
Abstract
The development of a high-throughput genotyping platform with high quality, flexibility, and affordable genotyping cost is critical for marker-assisted breeding. In this study, a genotyping by target sequencing (GBTS) platform was developed in maize, which can be realized for a small number of markers (several to 5 K) through multiplex PCR (GenoPlexs) and for a large number of markers (1 to 45 K) through in-solution capture. The later was used for development of four SNP marker panels (GenoBaits Maize) containing 20 K, 10 K, 5 K, and 1 K markers. Two genotype panels, one consisting 96 representative worldwide maize inbred lines and the other containing 387 breeding lines developed in our maize breeding programs, were used to test and validate the developed marker panels. First, a 20 K SNP panel, with markers evenly distributed across maize genome, was developed from a 55 K SNP array with improved genome coverage. From this single marker panel, 20 K, 10 K, 5 K, and 1 K SNP markers can be generated by sequencing the samples at the average sequencing depths of 50×, 20×, 7.5×, and 2.5×, respectively. Highly consistent marker genotypes were obtained between the four marker panels and the 55 K array (over 95%) and between two biological replications (over 98%). Also, highly consistent phylogenetic relationships were generated by using four marker panels and two genotype panels, providing strong evidence for the reliability of SNP markers and GBTS genotyping platform. Cost-benefit analysis indicated that the genotypic selection cost based on the GBTS in maize was lower than phenotypic selection, allowing GBTS an affordable genotyping platform for marker-assisted breeding. Integration of this affordable genotyping platform with other breeding platforms and open-source breeding network would greatly facilitate the molecular breeding activities in small- and medium-size companies and developing countries. The four marker panels could be used for many fields of marker application, including germplasm evaluation, genetic mapping, marker-assisted selection (including genomic selection), and plant variety protection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Conventional plant breeding has been largely based on phenotypic selection rather than genotypic selection. There is a certain relationship between individual plant phenotype and genotype, but the phenotype of a plant cannot be predicted accurately by its genotype. Factors such as environmental conditions, gene interaction, and genotype by environment interaction affect the response to phenotypic selection. With the development of molecular biology, a series of powerful tools has been invented for plant breeding, one of which is marker-assisted selection (MAS). Compared to phenotypic selection, MAS has the following advantages (Tanksley et al. 1989; Xu and Crouch 2008): (1) MAS can shorten breeding cycles, improve breeding efficiency, and save a lot of labor and resources, and thus increase the total genetic gain per year (Xu et al. 2017a); (2) molecular markers can be detected at all stages of crop growth, greatly facilitating selection; (3) MAS does not depend on specific environments or developmental stages that influence the expression of the target genes (for example, disease or pest resistance can be only identified phenotypically in specific environments or conditions); (4) MAS is cost-effective as some traits (such as quality) are very expensive to phenotype; and (5) MAS allows selection in off-season nurseries, making it possible to grow more crops per year and thus shorten the breeding cycle time. Because of these advantages, MAS can be used in evaluation of germplasm resources, gene transfer by backcrossing, pyramiding genes, and selection in early generations (Collard and Mackill 2008). Among all the types of molecular markers, SNPs have more advantages due to their whole genome coverage and high-throughput and automatic genotyping, becoming the first choice in MAS.
However, a major constraint to the implementation of MAS in breeding practice has been its perceived high relative cost (Kuchel et al. 2005; Xu and Crouch 2008). The high-genotyping cost is the main reason for the application of molecular marker technology to become the “patent” of a few multinational seed companies. These companies have gained technical and market advantages through the construction of comprehensive technical platform, large-scale application, equipment, reagents, and materials needed to obtain genotyping technology at lower cost (Bernardo 2008). Molecular marker technology and its breeding application have become the main weapon of multinational companies leading the world after genetically modified organism (GMO) (Prasanna et al. 2010). The wide application of molecular marker technology in small- and medium-sized seed companies and developing countries (except for molecular breeding studies supported by national grants) is largely dependent on low-cost marker techniques, molecular breeding networks (systems), and molecular markers with important functions.
