
Abstract
Maize (Zea mays L.), which is an important food crop in the word, displays large genetic diversity. Knowledge of the relationships among maize inbred lines is essential to the maize breeder because it directs the exploitation of germplasm in hybrid production. In this study, the genetic diversity, population structure, and relatedness between pairs of 157 elite maize inbred lines from the current breeding program of Sichuan province in Southwest China were assessed with 4976 polymorphic single-nucleotide polymorphisms (SNPs) developed by genotyping by sequencing (GBS). A total of 91.1% of the inbred lines were considered pure with < 5% heterogeneity, while the remaining 8.9% of the inbred lines had a heterogeneity ranging from 5.5 to 40.0%. Genetic distance between pairs of lines varied from 0.0000 to 2.0702, with 98.79% of the pairs distant. Relative kinship analysis showed that the kinship coefficients for 91.3% of the pairs of lines were above 0.500, which agrees with the pedigree. Cluster and model-based population structure analyses all divided the 157 lines into four groups, which were named Impro-local, Tem-tropic I A, Tem-tropic I B, and Impro-tropic, respectively, based roughly on genetic background of the parents used for breeding. Impro-local group consisted of lines primarily improved from local germplasm; Tem-tropic I A and Tem-tropic I B groups consisted of lines primarily developed from cross or backcross with introduced tropic germplasm, but with different combining ability that had demonstrated by the commercial hybrids; and Impro-tropic group contained lines primarily improved from continuous self-crossing of tropical hybrids and populations. Analysis of molecular variance showed 14.2% of the variation among groups, with the remaining 85.8% attributable to differences within groups. The differentiation between the groups was further validated by the pairwise FST value (0.0904–0.1520), which indicated the moderate genetic differentiation characterizing this panel. The genome-wide average linkage disequilibrium (LD) decay distance was 1.05–1.10 Mb and varied among different chromosomes. The genetic diversity and population structure revealed in this study will help breeders to better understand how to utilize the current maize germplasm in Sichuan province for hybrid breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maize (Zea mays L.), one of the most important cereal crops grown for food and feed globally, exhibits a large phenotypic and genotypic variability (Yan et al. 2009). A great diversity of agro-ecological environments for growing maize contributed to the development of divergent populations adapted to different edaphic and climatic conditions and biological factors, which ultimately manifested in a wide range of morphological, physiological, biochemical, agronomic, and genetic traits (Mikić et al. 2017). Maize has been the first grain crop that makes the greatest contribution to the total grain yield in China, since the rice (Oryza sative L.) production (204.236 million tonnes) was surpassed by that (205.614 million tonnes) of maize for the first time in 2012 (National Bureau of Statistics of China 2013). Harvesting of the 2018 maize crop finalized in October and total output of China is estimated at an average level of 216.500 million tonnes, slightly above last year’s record level (215.891 million tonnes) (FAO 2018). In recent years, new progress has been achieved in the selection and breeding of special purpose hybrid maize with high oil content, high protein content, and high lysine content as well as sweet maize and glutinous maize. These hybrids have not only played an important role in increasing the yield of maize, but also contributed to the development of diversification programs using maize varieties (Zuo 2001).
Sichuan province in Southwest China is located at north latitude 26°03′–34°19′ and east longitude 92°21′–108°12′ and is the fifth largest province in China (Wang et al. 2017). The topography over the whole province differs enormously from east to west with complex and various terrains, resulting in diverse ecological conditions that affect maize production. Maize breeding in Sichuan began in 1930 (Huang 1998a). On the base of collecting local maize germplasm resources, some hybrids were bred in 1950. Up to late 1960, maize breeding turned to single cross breeding and began to select hybrids which were suitable for regional ecological environments. Maize breeding developed faster in late 1970 to early 1980. Sichuan maize breeding program initiated with the 6th 5-year plan in 1981 and then the concept of super high yield breeding in accordance with the ecological diversity of Sichuan maize productive areas, in which new hybrids should be characterized by different maturity(e.g., mid-ripe maize hybrids for plain and hilly areas, early-ripe maize hybrids for summer and mountain areas), resistance to diseases and pests, drought tolerance, longer green persistent duration, full utilization of scattered light and soil fertility (e.g., low N), and suitable for high density plantation, was put forward on the basis of breakthroughs in the cross breeding of maize (Huang 1998b). Considering the narrow genetic base of the local maize germplasm, the breeding program introduced tropical or subtropical maize germplasm such as ETO, Tuxpeno, Suwan, Antigua, Dentado Amarillo, Celaya, Cateto, Hatian, and Tuson from Mexico, Indonesia, South Africa, Thailand and CIMMYT to broaden the genetic base of maize germplasm in the province. However, tropical or subtropical germplasm was not adapted well to the growing conditions in Sichuan, and breeders have to cross the tropical or subtropical germplasm with the local germplasm followed by backcross and self-cross. The introduction and improvement of a wide range of maize germplasm over the years have enabled the provincial maize breeding program to develop hundreds of maize inbred lines and release several hybrids for commercial production.
Hybrids, including inter-specific hybrids, double hybrids, three-way cross hybrids, and top cross hybrids, benefit mainly from the advantage of heterosis in the combination of two or more maize inbred lines. Inbred lines derived from certain unrelated or distantly related groups of breeding materials combine well and produce progeny with outstanding performance (Mumm and Dudley 1994). Groups of breeding materials defined in this manner represent a particular heterotic pattern and are referred to as heterotic groups. A heterotic group is a collection of related inbred lines which tend to produce vigorous hybrids when crossed with lines from a different group, but not when crossed to other lines of the same group (Ertiro et al. 2017). Thus, knowledge of the relationship is essential to the breeder because it directs the exploitation of germplasm. To fully exploit the potential of such breeding material and phenomenon of heterosis, the evaluation of germplasm resource diversities and the genetic relationships among germplasm have been an important part of maize breeding research. Accordingly, maize inbred lines of unknown heterotic response must be assigned to a heterotic group to facilitate both hybrid and line development. Such assignments can be made by assessing the genetic relationships of these inbred lines to lines of known heterotic response (Moll et al. 1965; Moll et al. 1962; Paternaini and Lonnquist 1963).
Molecular characterization of genetic diversity, population structure, and genetic relationships among maize germplasm developed by a given breeding program is useful for understanding how to use the assembled germplasm for further improvement, such as selecting parental lines, assigning heterotic groups, and creating a core set of germplasm (Wu et al. 2016). Simple sequence repeat (SSR) and single-nucleotide polymorphism (SNP) are the most commonly used molecular markers for molecular characterization and assignment of maize inbred lines to heterotic groups (Mir et al. 2013; Semagn et al. 2012b; Wen et al. 2012; Wu et al. 2014; Wu et al. 2016). Currently, SNPs are a marker of choice for a wide range of applications, including molecular characterization analyses, because of their low cost per data point, high genomic abundance, potential for high-throughput analysis, and lower genotyping error rates (Wu et al. 2016). SNP data can be generated using uniplex, multiplex (chip-based), and sequencing methods. Chip-based SNP genotyping methods may cause lower resolution in molecular characterization analyses, and most current chip-based SNPs were developed based on the sequencing information of a set of temperate maize lines, which leads to ascertainment bias of the allele frequency and affects the resolution of genetic diversity and population structure analysis of tropical germplasm collections (Lu et al. 2009).
