main mobile protein that responds to photoperiodic signals in leaves and then relocates to the shoot apex to promote floral transition (Lin et al. 2007; Corbesier et al. 2007; Mathieu et al. 2007). In Arabidopsis, NF-YA (NF-YA2) directly binds to the distal CCAAT box in the FT promoter as a complex with NF-YB/NF-YC, to induce FT expression and thus flowering (Siriwardana et al. 2016). The two soybean FT homologs GmFT2a and GmFT5a coordinately regulate flowering in soybean (Kong et al. 2010). Here, we provide evidence, in the form of transient infiltration assays in N. benthamiana, that GmNF-YA1b represses GmFT2a and GmFT5a expression under LD conditions in soybean (Fig. 4). Much of the focus on NF-YA10 and its homologs has related to its roles during high-salinity stress (Ma et al. 2015ab; Zhang et al. 2020), drought stress (Ma et al. 2015ab; Yu et al. 2020), and leaf growth (Zhang et al. 2017). Our results add to the known functions of NF-YA10 family members in soybean and provide a potential research direction.
Most ancestors of flowering plants had a simple genome, but that since, angiosperms have gone through multiple rounds of genome duplications (Masterston 1994). Therefore, many plants, especially domesticated crop species, are polyploid (Blanc and Wolfe 2004; Cui et al. 2006). As a typical flowering plant, soybean evolved numerous homologous genes following whole-genome duplication, which is associated with subsequent functional differentiation and weakening between homologs. The soybean genome encodes numerous NF-Y genes, with 21 GmNF-YA, 32 GmNF-YB, and 15 GmNF-YC genes (Quach et al. 2015). Although the function of many NF-Y genes has been studied in Arabidopsis, the function of GmNF-YA1b (Glyma.19G200800) in soybean remains to be elucidated, and it is still unknown whether it is a functional gene or a silent gene whose function can be covered by its homologs. In this study, we confirmed that GmNF-YA1b protein can negatively regulate GmFTs, and the detection of this QTL also demonstrated that GmNF-YA1b may be a real functional gene. At the same time, it also provides the foundation for the further study of this gene. We discovered that GmNF-YA1b may be a latitude adaptation gene, as determined by a geographical latitude analysis. The 154-G allele of GmNF-YA1b was frequent at high latitudes (Fig. 5b), indicative of the selection of GmNF-YA1b as a latitude adaptation gene. In addition, the early flowering allele 154-G increased in frequency from wild accessions to landraces and improved cultivars (Fig. 5b and c), indicating that this allele has been selected during breeding. This feature of GmNF-YA1b may be harnessed for breeding varieties that are adapted to high latitudes, broaden the geographic range of soybean cultivation, and increase yield.
In conclusion, we detected three QTLs related to flowering time and predicted their candidate genes. In particular, a more likely candidate gene of qR1-L, GmNF-YA1b, a potential latitude adaptability gene, might regulate flowering in soybean by influencing the expression of GmFTs. These results provide the theoretical basis to better understand the regulatory network controlling flowering in soybean and generate new materials to breed cultivars that are better adapted cultivars to high latitudes.
Data availability
Not applicable.