Although chip-based genotyping platform is widely used in genetics, its application in crop breeding is limited because of low customization efficiency, higher cost, expensive equipment, and less flexibility (Rasheed et al. 2017). The development of ultra-high-throughput, cost-effective genotyping platforms for practical breeding are becoming increasingly important. Next-generation sequencing (NGS) technologies have been used for genotyping through whole genome resequencing (Huang et al. 2010) and reduced-representation sequencing (Davey et al. 2011). In the latter case, genomic regions are selected from DNA samples before sequencing by restriction enzymes (Baird et al. 2008; Elshire et al. 2011) or targeted capture strategies (Davey et al. 2011; Mamanova et al. 2010). The restriction-enzyme-based strategies could produce unbiased, randomly-distributed, genome-wide markers. However, to simplify the genotyping pipeline, routinize bioinformatics procedure, and generate comparable marker data across laboratories, targeted-enrichment methods are a better option. High-throughput and low-cost sequencing will result in a full genome capture of genetic variation that will facilitate marker development (Xu et al. 2017a). Genotyping by target sequencing (GBTS), as one of the targeted sequence-capture strategies, has the characteristics of customized flexibility, high flux, low target sequencing cost, and accurate sequencing results. GBTS is mainly divided into multiplex PCR-based target sequencing and probe-in-solution-based target sequencing. Now, it includes three representative technologies, AmpliSeq (Li et al. 2017), NimbleGen (Krasileva et al. 2017), and SureSelect (Jiang et al. 2014). Both NimbleGen and SureSelect are probe-in-solution-based target enrichment. The probes in SureSelect are RNA with the length between 114 and 126 bp, while the probes in NimbleGen are DNA with the length between 55 and 105 bp. Both technologies could be used for whole exon capture. AmpliSeq, developed by Thermo Fisher, is multiplex PCR-based target enrichment, which is easier to use with lower cost but a smaller capture-region than the probe-in-solution-based. The targeted sequence technology has been widely used in medical applications (Yang et al. 2013), but rarely been reported in agriculture species.
Using GBTS, we developed in this report a series of high-quality SNP marker panels, including 20 K, 10 K, 5 K, and 1 K loci, from the previous 55 K SNP array with improved genome coverage and suitable for application in both temperate and tropical maize germplasms (Xu et al. 2017b). By in-solution capture, we evaluated these marker panels for their cost, efficiency, biological and sample replicabilities, and applications using 96 representative maize inbred lines and 387 maize breeding lines.
Materials and methods
Plant materials and DNA extraction
To evaluate the developed marker panels, two maize genotype panels were used. The first panel (96-genotype panel) contains 96 diverse maize inbred lines representing temperate (69) and tropical/subtropical (27) germplasms from China, USA, and CIMMYT. Several core inbred lines, B73, Mo17, Zi330, Huangzao4, Q319, and Ye478, were selected to represent the major heterotic groups identified in maize breeding programs (Liu et al. 2003; Lu et al. 2009; Wu et al. 2014; Xu et al. b). Tropical or subtropical materials were selected from diverse tropical populations or pools. For example, CML114, CML115, and CML116 are from Pop45; CML84 are from Pool32, and CML99 are from Pop42. The second genotype panel (387-genotype panel) contained 387 breeding materials from our ongoing breeding programs at Chinese Academy of Agricultural Sciences (CAAS). Some Chinese core inbred lines were also included as controls. Detailed information about the two genotype panels is provided in Supplementary Tables 1 and 2.
Eight seeds from each inbred line were seeded in one pot, and five leaves were sampled and mixed from different plants at three-leaf stage. The genomic DNA was extracted by cetyltrimethylammonium bromide (CTAB) method (Saghai-Maroof et al. 1984), and the quality and concentration of DNA were examined by agarose gel electrophoresis and NanoDrop. To evaluate biological replicability, four inbred lines, Ye478, Qi319, CML428, and DTMA241, were selected from the 96-genotype panel as biological replications for genotyping twice using the same DNA sample.
Selection of SNP loci for development of marker panels
A total of 24,495 SNP loci were first selected from the previous 55 K array, by the way to select 1 K core set (Xu et al. 2017b).