With the development of new next-generation sequencing (NGS) technologies, including restriction-site associated DNA marker sequencing (RAD) (Miller et al. 2007), reduced representation libraries (RRLs) (Van Tassell et al. 2008), whole-genome resequencing (WGR) (Huang et al. 2009), multiplexed shotgun genotyping (MSG) (Andolfatto et al. 2011), genotyping-by-sequencing (GBS) (Elshire et al. 2011), the discovery of SNP became easier and faster, and the cost per data point were reduced. GBS generates high-density genome-wide markers at a low per sample cost, which is a practical and cost-effective genotyping method for species with large genome size. This makes GBS as a powerful tool for different genetic applications, such as genetic diversity analysis, linkage mapping, association mapping, and genomic selection (Crossa et al. 2013; Poland and Rife 2012; Romay et al. 2013; Zhang et al. 2014).
Sichuan province is one of the main maize production regions in Southwest China. Breeders introduced maize inbred lines outside the province, which were different in their maturity, reaction to a wide range of abiotic and biotic stresses, and general combining ability (GCA). The introduced inbred lines were used by breeders in developing various new and improved maize germplasm with better adaptations to the different ecological conditions and complex geography and land features in the province. However, the pedigree information and heterotic groups of some introduce and improved inbred lines are not properly documented. Since groups of closely related parents tend to bring redundant genetic value to a breeding program, little is known about the genetic diversity, relatedness, and population structure of both the introduced and newly developed inbred lines at the molecular level. Such information is essential for optimizing breeding strategies in the future for germplasm conservation (Sachs 2009) and parent selection for new pedigree starts (Huang et al. 2014).
Few previous similar studies, which contain sufficient number of maize inbred lines used in Sichuan maize breeding program, had been conducted. Several years ago, characterization of the genetic diversity of 770 maize inbred lines obtained from breeding programs in Brazil (94), China (282), and CIMMYT (394) was performed with 1034 SNP markers using a 1536-SNP chip (Lu et al. 2009). Later, the genetic diversity, relatedness, and linkage disequilibrium (LD) decay of 367 inbred lines widely used in maize breeding of China were estimated with 41,819 informative SNPs using the MaizeSNP50 BeadChip with 56,110 SNPs (Wu et al. 2014). Recently, the genetic diversity, relatedness, and population structure among 362 maize inbred lines collected from the breeding program of Southwest China were characterized using 43,735 of the 56,110 SNPs assembled in the MaizeSNP50 BeadChip (Zhang et al. 2016). The two main concerns of the three studies were the high ascertainment bias of the SNP-chips (Schaefer and Bernardo 2013) used for genotyping and the inclusion of just 10, 10, and 33 lines from Sichuan maize breeding program, respectively. Moreover, the common lines are most of the old lines that were bred 10~20 years ago and were clustered into different groups by the three studies, such as 698-3 in Lancaster (Lu et al. 2009) vs. PA (Zhang et al. 2016), D340 in Lvda red cob (Lu et al. 2009) vs. Lancaster (Wu et al. 2014), and Suwan1611 in Tem-tropic I (Wu et al. 2014) vs. Tropical (Zhang et al. 2016). Here, a large number of newly developed maize inbred lines with tropical and subtropical backgrounds from eight breeding institutions, which were specifically and widely used in current Sichuan maize breeding program were genotyped using GBS. The objectives of the present study was to (1) assess the extent of genetic variation, relatedness, and population structure of 157 maize inbred lines and advanced breeding lines, which represent the current maize breeding program in Sichuan province, Southwestern China, using GBS; and (2) to evaluate the usefulness of GBS in predicting the most likely heterotic group membership of each line.
Materials and methods
Plant material
A panel of 157 maize inbred lines and advanced breeding lines (hereinafter referred as lines) from the current breeding program of Sichuan province, Southwest China, was used in this study (Supplementary Table S1). The panel comprised 21 lines from Maize Research Institute of Sichuan Agricultural University (CN), 17 lines from Crop Research Institute of Sichuan Academy of Agricultural Sciences (CZ), 28 lines from Mianyang Academy of Agricultural Sciences (MY), 19 lines from Yibin Academy of Agricultural Sciences (YB), 19 lines from Xichang Agricultural Science Research Institute of Sichuan (XC), 3 lines from Zoeve Seed Co. Ltd. (ZY), 16 lines from Sichuan Tonglu Agricultural Science and Technology Co. Ltd. (TL), 28 lines from Nanchong Academy of Agricultural Sciences (NC), and 6 lines (Dan340, Mo17, B73, Huangzao4, Qi319, and Ye478) with known heterotic groups. The six lines with known heterotic groups were used as testers to assess the heterotic group between these maize lines.
DNA extraction
The entire panel of maize lines was grown at Duoying farm of the Maize Research Institute of Sichuan Agricultural University, Ya’an (EY; 30°N, 103°E), China, in 2017 for DNA extraction. For each line, genomic DNA was extracted from a bulked leaf sample of six seedlings using the cetyltrimethylammonium bromide (CTAB) method (Semagn 2014). The concentration of each DNA sample was checked with Nanodrop 2000 spectrophotometer (Thermo Scientific) and Qubit Fluorometer 2.0 (Life Technologies) as described in the user’s manual and normalized to 100 ng μL−1. To check the quality of the genomic DNA, each DNA sample was digested with 0.5 μg/μL HindIII (Thermo Scientific) for 2 h, and the fragments were ran on 1% agarose gel that contained 0.5 μg/mL ethidium bromide. The final concentration of each DNA sample was adjusted to 100 ng μL−1 for library preparation.
Genotyping by sequencing
Six hundreds nanogram genomic DNA from each line was digested with 5000 units/mL ApeK1 restriction enzyme (4.5-bp recognition site 5′-GCWGC-3′) for 1 h. Then, the library preparation and sequencing were done by the Beijing Genomics Institute (BGI; Shenzhen, China) following the GBS protocol (Elshire et al. 2011). Raw sequence data from the machine were processed in four stringent steps to ensure that reads were reliable in the further analysis: firstly, we allocated the raw reads to its own origin sample according to the barcodes used for sequencing to distinguish samples; secondly, we discarded the raw reads with adapter/barcode sequences to exclude the contamination; thirdly, we ruled out those raw reads with more than 10% unidentified nucleotides (Ns); fourthly, we moved the raw reads with low Phred quality (Q ≤ 5) base number accounted for more than half of the whole reads. Next, the clean reads filtered from each line were aligned to the maize reference genome (Schnable et al. 2009) (B73 RefGen_v4, http://www.maizesequence.org), which has an effective genome length of ~ 2500 Mb (Li et al. 2014), using Short Oligo-nucleotide Alignment Program 2 (SOAP2) software (Li et al. 2009b) (version 2.18, http://soap.genomics.org.cn/). SOAPsnp software (version 1.03) (Li et al. 2009a) was used to call SNPs. For statistical analysis described below, we selected 4976 SNPs with the following criteria: (1) SNPs with missing data points smaller than 50% of the total sample size; (2) reads depth of same position of SNP for each samples at least 5 (Bird et al. 2017); (3) class of genotype (i.e., SNP alleles among all samples) for each positions must be no less than 2; (4) SNPs with a minor allele frequency (MAF) exceeding 0.05.