References
Bernard RL (1971) Two major genes for time of flowering and maturity in soybeans. Crop Sci 11:242–247
Bernard RL (1972) Two genes affecting stem termination in soybeans. Crop Sci 12:235–239
Blanc G, Wolfe KH (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16:1667–1678
Bonato ER, Vello NA (1999) E6, a dominant gene conditioning early flowering and maturity in soybeans. Genet Mol Biol 22:229–232
Buzzell RI (1971) Inheritance of a soybean flowering response to fluorescent day length conditions. Can J Genet Cytol 13:703–707
Buzzell RI, Voldeng HD (1980) Inheritance of insensitivity to long daylength. Soybean Genet Newslett 7:26–29
Cheng W, Liu F, Li M, Hu X, Chen H, Pappoe F (2015) Variation detection based on next-generation sequencing of type Chinese 1 strains of Toxoplasma gondii with different virulence from China. BMC Genomics 16:888
Cober ER, Voldeng HD (2001) A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Sci 41:698–701
Cober ER, Molnar SJ, Charette M, Voldeng HD (2010) A new locus for early maturity in soybean. Crop Sci 50:524–527
Corbesier L, Vincent C, Jang S, Fornara F, Fan Q, Searle I et al (2007) FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316(5827):1030–1033
Cui L, Kerr Wall P, James H et al (2006) Widespread genome duplications throughout the history of flowering plants. Genome Res 16:738–749
Davey JW, Cezard T, Fuentes-Utrilla P, Eland C, Gharbi K, Blaxter ML (2013) Special features of RAD sequencing data: implications for genotyping. Mol Ecol 22:3151–3164
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLOS ONE 6(5):e19379
Fang C, Liu J, Zhang T, Su T, Li S, Cheng Q, Kong L, Li X et al (2020) A recent retrotransposon insertion of J caused E6 locus facilitating soybean adaptation into low latitude. J Integr Plant Biol. https://doi.org/10.1111/jipb.13034
Fehr WR, Caviness CE, Burmood DT, Pennington JS (1971) Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci 11:929–931
Hartwig EE, Kiihl RA (1979) Identification and utilization of a delayed flowering character in soybeans for short-day conditions. Field Crop Res 2:145–151
Heatherly LG, Smith JR (2004) Effect of soybean stem growth habit on height and node number after beginning bloom in the MIDSOUTHERN USA. Crop Sci 44:1855–1858
Hoeck JA, Fehr WR, Shoemaker, RC, Welke, GA, Cianzio SR (2003) Molecular marker analysis of seed size in soybean. Crop Science 43:68–74
Hou X, Zhou J, Liu C, Liu L, Shen L, Yu H (2014) Nuclear factor Y-mediated H3K27me3 demethylation of the SOC1 locus orchestrates flowering responses of Arabidopsis. Nat Commun 5:4601
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan J, Fan D, Weng Q, Huang T, Dong G, Sang T, Han B (2009) High-throughput genotyping by whole-genome resequencing. Genome Res 19:1068–1076
Jiang B, Nan H, Gao Y, Tang L, Yue Y, Lu S, Ma L, Cao D, Sun S, Wang J, Wu C, Yuan X, Hou W, Kong F, Han T, Liu B (2014) Allelic combinations of soybean maturity loci E1, E2, E3 and E4 result in diversity of maturity and adaptation to different latitudes. Plos One 9:e106042
Kong F, Liu B, Xia Z, Sato S, Kim BM et al (2010) Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiol 154:1220–1231
Kong F, Nan H, Cao D, Li Y, Wu F, Wang J, Lu S, Yuan X, Cober ER, Abe J, Liu B (2014) A new dominant gene coditions early flowering and maturity in soybean. Crop Sci 54:2529–2535
Kong L, Lu S, Wang Y, Fang C, Wang F, Nan H et al (2018) Quantitative trait locus mapping of flowering time and maturity in soybean using next-generation sequencing-based analysis. Front Plant Sci 9:995
Kuluev B, Avalbaev A, Nurgaleeva E, Knyazev A, Nikonorov Y, Chemeris A (2015) Role of AINTEGUMENTA-like gene NtANTL in the regulation of tobacco organ growth. J Plant Physiol 189:s11-23