We then designed a set of the probes each with 120 nt long to cover marker regions. The specificity of all probes from each marker region on the reference genome was assessed. At the end, each marker locus was captured by 3~4 cross-covered probes. The probe set was synthesized by semiconductor based in-situ-synthesis process. According to the results of 96-genotype panel, the 24,495 SNP loci were ranked by the average missing rate per locus and the average sequencing depth. The last 3495 loci were removed, leaving 21,000 loci. Finally, according to uniform distribution, 20 K loci were selected as the 20 K SNP panel.
From the 20 K SNP marker panel, we developed a series of marker panels including 10 K, 5 K, and 1 K SNP markers by the following the same steps above. We randomly selected 200 samples from the 483 tested genotype samples, which were used to select 10 K, 5 K, and 1 K SNP markers according to their sequencing depths and distributions on maize chromosomes. The higher sequencing depths can be captured at a marker locus, the less overall sequencing depths will be required in future genotyping and thus the lower minimum genotyping cost will be required to have the marker missing scores controlled under the threshold. That means, the same set of 20 K marker probes can be used to generate 10 K, 5 K, and 1 K SNP markers, by sequencing at different depths. Generating more markers will need higher sequencing depths.
DNA library construction and probe hybridization
We followed Samorodnitsky et al. (2015), Mamanova et al. (2010), and Jianan Zhang (personal communication) for DNA library construction and probe hybridization.
DNA library was constructed from each plant sample mainly by the following four steps:
-
1.
DNA fragmentation: which was processed by Ultrasonic;
-
2.
End Repair and adding A-tail: use End Repair reagent to repair the end of fragmented DNA and add A tail;
-
3.
Adapter ligation: Y-type adapter was ligated with fragmented DNA by T4 ligase;
-
4.
Library amplification: ligated DNA was amplified by barcode primers and a high-fidelity DNA polymerase. Samples were distinguished by labeling with different barcode primers.
Probe hybridization included the following six steps:
-
1.
Library mixture: DNA libraries, maize special blocker, and universal adapter blocker were mixed in a tube, which was vacuumed into dry powder;
-
2.
Library hybridization: probes and hybridization buffer were put in the tube, and placed on PCR at 65° to hybrid 16 h;
-
3.
Target capture: Dynabeads® MyOne™ Streptavidin C1 and binding buffer were put in hybrid product to selectively pick up the target fragments but remove the non-target ones;
-
4.
Library amplification: the selected fragments were amplified by library amplification primer and a high-fidelity DNA polymerase;
-
5.
Product purification: two rounds of purification were performed using Beckman AMPure Beads;
-
6.
Library quality check: qubit and qPCR were used to quantify the library concentration and sequencing was done with illumina HiSeq X.
In silico analysis of sequence data
Sequence data generated by probe-in-solution target sequencing are subjected to in silico analysis as follows: the sequencing data were first checked for quality control; two-terminal reads were merged using FLASH, and sequencing data were then compared with the reference B73 genome using BBMap. The alignment results were saved in SAM/BAM (binary alignment map) format. To compare the repetitive sequences generated by PCR, the alignment results were processed using Picard software, and the final results were saved as BAM files for further analyses. Finally, SNP variants were detected by FreeBayes.
Marker data analysis
The frequency of heterozygotes reflects the proportion of heterozygous loci detected in each maize line. The polymorphic information content (PIC), described by Botstein et al. (1980), was used to refer to the relative value of each marker with respect to the amount of polymorphism exhibited. PIC is estimated as:
where Pi and Pj are the population frequencies of the ith and the jth allele. Gene diversity (GD) is relevant to the sum of squares of allele frequencies and estimated as:
where Pi is frequencies of the ith allele. Distribution of markers on chromosomes was mapped using “CMplot” package in R language.
Neighbor-joining (NJ) trees (Saitou and Nei 1987) were constructed by software TASSEL 5.2.23, and heterotic groups were identified according to the phylogenetic tree. The genetic distance between heterotic groups was estimated based on the average pairwise nucleotide diversity of all lines within each group.