Genetic diversity analysis and phylogenetic tree
Allele frequency, gene diversity, and polymorphism information content (PIC) for each polymorphic SNP, the proportion of observed heterozygosity in each line, and Nei’s genetic distance (Nei 1972) between pair of inbred lines were calculated using PowerMarker V3.25 (Liu and Muse 2005). A relative kinship matrix was calculated between each pair of lines in the study using Dominance Normalized IBS method implemented in TASSEL 5.2.43 (Bradbury et al. 2007) software. A relative kinship close to 0 indicates no relationship, and a value close to 1 indicates a closer relationship. Matrices of Nei’s genetic distance were used as an input for neighbor-joining (NJ) cluster analysis with Interactive Tree Of Life (iTOL) V4.2 (Letunic and Bork 2016). The genetic distance between pair of groups was estimated based on the average genetic distance of all lines within each group.
Population structure analysis
The population structure of the panel was determined using STRUCTURE 2.3.4 (Pritchard et al. 2000). STRUCTURE was ran using an admixture model by varying the number of groups (K) from 1 to 8, each K repeated five times with a burn-in period of 10,000 and 10,000 MCMC (Markov Chain Monte Carlo) replications after burn-in. Based on the output log likelihood of data (LnP(D)) of STRUCTURE, the ad hoc statistic delta K (ΔK) was used to determine the optimal number of groups (Evanno et al. 2005). Results of five replicate files were integrated using the CLUMPP software (Jakobsson and Rosenberg 2007). Lines with probability of membership > 50% were assigned to the same group, while those lines with < 50% probabilities with any group were assigned to a “mixed group” (Wu et al. 2014).
FST-based pairwise genetic distance matrices (Holsinger and Weir 2009) were computed among different groups produced by STRUCTURE at K = 4 using ARLEQUIN v.3.5.2.2 (Excoffier and Lischer 2010). FST values are indicative of the evolutionary processes that influence the structure of genetic variation among groups, where an FST of 0 indicates no differentiation between groups and a value of 1 indicates complete differentiation. Groups were considered to have little differentiation when FST values were less than 0.05, moderate differentiation when FST values were between 0.05 and 0.15, strong differentiation when FST values were between 0.15 and 0.25, and very strong genetic differentiation when FST values were greater than 0.25 (Ndjiondjop et al. 2017). Analysis of molecular variance (AMOVA) (Excoffier et al. 1992) was also computed using ARLEQUIN v.3.5.2.2 to partition the variation of lines among and within groups.
Linkage disequilibrium analysis
The average linkage disequilibrium (LD) between SNPs on each chromosome was measured using TASSEL 5.2.43 (Bradbury et al. 2007). Squared Pearson correlation coefficient (r2) between vectors of SNP alleles was used to assess the level of LD decay on each chromosome, and The LD decay distance was evaluated as the interval with the mean r2 value decreased below a threshold level r2 < 0.1 (Zhang et al. 2016). The LD decay distances were calculated separately for the entire panel and each group.
Results
Genotypic data and genetic purity
A total of 4976 genome-wide high-quality SNPs were retained for genotyping and genetic characterization analyses, with less than 50% missing data in 157 maize inbred lines. The number of alleles per marker loci among all lines ranged from three to five, transitions and transversions included, resulting actually polymorphic. The 4976 polymorphic SNPs were evenly distributed across the whole maize genome, with coverage ranging from 314 SNPs on chromosome 6 to 894 SNPs on chromosome 1, accounting for 6.3% and 18.0% of the polymorphic SNPs, respectively (Table 1). The physical length of each chromosome varied from 149,802 kb on chromosome 10 to 306,117 kb on chromosome 1, with an average density of one SNP per 440.63 kb (Table 1). Minor allele frequency for the 4976 SNPs varied from 0.05 to 0.50, with an overall average of 0.3647; approximately 88% of the markers (4399 of 4976 SNPs) had a minor allele frequency between 0.25 and 0.50. The PIC values varied from 0.1371 to 0.7107 with a mean value of 0.4132; about 81% of the markers (4008 of 4976 SNPs) had PIC value between 0.25 and 0.50 (Supplementary Table S2).
Using 4976 SNPs, observed heterozygosity among the 157 inbred lines ranged from 0.0 to 40.0%, with an overall average of 4.1%; only 14 lines (8.9% of the 157 lines) had observed heterozygosity > 5.0% (Supplementary Table S1). For different institutions in the breeding program, 16 inbred lines of Sichuan Tonglu Agricultural Science and Technology Co. Ltd. (TL) are all considered pure or fixed with the genetic purity at least 97.3%, followed by Mianyang Academy of Agricultural Sciences (MY), of which 28 inbred lines with the genetic purity at least 93.2% (Fig. 1, Supplementary Table S1).

Genetic purity of 157 inbred lines based on 4976 polymorphic SNPs. The number of inbred lines is shown in brackets on X-axis
Genetic relatedness
Genetic distance between pair of the 157 inbred lines computed from the 4976 polymorphic SNPs ranging from 0.0000 to 2.0702 (Supplementary Table S3), with an overall average of 0.5012. The majority (98.79%) of the pairs of lines had a genetic distance value > 0.400, which is concentrated between 0.501 and 0.800, and is extremely large as compared with just 1.21% of the pairs of lines that had genetic distance value < 0.400 (Fig. 2).

Frequency distribution categories of pairwise genetic distance of 157 maize inbred lines based on 4976 polymorphic SNPs
A neighbor-joining tree generated from the genetic distance matrix of the 4976 SNPs clustered the 157 lines into four groups (Fig. 3, Supplementary Table S1). The groups clustered somewhat according to genetic relationship (i.e., the common ancestral parents for breeding), but not institute/company of origin. The first group (group 1) consisted of 12 lines, which were mostly improved from 08-641 breeding by Maize Research Institute of Sichuan Agricultural University, including T295, T119, Mian 2652, and 08-641 itself; these lines originated from 5 institutes, and the vast majority (41.7%) was from Sichuan Tonglu Agricultural Science and Technology Co. Ltd. The second group (group 2) consisted of 47 lines originating from 8 institutes, of which 17.0% were equal from Maize Research Institute of Sichuan Agricultural University and Nanchong Academy of Agricultural Sciences. The third group (group 3) consisted of 54 lines originating from 8 institutes, and most lines (27.8%) were from Nanchong Academy of Agricultural Sciences. Lines in groups 2 and 3 were manly developed from foreign hybrids with tropical background (e.g., Y78698, Y78573, PN78599, Y7865, CGT-2, CGT-15, Xianyu335, and Xianyu508) and tropical and subtropical germplasm (e.g., TI5604, QR273, AP5, SUWAN1, and CML384). The two groups were separated from each other that is may be due to the genetic distance, which were yielded by crossing or backcrossing in different generations. Group 4 (G4) had a total of 44 lines from 8 institutes, and Mianyang Academy of Agricultural Sciences accounted for the most (34.1%), followed by Yibin Academy of Agricultural Sciences (20.5%). Lines in group 4 were mostly improved from direct self-fertilization and selection of tropical germplasm introduced from Thailand, Mexico, Syngenta, Pioneer, and CIMMYT.