Kusnetsov V, Landsberger M, Meurer J, Oelmuller R (1999) The assembly of the CAAT-box binding complex at a photosynthesis gene promoter is regulated by light, cytokinin, and the stage of the plastids. J Biol Chem 274(50):36009–36014
Langewisch T, Zhang H, Vincent R, Joshi T, Xu D, Bilyeu K (2014) Major soybean maturity gene haplotypes revealed by SNPViz analysis of 72 sequenced soybean genomes. PLoS ONE 9(4):E94150
Langewisch T, Lenis J, Jiang G-L, Wang D, Pantalone V, Bilyeu K (2017) The development and use of a molecular model for soybean maturity groups. BMC Plant Biol 17(1):91
Li X, Fang C, Xu M, Zhang F, Lu S, Nan H, Su T, Li S, Zhao X, Kong L, Yuan X, Liu B, Abe J, Cober ER, Kong F (2017) Quantitative trait locus mapping of soybean maturity gene E6. Crop Sci 57(5):2547
Lin MK, Belanger H, Lee YJ, Varkonyi-Gasic E, Taoka K, Miura E et al (2007) FLOWERING LOCUS T protein may act as the long-distance florigenic signal in the cucurbits. Plant Cell 19(5):1488–1506
Lin X, Liu B, Weller JL, Abe J, Kong F (2020) Molecular mechanisms for the photoperiodic regulation of flowering in soybean. J Integr Plant Biol. https://doi.org/10.1111/jipb.13021
Liu B, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J (2008) Genetic redundancy in soybean photo responses associated with duplication of the Phytochrome A gene. Genetics 180:995–1007
Liu B, Watanabe S, Uchiyama T, Kong F, Kanazawa A, Xia Z et al (2010) The soybean stem growth habit gene dt1 is an ortholog of Arabidopsis TERMINAL FLOWER1. Plant Physiol 153(1):198–210
Lu S, Zhao X, Hu Y, Liu S, Nan H, Li X, Fang C, Cao D, Shi X, Kong L, Su T, Zhang F, Li S, Wang Z, Yuan X, Cober ER, Weller JL, Liu B, Hou X, Tian Z, Kong F (2017) Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat Genet 49:773–779
Lu S, Dong L, Fang C, Liu S, Kong L, Cheng Q, Chen L, Su T, Nan H, Zhang D, Zhang L, Wang Z, Yang Y, Yu D, Liu X, Yang Q, Lin X, Tang T, Zhao X, Yang X, Tian C, Xie Q, Li X, Yuan X, Tian Z, Liu B, Weller J, Kong F (2020) Stepwise selection on homologous PRR genes controlling flowering and maturity during soybean domestication. Nat Genet 52:428–436
Ma X, Li C, Wang M (2015a) Wheat NF-YA10 functions independently in salinity and drought stress. Bioengineered 6(4):245–247
Ma X, Zhu X, Li C et al (2015b) Overexpression of wheat NF-YA10 gene regulates the salinity stress response in Arabidopsis thaliana. Plant Physiol Biochem 86:34–43
Mantovani R (1999) The molecular biology of the CCAAT-binding factor NF-Y. Gene 239(1):15–27
Masterston J (1994) Stomatal size in fossil plants: evidence for polyploidy in majority of angiosperms. Science 264:421–423
Mathieu J, Warthmann N, Kuttner F, Schmid M (2007) Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Current Biology CB 17(12):1055–1060
McBlain BA, Bernard RL (1987) A new gene affecting the time of flowering and maturity in soybeans. J Hered 78(3):160–162
Meng L, Li H, Zhang L, Wang J (2015) QTL icimapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Ogiso-Tanaka E, Shimizu T, Hajika M, Kaga A, Ishimoto M (2019) Highly multiplexed AmpliSeq technology identifies novel variation of flowering time-related genes in soybean (Glycine max). DNA Res 26:243–260
Okamuro JK, Caster B, Villarroel R, VanMontagu M, Jofuku KD (1997) The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc Natl Acad Sci USA 94:7076–7081
Pareek CS, Smoczynski R, Tretyn A (2011) Sequencing technologies and genome sequencing. J Appl Genet 52(4):413–435
Quach TN, Nguyen HT, Valliyodan B, Joshi T, Xu D, Nguyen HT (2015) Genome-wide expression analysis of soybean NF-Y genes reveals potential function in development and drought response. Mol Genet Genomics : MGG 290(3):1095–1115
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5(2):94–100
Ray JD, Hinson K, Mankono JEB, Malo MF (1995) Genetic control of a long-juvenile trait in soybean. Crop Sci 35:1001–1006