Cost-benefit analysis
The costs associated with the use of conventional phenotypic selection and genotypic selection with different marker panels were estimated by following Dreher et al. (2003). The genotypic selection cost for different marker panels includes DNA extraction, construction of library, probe hybridization, sequencing, bioinformatics analysis, depreciation, and field and laboratory labors. The phenotypic selection cost includes land rent, chemicals, fuel, tractor plowing, travel costs, field management, supplies, irrigation, and labor. Three experimental stations in China, Shunyi (Beijing), Xinxiang (Henan), and Sanya (Hainan), where phenotyping was performed, were taken as field trial examples to calculate the cost for phenotypic selection.
Results
High-quality genotyping achieved
Firstly, we compared the genotyping results of the 96 representative germplasms among different SNP marker panels and the previous 55 K SNP array. For the 20 k marker panel, the average consistency with the 55 K SNP array was 95.31%, ranging from 93.11 to 98.91%, compared to 97.35% (95.78–99.10%), 97.82 (96.08–98.97%), and 99.03% (97.34–99.87%) for 10 K, 5 K, and 1 K panels, respectively, indicating that the current genotyping platform is highly reliable and the consistency increased with the SNP panels captured by higher sequence depths and containing less markers. A high level of concordant genotype calls (97.96 to 98.44%) was observed between two biological replications for each of the four tested inbred lines. For the 20 K marker panel, the average heterozygosity is 0.56% (0.01–2.56%), compared to 0.49% (0.06–2.34%), 0.44% (0.04–2.11%), and 0.36% (0–1.94%) for 10 K, 5 K, and 1 K panels, respectively. The proportion of the markers with MAF (minor allele frequency) less than 0.1 was 6.30% for the 20 K marker panel (Supplementary Fig. 1), compared to 4.27%, 3.85%, and 4.94% for 10 K, 5 K, and 1 K panels, respectively. The proportion of the markers with MAF less than 0.05 is 1.5% for the 20 K marker panel, compared to 0.30%, 0.29%, and 0.41% for 10 K, 5 K, and 1 K panels, respectively. The results indicate that our marker panels produced much lower missing and heterozygous rates than the 55 K array, which had 1.83% and 0.70% missing and heterozygous rates when different experimental batches were combined. On the other hand, our marker panels generated much higher MAFs.
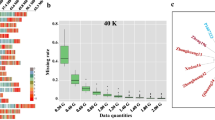
For different marker panels, the missing rate was calculated for different data/sequence quantities (Fig. 1). In general, as the sequencing quantity increased, the missing rate gradually decreased. For the same data quantities, the missing rate decreased with the decrease of marker numbers included in the marker panels. Considering 20 K SNPs, each requiring a 300 bp read to capture the marker region, and the desirable sequencing depths of 100×, the sequence quantity required is 600 Mb (20 K × 300 bp × 100). To control the missing rate below 2.0%, 1000 M bp must be sequenced for the 20 K marker panel, compared to 300 M, 200 M, and 100 M bp for 10 K, 5 K, and 1 K panels, respectively (Fig. 1). When the number of markers was reduced by half in the marker panels, sequencing data could be reduced to as low as 30–40%.

Data missing rates for different sequencing depths and four marker panels using the 200 selected maize inbred lines. The x axis represents the amount of data quantities, and the y axis is the missing rate
Chromosomal distribution of SNP markers and their polymorphic information
The distribution of markers on chromosomes was mapped on the maize genome B73 RefGen_v2. Each chromosome was divided into windows with 1 Mb, and then the number of SNP was counted in each window. For each marker panel, SNP markers almost covered the whole genome, while they were relatively abundant near telomeric regions and relatively sparse around centromeres (Fig. 2). This is consistent with the distribution of genes on chromosomes. The numbers of informative SNPs across 10 chromosomes ranged from 1396 (chromosome 10) to 3249 (chromosome 1) for the 20 K marker panels, compared to 694 to 1559, 347 to 774, and 64 to 149 for 10 K, 5 K, and 1 K marker panels, respectively.