The neighbor joining tree of 157 maize inbred lines based on the pairwise distance matrix using 4976 polymorphic SNPs (G1 = group 1, G2 = group 2, G3 = group 3, G4 = group 4)
The relative kinship reflects the approximate degree of identity between two given individuals. Relative kinship values between pairs of the 157 inbred lines are summarized in Fig. 4. The results showed that no paired relative kinships were equal to zero (less than 0.05); 8.7% of the relative kinship values ranged between 0.101 and 0.500, and the remaining 91.3%, which were concentrated between 0.701 and 0.800, were above 0.500 (Fig. 4, Supplementary Table S4). This analysis indicated that moderate relationship existed in the whole panel of maize germplasm, which was consistent with known pedigree/sources of the 157 lines.

Summary of kinship coefficients between pair of 157 maize inbred lines based on 4976 polymorphic SNPs
Population structure analysis
We explored population structure in the current maize germplasm from Sichuan breeding program using the model-based population structure. The log probability of the data (LnP(D)) showed a linear increase between K = 1 and K = 4 (Fig. 5a), which is also evident from the high ΔK value at K = 4 in Fig. 5b. Based on these two results, the 157 inbred lines can be divided in to four groups (Fig. 5c). The proportion of lines assigned to the four groups predicted from STRUCTURE varied from 5.1% in group 1 to 40.1% in group 2 (Supplementary Table S1). Overall, 148 of 157 lines (94.3%) could be confidently placed within these four groups.

Population structure of 157 maize inbred lines estimated with 4976 SNPs. a The plots of LnP(D) for each K calculated by STRUCTURE, the LnP(D) data was shown as mean ± SD (standard deviation). b Delta K (ΔK) values for K ranging from 2 to 7 according to Evanno et al. c Population structure of the panel when K = 2–6. Each of the 157 lines is represented by a thin vertical bar, which is partitioned into K colored segments on the x-axis, with lengths proportional to the estimated probability membership in each of the K-inferred clusters (y-axis)
Group 1 was the smallest group (8) and contained six lines predominantly sourced from Mianyang Academy of Agricultural Sciences and Sichuan Tonglu Agricultural Science and Technology Co. Ltd.; lines in this group had no unified genetic background, but as we know, they were basically developed from local elite germplasm in China, such as zheng58, 65232, 698-3, and R08. Group 2 consisted of 63 lines, 11 of which were sourced from Xichang Agricultural Science Research Institute of Sichuan, and ten lines were sourced from Nanchong Academy of Agricultural Sciences; many of the lines in this group had tropical or subtropical backgrounds, one or both of their parents used for breeding were from tropical or subtropical germplasm, including PN78599, Y78698, Y78573, QR273, and S37. Group 3 was composed of 57 lines, which were mainly sourced from Nanchong Academy of Agricultural Sciences; lines in this group most were derived from tropical and subtropical germplasm, hybrids of Syngenta, Pioneer and Yogoslavic, population of Suwan1 and CIMMYT included. Group 4 had 20 lines in total; most were sourced from Mianyang Academy of Agricultural Sciences; many of the lines in this group were improved directly from tropical germplasm without circles of cross or backcross and integration of several populations; for example, Mian1838 was selected from the F1 of 652323 × CIMMYT tropical germplasm with 5 generations of continuous self-cross. In addition to the inbred lines that were clearly assigned with membership probability ≥ 0.50 to a single group, 9 inbred lines (5.7% of the total) with lower membership possibility < 0.50 could not be clearly assigned to any of these groups. These lines were placed in a mixed group which sourced mainly from Mianyang Academy of Agricultural Sciences (Supplementary Table S1).
Compassions of groups predicted based on STRUCTURE and cluster analyses
Group membership obtained from STRUCTURE agreed with that of cluster analysis for 112 lines (71.3%), the remaining 45 lines (28.7%) differed in their group membership between cluster analysis and STRUCTURE. Of these, nine lines were assigned in to a mixed group in STRUCTURE. The other 36 lines grouped into different STRUCTURE groups than would have been predicted by cluster analysis, including lines that belong to the STRUCTURE group 1 (1 line), group 2 (20 lines), group 3 (14 lines), and group 4 (1 line) (Supplementary Table S1). Excluding lines that were assigned in to a mixed group, nearly 49% of the lines that showed discrepancy between the cluster analysis and STRUCTURE came from group 4.
The phylogenetic clusters helped to disentangle the genetic background of lines with unknown pedigree. Yi14A2 and Yi12A4 in group 1 of phylogenetic tree were clustered with Mian0232 and T119, respectively, telling us they had the same or nearest ancestral parents; also, hot resistance 67 and 1316 in group 2 of phylogenetic tree were clustered with C381 and (Qi319*CML187)-0(Fig. 3, Supplementary Table S1). However, model-based STRUCTURE analysis in some extent separated lines to different groups according to their heterotic pattern, although some of them are sister lines. Here, tens of commercial hybrids breeding by single cross with lines in our study were listed out (Table 2). From Table 2, we know that hybrids in recent years were developed by crossing lines among groups but not within groups.
With the known pedigree information and group membership based on cluster analysis and the model-based STRUCTURE, group 1 was named Impro-local which indicated that lines in this group were manly improved from local germplasm; group 4 was named Impro-tropic which indicated that lines in this group were manly improved from tropical germplasm; and groups 2 and 3 were named Tem-tropic I A and Tem-tropic I B, respectively, which indicated that lines in the two groups were both developed by cross and backcross with the introduced tropical germplasm in Sichuan maize breeding history, but with different combining ability in heterotic pattern.
Molecular diversity and genetic differentiation
Table 3 summarizes the average PIC, observed heterozygosity, and gene diversity values computed for each group. The Impro-local group showed the highest PIC value; the PICs of the Tem-tropic I A and Impro-tropic groups were similar, and the values were lower than that of the Impro-local and Tem-tropic I B groups. In contrast to the 35.8% heterozygosity rate in the Impro-local group, the heterozygosity rate in the other three groups was 2.5%, 2.5%, and 1.9%, well within the expected ranges for residual heterozygosity found in maize inbred lines. The GDs of the Tem-tropic I B and Impro-tropic groups were similar and higher than that in the Impro-local and Tem-tropic I A groups, which were similar too; the Tem-tropic I A group showed the lowest diversity (0.4475) although the sample size was bigger, and the Tem-tropic I B group showed the highest diversity (0.4573) among groups.