Saindon G, Beversdorf WD, Voldeng HD (1989) Adjusting of the soybean phenology using the E4 loci. Crop Sci 29:1361–1365
Samanfar B, Molnar SJ, Charette M, Schoenrock A, Dehne F, Golshani A, Belzile F, Cober ER (2016) Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean. Theor Appl Genet 130:377–390
Schaarschmidt S, Gresshoff PM, Hause B (2013) Analyzing the soybean transcriptome during autoregulation of mycorrhization identifies the transcription factors GmNF-YA1a/b as positive regulators of arbuscular mycorrhization. Genome Biol 14(6):R62–R62
Schmid M, Uhlenhaut H, Godard F, Demar M, Bressan R, Weigel D, Lohmann J (2003) Dissection of floral induction pathways using global expression analysis. Development 130(24):6001–6012
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Semagn K, Bjrnstad NMN (2006) An overview of molecular marker methods for plants. Afr J Biotech 525(25):2540–2568
Sinha S, Maity SN, Lu J, Crombrugghe BD (1995) Recombinant rat CBF-C, the third subunit of CBF/NFY, allows formation of a protein-DNA complex with CBF-A and CBF-B and with yeast HAP2 and HAP3. Proc Natl Acad Sci USA 92(5):1624–1628
Sinha S, Kim IS, Sohn KY, de Crombrugghe B, Maity SN (1996) Three classes of mutations in the A subunit of the CCAAT-binding factor CBF delineate functional domains involved in the three-step assembly of the CBF-DNA complex. Mol Cell Biol 16:328–337
Siriwardana CL, Gnesutta N, Kumimoto RW, Jones DS, Iii BFH (2016) Nuclear Factor Y, subunit A (NF-YA) proteins positively regulate flowering and act through flowering locus T. Plos Genet 12(12):e1006496
Sonah H, Bastien M, Iquira E et al (2013) An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. Plos One 8:e54603
Specht JE, Chase K, Macrander M, Graef GL, Chung J, Markwell JP, Germann M, Orf JH, Lark KG (2001) Soybean response to water: a QTL analysis of drought tolerance. Crop Sci 41:493–509
Sun F, Xu M, Park C, Dwiyanti MS, Nagano AJ, Zhu J, Watanabe S, Kong F, Liu B, Yamada T, Abe J (2019) Characterization and quantitative trait locus mapping of late-flowering from a Thai soybean cultivar introduced into a photoperiod-insensitive genetic background. PLoS ONE 14:e0226116
Takayuki Y, Hiroki S, Yoshihiro Y, Quan X, Takehito A, Takuji T et al (2014) Se14, encoding a jmjc domain-containing protein, plays key roles in long-day suppression of rice flowering through the demethylation of h3k4me3 of rft1. Plos One 9(4):e96064
Takeshima R, Hayashi T, Zhu J, Zhao C, Xu M, Yamaguchi N, Sayama T, Ishimoto M, Kong L, Shi X, Liu B, Tian Z, Yamada T, Kong F, Abe J (2016) A soybean quantitative trait locus that promotes flowering under long days is identified as FT5a, a FLOWERING LOCUS T ortholog. J Exp Bot 67:5247–5258
Tasma IM, Lorenzen LL, Green DE, Shoemaker RC (2001) Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean. Mol Breeding 8(1):25–35
Thakare D, Kumudini S, Dinkins RD (2010) Expression of flowering-time genes in soybean E1 near-isogenic lines under short and long day conditions. Planta 231(4):951–963
Wang SC, Basten CJ, Zeng ZB (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC
Wang Y, Wang L, Zou Y, Chen L, Cai Z, Zhang S et al (2014) Soybean miR172c targets the repressive AP2 transcription factor NNC1 to activate ENOD40 expression and regulate nodule initiation. Plant Cell 26:4782–4801
Wang F, Nan H, Chen L, Fang C, Zhang H, Su T, Li S, Cheng Q, Dong L, Liu B, Kong F, Lu S (2019) A new dominant locus, E11, controls early flowering time and maturity in soybean. Mol Breeding 39:70
Watanabe S, Hideshima R, Xia Z, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T, Tabata S, Harada K (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182:1251–1261