Distribution of SNP markers on 10 maize chromosomes for four marker panels. The marker density is indicated by different bar colors, and each bar represents 1-Mb window size
We calculated PIC for each marker and marker panels. The average of PIC was 0.298 (0.000–0.499), 0.303 (0.000–0.473), 0.303 (0.000–0.473), and 0.303 (0.000–0.422) for 20 K, 10 K, 5 K, and 1 K marker panels, respectively (Supplementary Fig. 2). No significant difference in PIC was found among chromosomes (0.274–0.304) or between the germplasms from tropics (0.278) and temperate (0.293), which are comparable to those revealed by our previous 55 K SNP array with an averaged PIC of 0.281. For the 20 K marker panel, 57.5% of the markers had PIC values greater than 0.300 while only 0.84% of the markers had PIC values less than 0.050. Average gene diversity among inbred lines were 0.375 (0.021–0.500) for the 20 K marker panel, compared to 0.380 (0.000–0.500), 0.381 (0.000–0.500), and 0.380 (0.000–0.500) for 10 K, 5 K, and 1 K marker panels, respectively (Supplementary Fig. 3). The results indicate that these marker panels can be used in genetics and breeding studies with diverse maize germplasms. For example, when the 20 K marker panel is used for gene mapping with biparental populations, the number of polymorphic markers will be 36.16% on the average, ranging from 2.1 to 50.0%, which could provide a relatively fine mapping result. The 10 K and 5 K marker panels, which had averaged polymorphic markers of 38.0% and 38.1%, respectively, can be used for preliminary mapping.
Phylogenetic relationship and heterotic groups revealed by different marker panels
Neighbor-joining trees were constructed with the 96-genotype panel and different marker panels. Almost an identical phylogenetic relationship was identified by the four marker panels with eight heterotic groups revealed, that is, Reid, SPT, LRC, Lancaster, PA, PB, Iodent, and Tropical (Fig. 3). The phylogenetic tree and heterotic groups are consistent with those established based on the pedigree information and breeders’ experience on inbreds’ combining ability. As the 1 K marker panel provided almost identical results to other marker panels with much more markers, it can be used as the best marker panel for construction of phylogenetic trees and heterotic groups to minimize the genotyping cost as we will discuss later (Fig. 3).

Phylogenetic trees constructed for the 96 representative maize inbred lines (96-genotype panel) using four marker panels. The phylogenetic relationship reveals the heterotic groups existing in Chinese maize breeding programs
The genetic diversity as an important index in evaluating marker performance was examined using average nucleotide difference for different maize groups identified by marker panels (Supplementary Table 3). For the 20 K marker panel, the average genetic diversity ranged from 0.346 (between PA and PB) to 0.408 (between Reid and SPT). The genetic diversity between Lancaster and Reid is also very high (0.405), which matches up with the typical heterotic pattern with two most widely used germplasm groups in breeding and production in the USA. Compared with the 20 K marker panel, other marker panels containing less markers detected higher levels of genetic diversity, and the overall nucleotide diversities across 96 representative inbred lines for 20 K, 10 K, 5 K, and 1 K marker panels are 0.362, 0.382, 0.383, and 0.387, respectively (Supplementary Fig. 4). As reveal by the pairwise nucleotide diversity within group, the tropical germplasm had the highest level of genetic diversity (Supplementary Table 3), which is in line with the fact that tropical maize hosts a higher level of genetic diversity.
The marker panels validated using breeding materials
The marker panels were validated for their values using the 387-genotype panel containing breeding materials. The missing rate per sample ranged from 0 to 10.0%, with an average of 3.2%, which is not significantly different from what was revealed by the 96-genotype panel (1.6%). The average heterozygosity is 4.3% (0.1–43.2%), higher than that of the 96-genotype panel (0.6%), indicating a much higher level of remaining heterozygosity in the breeding materials. Clustering analysis using different marker panels showed that the composition of the breeding materials was more complex and could be classified into more diverse groups (Supplementary Fig. 5). Firstly, the 96 inbred lines, when clustered along with 387 maize lines, were also clearly classified into eight separate groups, which is corresponding to the groups identified by the 96-genotype panel alone. Secondly, some breeding materials were also fit into these eight groups. For example, WH119, WH120, WH121, WH122, and WH123 were classified into the same tropical group with different marker panels, which is consistent with the fact that these germplasm accessions were bred with tropical lines as parents. WH016, WH099, and WH310, as inbred lines bred from B73, were all classified into Reid group, while WH217, WH224, and WH380, as inbred lines bred from a widely used inbred Huangzao 4, were all classified into SPT group, by different marker panels (Supplementary Fig.5). Therefore, the marker panels have been validated successfully with the 387-genoptype panel containing diverse breeding materials.