The genetic distance among different groups was estimated with the 4976 SNPs (Table 4); results indicated that the genetic distance between the Impro-local group and other three groups (Tem-tropic I A, Tem-tropic I B, and Impro-tropic) is greater than the genetic distance among Tem-tropic I A, Tem-tropic I B, and Impro-tropic groups. The greatest genetic distance was observed between the Impro-local and the Impro-tropic groups with a value of 0.2126, and the smallest genetic distance was observed between the Tem-tropic I A and the Tem-tropic I B groups with a value of 0.1026. The genetic distance among different groups is consistent with the kinship results.
When pairwise fixation indices (FST) values were compared to understand the extent of genetic differentiation (divergence) between pairs of groups (Table 4), the values were highly variable ranging from 0.0904 (moderate genetic differentiation) to 0.6372 (very great genetic differentiation). Lines among the categorical groups showed moderate genetic differentiation (0.0904–0.1418), except that lines in Impro-local showed great differentiation in Tem-tropic I B (0.1520). Results from partitioning of the overall molecular variance into different hierarchical levels indicated that 14.2% of the total genetic variation occurred among the groups, while a larger amount of variation (85.8%) was within groups.
Linkage disequilibrium decay distance
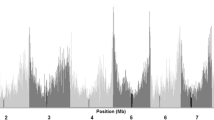
The average r2 of each distance interval was calculated for the entire panel and within chromosomes (Fig. 6). LD decay distance varied across the ten chromosomes. The average LD decay distance over all ten chromosomes in the entire panel with r2 < 0.1 was 1.0–1.5 Mb (~ 1050 kb). Among the chromosomes in the panel, Chr 1 showed the smallest distance and Chr 9 showed the largest. In Tem-tropic I A, Tem-tropic I B, and Impro-tropic group, the average LD decay distances over all ten chromosomes with r2 < 0.1 were 0.50–0.75 Mb, 0.25–0.50 Mb, and 1.00–1.50 Mb, respectively. The Impro-tropic group has the largest LD decay distance, whereas the Tem-tropic I B group has the smallest LD decay distance. Due to limited number of samples in the Impro-local group, the LD decay distance of this group was not estimated.

Mean r2 over different intervals of LD decay distance for the entire panel and each chromosome
Discussion
Genetic purity, genetic distance, and kinship
Genetic purity of inbred lines is an important quality control criteria in maize breeding and seed system, which directly affects both the quality of hybrid seed and development of new inbred lines (Ertiro et al. 2017). Currently, most maize breeding programs consider S4 or later generation as a fixed inbred line for evaluation in hybrid combination. Inbred lines are considered pure or fixed when the proportion of heterozygous SNP loci does not exceed 5% (Semagn et al. 2012a). As a result, most lines were expected to display less than the average observed heterozygosity expected in S5 breeding lines derived from biparental crosses (4.1%). This was seen with ∼ 90% of the lines, but the remaining ∼ 9% (14 of 157 accessions) had observed heterozygosity exceeding 5% (Supplementary Table S1), which is higher than expected in the absence of human error. Inbred lines with higher than 5% heterogeneous SNP loci are likely to have some changes in allele frequencies occurred during seed regeneration, maintenance breeding, and possible contamination with seeds or pollen of other samples (Heckenberger et al. 2002; Warburton et al. 2010). Overall, approximately 93% of lines from Mianyang Academy of Agricultural Sciences, 88% of lines from Crop Research Institute of Sichuan Academy of Agricultural Sciences, 86% of lines from Maize Research Institute of Sichuan Agricultural University, 96% of lines from Nanchong Academy of Agricultural Sciences, 100% of lines from Sichuan Tonglu Agricultural Science and Technology Co. Ltd., 79% of lines from Xichang Agricultural Science Research Institute of Sichuan, 95% of lines from Yibin Academy of Agricultural Sciences, 67% of lines from Zoeve Seed Co. Ltd. were considered fixed. Excluding the higher level of heterogeneity was observed in lines from Zoeve Seed Co. Ltd., of which the total lines were only 3, lines sourced from Xichang Agricultural Science Research Institute of Sichuan had the highest level of heterogeneity, which vary from 12.4 to 40.0%. We therefore suggest additional generations of selfing in order to fix these inbred lines (with the heterozygosity exceeding 5%) to achieve a number of advantages from the use of pure lines, including ease of maintenance of parental lines, high heterosis in hybrids, and ease in quality control during hybrid seed production (Ertiro et al. 2017).
Genetic distance is a measure of the genetic divergence between pairs of lines or populations, with pairs that share many alleles having small genetic distance. Results from the present study showed highly variable levels of genetic distances between pairs of maize inbred lines, with 0.3% of the pairs nearly identical(< 0.05), 0.3% of the pairs similar (0.05–0.20), 5.4% of the pairs moderately distant (0.20–0.50), and 94.0% of the pairs highly distant (> 0.50) (Fig. 1). Our results clearly suggest the presence of a low percentage of redundant lines that contribute very little to the observed genetic variation and genetic divergence in the maize germplasm from the current breeding program of Sichuan. Cluster analysis also demonstrated that lines with small genetic distance were more closely related to each other and showed a near branch on the tree, such as CZ205-1-1 and CZ205-22 in G3, which were both derived from Y7865_F2 × qi205; (Qi319*CML187)-0 and 1316 in G2, although the pedigree of 1316 was unknown (Fig. 3). Selection of parents with good phenotypic performance and wide genetic base is one of the most important steps in the development of new hybrid varieties. Generally, progeny variance increases in crosses between genetically distant parents, which provided opportunities to generate progenies with maximum segregation for target traits that are desired (Cox et al. 1985).
The pairwise relative kinship coefficients among the 157 inbred lines ranged between 0.2495 and 0.8312, where values close to zero indicate lack of relationship, while those close to 2 indicate complete relationship. A total of 91.3% of the paired lines were above 0.5000. This information reflected a moderate relationship among inbred lines in the current breeding program. We infer that different maize breeding institutes/companies in Sichuan province are selecting and improving maize lines from some similar resources. Our kinship coefficient results are relatively higher than those of 450 maize inbred lines developed and released mainly by CIMMYT’s eastern and southern Africa maize breeding programs, among which 79% of the kinship coefficients ranging from 0.05 to 0.50 (Semagn et al. 2012b), and was higher than that of 367 elite inbred lines collected from research institutions or companies in China, among which 94.97% of paired relative kinship ranged from 0.05 to 0.28 (Wu et al. 2014), and much higher than those of 359 inbred maize lines, among which about 60% of the pairwise kinship coefficients were close to zero (Wen et al. 2011), and 544 inbred maize lines, among which 62% of the paired kinship coefficients were equal to zero and only 2% of them were above 0.05 (Wu et al. 2016). Assemblage of maize germplasm with wide genetic divergence developed from different environmental adaptation might have contributed to the observed low level of relatedness among inbred lines in these studies.