Watanabe S, Xia Z, Hideshima R, Tsubokura Y, Sato S, Yamanaka N et al (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188(2):395–407
Weigel D (1995) The APETALA2 domain is related to a novel type of DNA-binding domain. Plant Cell 7:388–389
Wenkel S, Turck F, Singer K, Gissot L, Gourrierec JL, Samach A, Coupland G (2006) CONSTANS and the CCAAT box binding complex share a functionally important domain and interact to regulate flowering of Arabidopsis. Plant Cell 18(11):2971–2984
Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H et al (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci USA 109(32):E2155-2164
Xu M, Zhang L, Li W, Hu X, Wang M, Fan Y et al (2004) Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J Exp Bot 65(1):89–101
Xu M, Xu Z, Liu B, Kong F, Tsubokura Y, Watanabe S, Xia Z, Harada K, Kanazawa A, Yamada T, Abe J (2013) Genetic variation in four maturity genes affects photoperiod insensitivity and PHYA-regulated post-flowering responses of soybean. BMC Plant Biol 13:91
Yin Z, Qi H, Mao X, Wang J, Hu Z, Wu X et al (2018) QTL mapping of soybean node numbers on the main stem and meta-analysis for mining candidate genes. Biotechnology & Biotechnological Equipment 32(4):315-922
Yu Y, Bai Y, Wang P et al (2020) Soybean nuclear factor YA10 positively regulates drought resistance in transgenic Arabidopsis thaliana. Environ Exp Bot 180:104249
Yue L, Li X, Fang C, Chen L, Yang H, Yang J et al (2021) FT5a interferes with the Dt1-AP1 feedback loop to control flowering time and shoot determinacy in soybean. J Integr Plant Biol. https://doi.org/10.1111/jipb.13070
Zhang W, Wang Y, Luo G, Zhang JS, He C, Wu X et al (2004) QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theor Appl Genet 108(6):1131–1139
Zhang L, Kyei-Boagen S, Zhang J, Zhang M, Freeland T, Watson C (2007) Modifications of optimum adaptation zones for soybean maturity groups in the USA. Crop Manage 6:1
Zhang M, Hu X, Zhu M et al (2017) Transcription factors NF-YA2 and NF-YA10 regulate leaf growth via auxin signaling in Arabidopsis. Sci Rep 7(1):1395
Zhang Q, Zhang J, Wei H et al (2020) Genome-wide identification of NF-YA gene family in cotton and the positive role of GhNF-YA10 and GhNF-YA23 in salt tolerance. Int J Biol Macromol 165(Pt B):2103–2115
Zhao L, Xu S, Chai T, Wang T (2006) OsAP2-1, an AP2-like gene from Oryza sativa, is required for flower development and male fertility. Sex Plant Reprod 19:197–206
Zhao C, Takeshima R, Zhu J, Xu M, Sato M, Watanabe S, Kanazawa A, Liu B, Kong F, Yamada T, Abe J (2016) A recessive allele for delayed flowering at the soybean maturity LOCUS E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog. BMC Plant Biol 16:20
Zhao L, Li M, Xu C, Yang X, Li W (2018) Natural variation in GmGBP1 promoter affects photoperiod control of flowering time and maturity in soybean. Plant J 96(1):147–162
Zhao L, Luo Q, Yang C, Han Y, Li W (2018b) A RAV-like transcription factor controls photosynthesis and senescence in soybean. Planta 227:1389–1399
Funding
This work was supported by the National Natural Science Foundation of China (31930083), the Natural Key R&D Program of China (2017YFE0111000), the Major Program of Guangdong Basic and Applied Research (2019B030302006), the Open Project Foundation of National Key Laboratory for Crop Genetics and Germplasm Enhancement (ZW201901), the Heilongjiang Academy of Agricultural Sciences Fund for Distinguished Young Scholars (2020JCQN005), and the Heilongjiang Provincial Science and Technology Major Project (2019ZX16B01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, S., Su, T., Wang, L. et al. Rapid excavating a FLOWERING LOCUS T-regulator NF-YA using genotyping-by-sequencing . Mol Breeding 41, 45 (2021). https://doi.org/10.1007/s11032-021-01237-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-021-01237-w