Population structure
Using model-based population structure analysis, we observed four groups. It is rather remarkable that the likelihood (Ln) value of this analysis increased to a maximal value, then holding on a stable level no matter how the given Pritchard’s K (the supposed number of subpopulations based on the model) was changed (Fig. 5a). However, likelihood value for each K of previous studies increases continuously with no obvious inflection point. This could imply that the lines included in the analysis were very specific as well as highly homozygous or contained genetically distinct groups. Most lines that are related by heterotic pattern tended to cluster into the same group but not by sources (institute/company), this was in agreement with previous study, where the authors reported a lack of clear clustering patterns in the CIMMYT germplasm based on environmental adaptation or mega-environment (Semagn et al. 2012b). If we rely only on pedigree information, inbred lines in groups 2 and 3 should belong to the same heterotic group (Tem-tropic I). However, the magnitude of heterosis supports the molecular marker grouping. This was supported by tens of commercial hybrids which were bred by single cross with lines among groups (Table 2). The results of these analyses revealed the population structure and clear genetic divergence between Impro-local, Tem-tropic I A, Tem-tropic I B, and Impro-tropic inbred lines. However, our results were different from those of several previous studies as follows: 367 elite maize inbred lines with temperate adaptation collected from research institutions or companies in China were separated into two groups which included local and introduced germplasm and five subgroups, including Reid, Tem-tropic I, P, TSPT, and Lancaster (Wu et al. 2014); 362 diverse inbred lines from the current Southwest China breeding program were divided into two (tropic and temperate), three (tropic, SS and NSS), four (tropic, PA, PB, and Reid), and six (BSSS, Reid, PA, PB, and North) subgroups (Zhang et al. 2016). These differences could be explained by our use of specific germplasm, which were sourced from Sichuan province rather than Southwest China or whole China, resulting in the particular genetic characterization. According to the AMOVA results, 14.2% of the marker variation was explained by the population structure of the panel. This result also suggests the absence of a complicated population structure in our study. The differentiation between the groups was further validated by the pairwise FST value, which indicated that there were moderate and strong genetic differentiations among the four groups (Table 3).
Linkage disequilibrium decay distance
Previous studies have measured LD decay distance in different germplasm collections with various kinds of low-to-medium density genotyping platforms. Compared with previous published values in maize, the average LD decay in this study was slower than that (391 kb) in a collection of 367 inbred lines widely used in maize breeding of China (Wu et al. 2014), and that (0.50–0.75 Mb) of 362 diverse inbred lines from the current Southwest China breeding program (Zhang et al. 2016), with an average LD decay distance of 1.0–1.50 Mb in the entire panel. The LD decay distance measured in this study was much larger than that reported in the China maize collection, this may be caused by the similar population ancestor in maize germplasm in our study, and lines in previous studies are more diverse with sources and contain more rare alleles. Also, the panel of 157 lines in our study contained maize germplasm which consisting several elite lines and its descendants, the increasing relatedness would weaken the LD decay. However, LD level in this study was smaller than that (2.74 cM) in 1537 commercial maize germplasm (Van Inghelandt et al. 2011), because 1 cM in maize genome averagely corresponds to 1460 kb (Civardi et al. 1994). LD is affected by recombination, genetic drift, selection within population, population admixture, and relatedness (Remington et al. 2001). Generally, the LD decay became slow when the population has experienced a long-term domestication history and undergone high selection pressure on different development goals in breeding.
Heterotic group classification with molecular marker
In the current collection, only about 20% of the lines have heterotic information estimated based on pedigree and combining ability information from the provider. Moreover, only a limited number of lines can be included in each combining ability test experiment; it is not possible to estimate the heterotic group and genetic relatedness of all maize lines via one general combining ability test, as heterotic group assignment is made based on combining ability from diallel or line by tester experiments. Molecular marker analyses provide an alternative approach for large-scale genetic diversity characterization within a given germplasm collection. However, it has been reported that the heterotic patterns estimated based on molecular markers are not fully consistent with those estimated based on combining ability tests and pedigree information (Lu et al. 2009; Semagn et al. 2012b; Wen et al. 2012; Wu et al. 2016). In this study, we measured the genetic relatedness among all lines and separate them into four groups, but the results are not consistent with the population structure and genetic differentiation for inbred lines within heterotic groups such as Lancaster (Lancaster Surecrop), Reid (Reid Yellow Dent), Sipingtou (SPT, derivatives from Tangshan Sipingtou Chinese landrace), LRC (derivatives from Lvda red coda Chinese landrace), PA (group A germplasm derived from modern US hybrids), PB (group B germplasm derived from modern US hybrids), and BSSS (Iowa Stiff Stalk Synthetic population) that were established based on combining ability tests by maize breeders. For example, population structure analysis separated testers Mo17, Qi319, and Ye478, which were representative of Lancaster, PB and PA heterotic group, respectively, into one group (Tem-tropic I A), and testers Dan340, B73, and Huangzao4, which were representative of LRC, Reid and SPT heterotic group, respectively, into another group (Tem-tropic I B). Moreover, lines such as 698-3 included in Lancaster heterotic group (Lu et al. 2009) were classified with PA heterotic group (Zhang et al. 2016), resulting in the confused conclusion by different previous studies. The difficulties of assigning lines to different heterotic groups are due to the diverse original and incomplete pedigree information, regardless of the marker system used. Using of different testers across breeding programs are probably the other important reasons, although the development of heterotic groups in temperate maize started around 100 years ago. The same line can be heterotic group A or B depending on the tester used, which may result in mixing up of heterotic groups. But we also found that the lines with the same genetic background tend to cluster together. 78599-211 and Qi319, for instance, which were both derived from Pioneer hybrid 78599, were both clustered into Tem-tropic I A subgroup. This confirmed that heterotic grouping with molecular markers was also somewhat in accordance with the conventional heterotic grouping based on the pedigree. Therefore, combining the current heterotic information based on combining ability tests and the genetic relationships inferred from molecular marker analyses may be the best strategy to define heterotic groups for future maize improvement.
Potential utilization of this heterotic grouping for hybrid breeding
Maize has been grown in China for nearly 500 years since its first introduction into this second biggest production country in the world (Liu et al. 2010). Currently, there are three major production regions throughout the country according to the ecological conditions and farming systems, including the North Spring Maize Region, the Huang-Huai-Hai Summer Maize Region, and the Southwest Maize Region (Fig. 7). In this study, a core set of 157 maize inbred lines (Supplementary Table S1) were specifically collected from Sichuan province, the first big province regarding of maize planting area in the Southwest Maize Region, and divided into Impro-local, Tem-tropic I A, Tem-tropic I B, and Impro-tropic groups for the first time, and lines in the Tem-tropic I groups accounted for the vast majority of proportion. This was consistent with the regional division of maize regions by Monsanto Company, which creatively separated the Southwest Maize Region into sub-tropic (here we called Tem-tropic) and tropic area (Fig. 7).

Maize regions of China. Three main regions to a certain degree can be described by distinct natural and agronomical conditions as far as maize production is concerned. The North Spring Maize Region (upper green part of map) includes the provinces Heilongjiang, Jilin, Liaoning, Inner Mongolia, Gansu, and Xinjiang. The Huang-Huai-Hai Summer Maize Region (central yellow part of map) includes the provinces Tianjin, Hebei, Henan, Shaanxi, Shandong, Jiangsu, Anhui, Hubei, and Shanxi. The Southwest Maize Region (lower red and orange parts of map) includes the provinces Sichuan, Yunnan, Guizhou, Guangxi, Hunan, Hubei and Shaanxi
The original heterotic groups including local and introduced germplasm, Temperate and tropic germplasm was mainly established based on the origin of maize, which were used directly as local × introduced and temperate × tropic without little improvement. Then, the major heterotic groups were proposed as Lancaster, Reid, SPT, Zi330, and E28 in the early 1990s in Northern China, the corresponding heterotic patterns were Reid × SPT, Zi330 × Lancaster, Lancaster × SPT, Lancaster × E28, and Reid × Zi330; subsequently, they became Reid, Tem-tropic I, Zi330, SPT, and Lancaster in the early twenty-first century, where Tem-tropic I was a new heterotic group, which contained tropic maize germplasm; and the corresponding heterotic patterns were Reid × Tem-tropic I, Zi330 × Tem-tropic I, Reid × Zi330, Reid × SPT, and Lancaster × SPT (Teng et al. 2004). Previous study had reported that the widely used heterotic patterns in Northern China such as Lancaster × Reid, Reid × SPT, and Lancaster × SPT were not suitable for hybrid breeding in the Southwest Maize Region such as Sichuan province, and hybrids in this area were mostly bred by the pattern temperate × tropic, in which the Reid, Lancaster, and LRC were included in the temperate (Li and Pan 2005). The results of present study classified Dan340, B73, Huangzao4, Mo17, Qi319, and Ye478, which represent LRC, Reid, SPT, Lancaster, PA, and PB heterotic group in Northern China, respectively, into Tem-tropic I heterotic group with A and B subgroups, since tropical germplasm from CYMMIT and SUWAN has been introduced for temperate germplasm improvement in this area. So, we propose that the Tem-tropic × Tropic and Tem-tropic × Tem-tropic instead of temperate × tropic were the current main heterotic pattern for hybrid maize breeding in Sichuan province, and the A and B subgroups in the Tem-tropic I heterotic group should be properly utilized in the selection of parental germplasm and population improvement.
Conclusions
A subset of 4976 high-quality SNPs were generated by GBS from this work. The SNP markers were used to evaluate the population structure, genetic diversity, and relatedness of 157 important inbred lines specifically collected from the current breeding program of Sichuan province, Southwest China. We identified four groups according to the bioinformatic study and breeding experience; it illustrated that germplasm of different heterotic groups is not mutually exclusive. The genomic characterization presented here depicted a clear insight about the genetic diversity of maize germplasm from the breeding program, and analysis in our study is an essential step towards a future exploitation of these germplasm resources in hybrid breeding. Overall, we concluded and recommended that GBS is a cost-effective primary tool in application to genetic diversity analysis for maize. In future work, we intend to apply GBS routinely to genotype and select among full number of advanced maize breeding lines.
References
Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, Stern DL (2011) Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res 21:610–617
Bird KA, An H, Gazave E, Gore MA, Pires JC, Robertson LD, Labate JA (2017) Population structure and phylogenetic relationships in a diverse panel of Brassica rapa L. Front Plant Sci 8
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Civardi L, Xia Y, Edwards KJ, Schnable PS, Nikolau BJ (1994) The relationship between genetic and physical distances in the cloned a1-sh2 interval of the Zea mays L. genome. Proc Natl Acad Sci 91:8268–8272
Cox TS, Kiang YT, Gorman MB, Rodgers DM (1985) Relationship between coefficient of parentage and genetic similarity indices in the Soybean1. Crop Sci 25:529–532
Crossa J, Beyene Y, Kassa S, Perez P, Hickey JM et al (2013) Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3: genes, genomes. Genetics 3:1903–1926
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Ertiro BT, Semagn K, Das B, Olsen M, Labuschagne M, Worku M, Wegary D, Azmach G, Ogugo V, Keno T, Abebe B, Chibsa T, Menkir A (2017) Genetic variation and population structure of maize inbred lines adapted to the mid-altitude sub-humid maize agro-ecology of Ethiopia using single nucleotide polymorphic (SNP) markers. BMC Genomics 18:777
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour 10:564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restrictiondata. Genetics 131:629–639
FAO (2018) GIEWS-Global Information and Early Warning System/Country Briefs/China (2018). http://www.fao.org/giews/countrybrief/country.jsp?code=CHN (accessed 01.12 18)
Heckenberger M, Bohn M, Ziegle JS, Joe LK, Hauser JD, Hutton M, Melchinger AE (2002) Variation of DNA fingerprints among accessions within maize inbred lines and implications for identification of essentially derived varieties. Mol Breed 10:181–191
Holsinger KE, Weir BS (2009) Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet 10:639–650
Huang Y (1998a) Achievement and Progress of maize cross breeding in Sichuan Province. Southwest China J Agr Sci 11:31–37
Huang Y (1998b) Progrees in Sichuan maize breeding and exploration of super high yield breeding. Southwest China J Agr Sci 11:47–53
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan J, Fan D, Weng Q, Huang T, Dong G, Sang T, Han B (2009) High-throughput genotyping by whole-genome resequencing. Genome Res 19:1068–1076
Huang P, Feldman M, Schroder S, Bahri BA, Diao X, Zhi H, Estep M, Baxter I, Devos KM, Kellogg EA (2014) Population genetics of Setaria viridis, a new model system. Mol Ecol 23:4912–4925
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245
Li G, Pan G (2005) The utilization present situation and study advances of the germplasm in southwest maize zone. J Maize Sci 13:3–7
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J (2009a) SNP detection for massively parallel whole-genome resequencing. Genome Res 19:1124–1132
Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, Wang J (2009b) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li Q, Eichten SR, Hermanson PJ, Zaunbrecher VM, Song J, Wendt J, Rosenbaum H, Madzima TF, Sloan AE, Huang J, Burgess DL, Richmond TA, McGinnis KM, Meeley RB, Danilevskaya ON, Vaughn MW, Kaeppler SM, Jeddeloh JA, Springer NM (2014) Genetic perturbation of the maize Methylome. Plant Cell 26:4602–4616
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu Z-Z, Guo R-h, J-r Z, Y-l C, F-g W et al (2010) Analysis of genetic diversity and population structure of maize landraces from the south maize region of China. Agric Sci China 9:1251–1262
Lu Y, Yan J, Guimarães CT, Taba S, Hao Z, Gao S, Chen S, Li J, Zhang S, Vivek BS, Magorokosho C, Mugo S, Makumbi D, Parentoni SN, Shah T, Rong T, Crouch JH, Xu Y (2009) Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet 120:93–115
Mikić S, Kondić-špika A, Brbaklić L, Stanisavljević D, Ćeran M, Trkulja D, Mitrović B (2017) Molecular and phenotypic characterisation of diverse temperate maize inbred lines in Southeast Europe. Zemdirbyste-Agriculture 104:31–40
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17:240–248
Mir C, Zerjal T, Combes V, Dumas F, Madur D, Bedoya C, Dreisigacker S, Franco J, Grudloyma P, Hao PX, Hearne S, Jampatong C, Laloë D, Muthamia Z, Nguyen T, Prasanna BM, Taba S, Xie CX, Yunus M, Zhang S, Warburton ML, Charcosset A (2013) Out of America: tracing the genetic footprints of the global diffusion of maize. Theor Appl Genet 126:2671–2682
Moll RH, Salhuana WS, Robinson HF (1962) Heterosis and genetic diversity in variety crosses of maize. Crop Sci 2:197–198
Moll RH, Lonnquist JH, Velez Fortuno J, Johnson EC (1965) The relationship of Heterosis and genetic divergence in maize. Genetics 52:139–144
Mumm RH, Dudley JW (1994) A classification of 148 U.S. maize Inbreds: I. Cluster analysis based on RFLPs. Crop Sci 34:842–851
National Bureau of Statistics of China N (2013) China Statistic Yearbook. 13–2 Output of Agriculture, Animal Husbandry and Fishery http://www.stats.gov.cn/tjsj/ndsj/2013/indexeh.htm
Ndjiondjop M-N, Semagn K, Gouda AC, Kpeki SB, Dro Tia D, Sow M, Goungoulou A, Sie M, Perrier X, Ghesquiere A, Warburton ML (2017) Genetic variation and population structure of Oryza glaberrima and development of a mini-Core collection using DArTseq. Front Plant Sci 8
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Paternaini E, Lonnquist JH (1963) Heterosis in interracial crosses of corn (Zea mays L.). Crop Sci 2:504–507
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. The Plant Genome 5:92–102
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci 98:11479–11484
Romay MC, Millard MJ, Glaubitz JC, Peiffer JA, Swarts KL, Casstevens TM, Elshire RJ, Acharya CB, Mitchell SE, Flint-Garcia SA, McMullen MD, Holland JB, Buckler ES, Gardner CA (2013) Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol 14:R55
Sachs MM (2009) Cereal germplasm resources. Plant Physiol 149:148–151
Schaefer CM, Bernardo R (2013) Population structure and single nucleotide polymorphism diversity of historical Minnesota maize Inbreds. Crop Sci 53:1529–1536
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ, McMahan L, van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS, Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z, Nagel DH, McCann MC, SanMiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC, Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, McCombie WR, Wing RA, Wilson RK (2009) The B73 maize genome: complexity, diversity, and dynamics. Science (New York, NY) 326:1112–1115
Semagn K (2014) Leaf tissue sampling and DNA extraction protocols. In: Besse P (ed) Molecular plant taxonomy: methods and protocols, vol 1115. Human Press, New York
Semagn K, Beyene Y, Makumbi D, Mugo S, Prasanna BM, Magorokosho C, Atlin G (2012a) Quality control genotyping for assessment of genetic identity and purity in diverse tropical maize inbred lines. Theor Appl Genet 125:1487–1501
Semagn K, Magorokosho C, Vivek BS, Makumbi D, Beyene Y, Mugo S, Prasanna B, Warburton ML (2012b) Molecular characterization of diverse CIMMYT maize inbred lines from eastern and southern Africa using single nucleotide polymorphic markers. BMC Genomics 13:113
Teng W, Cao Q, Chen Y, Liu X, Men S, Jing X, Li J (2004) Analysis of maize heterotic groups and patterns during past decade in China. Sci Agric Sin 37:1804–1811
Van Inghelandt D, Reif JC, Dhillon BS, Flament P, Melchinger AE (2011) Extent and genome-wide distribution of linkage disequilibrium in commercial maize germplasm. Theor Appl Genet 123:11–20
Van Tassell CP, Smith TPL, Matukumalli LK, Taylor JF, Schnabel RD et al (2008) SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods 5:247–252
Wang Q, Huang C, Sun Y(2017) Selection of a research area to construct a virtual case for integrated risk assessment of earthquake and flood. In: RISK ANALYSIS AND MANAGEMENT–TRENDS, CHALLENGES AND EMERGING ISSUES, p 131
Warburton ML, Setimela P, Franco J, Cordova H, Pixley K, Bänziger M, Dreisigacker S, Bedoya C, MacRobert J (2010) Toward a cost-effective fingerprinting methodology to distinguish maize open-pollinated varieties. Crop Sci 50:467–477
Wen W, Araus JL, Shah T, Cairns J, Mahuku G, Bänziger M, Torres JL, Sánchez C, Yan J (2011) Molecular characterization of a diverse maize inbred line collection and its potential utilization for stress tolerance improvement. Crop Sci 51:2569–2581
Wen W, Franco J, Chavez-Tovar VH, Yan J, Taba S (2012) Genetic characterization of a Core set of a tropical maize race Tuxpeño for further use in maize improvement. PLoS One 7:e32626
Wu X, Li Y, Shi Y, Song Y, Wang T, Huang Y, Li Y (2014) Fine genetic characterization of elite maize germplasm using high-throughput SNP genotyping. Theor Appl Genet 127:621–631
Wu Y, San Vicente F, Huang K, Dhliwayo T, Costich DE, Semagn K, Sudha N, Olsen M, Prasanna BM, Zhang X, Babu R (2016) Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theor Appl Genet 129:753–765
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One 4:e8451
Zhang X, Pérez-Rodríguez P, Semagn K, Beyene Y, Babu R et al (2014) Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity 114:291
Zhang X, Zhang H, Li L, Lan H, Ren Z, Liu D, Wu L, Liu H, Jaqueth J, Li B, Pan G, Gao S (2016) Characterizing the population structure and genetic diversity of maize breeding germplasm in Southwest China using genome-wide SNP markers. BMC Genomics 17:697
Zuo M (2001) CROP DIVERSIFICATION IN CHINA. Paper presented at the Crop Diversification in the Asia-Pacific Region, Bangkok, Thailand
Acknowledgments
We thank the team members of the Disruptive Materials and Methods Innovation in Maize Breeding in the Sichuan Science and Technology Support Project for providing the maize inbred lines.
Funding
Financial support was received from the National Key Research and Development Program of China (2016YFD0101206) and the Sichuan Science and Technology Support Project (2016NZ0054).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Leng, Y., Lv, C., Li, L. et al. Heterotic grouping based on genetic variation and population structure of maize inbred lines from current breeding program in Sichuan province, Southwest China using genotyping by sequencing (GBS). Mol Breeding 39, 38 (2019). https://doi.org/10.1007/s11032-019-0946-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0946-y