Cost advantages of MAS over phenotypic selection
The objectives of MAS are not only to improve selection efficiency, but also to reduce selection cost. The cost associated with MAS discussed in this study includes four major components, that is, DNA library construction, probe hybridization, sequencing, and bioinformatic analysis (Table 1). For different marker panels, the cost involved in the first two steps was almost the same ($6.61 and $3.00 per sample, respectively) and the cost for bioinformatics analysis can be largely ignored once the server and hard facilities have been set up, but we considered a depreciation cost ($0.40 per sample) (Table 1). So, the cost difference among the marker panels is largely determined by sequencing, the generation of sequence data. High-throughput facilities available have significantly reduced the cost in DNA extraction. The costs per sample for the four marker panels currently involved in sequencing are $15.01, $6.01, $2.25, and $0.75, respectively (Table 1). The total costs per sample for the four marker panels ranged from $12.76 to $27.02 (Table 1).
The cost of phenotypic evaluation is location-dependent, largely due to the differences in land rent, labor, and traveling (Supplementary Table 4). Taking our Fusarium verticillioides ear rot resistance screening as an example, 509 maize inbreds were screened in the field, each in a two-row plot with two replications. Field testing costs in Shunyi, Xinxiang, and Sanya are $6.29, $6.03, and $10.22 per row, respectively (Supplementary Table 4), and total costs for phenotypic evaluation in the three locations are $25.18, $24.11, and $40.89 per inbred line (four rows), respectively (Supplementary Table 5). On the average, using the 10 K marker panel to replace the phenotypic evaluation in the three locations can reduce the cost by 28.43%, 25.25%, and 55.93%, respectively (Supplementary Table 5). By integrating the 1 K marker panel with functional markers such as for resistance to Fusarium verticillioides ear rot, or using multiplexing PCR (GenoPlexs) containing less markers, will significantly reduce the genotypic selection cost further. Although GBTS can be used at any breeding stage, selection in early stages/generations can save more, compared to multicycle phenotypic selection that is often required in breeding programs.
Discussion
With the availability of high-throughput molecular markers, several genotyping platforms, including GBS (Huang et al. 2014), marker chips or arrays (Fan et al. 2003; Ganal et al. 2011; Hyten et al. 2008; Unterseer et al. 2014; Yan et al. 2010) and KASP (Semagn et al. 2013), have been developed to meet requirements of different applications. Although these platforms have been playing a significant role in genetic mapping and gene discovery, with the time and cost saved for genotyping large populations, their applications in crop improvement are very limited (Rasheed et al. 2017). This is largely attributed to the cost that goes beyond breeders’ affordability. On the other hand, one genotyping platform does not always satisfy the needs of different applications. For example, map-based cloning requires low-density and widespread markers at the beginning but high-density and regional markers by the end. To meet the different requirements, each laboratory or breeding company usually needs two or more genotyping platforms, which is cost-ineffective. Therefore, a flexible genotyping platform that can handle various marker numbers is desired. In this research, we developed a series of marker panels with different marker numbers in maize using GBTS technique (Fig. 2) and validated for their efficiency. Our cost-benefit analysis indicated that the GBTS platform is not only suitable for different applications but also affordable to breeders, particularly in small- and medium-sized companies and developing countries (Table 1; Supplementary Tables 4 and 5). Compared with other gene capture technologies, GBTS has several advantages. Firstly, it is suitable for genotyping of many types of molecular markers, including SNPs, SSRs, and InDels, for a wide range of marker numbers (50 to 45,000) through either multiplex-PCR or in-solution capture. Secondly, it can be optimized for specific species. For example, species-specific enhancer and universal adapter blocker can be used to achieve improved capture. Thirdly, it is upgradable so that newly discovered important or functional loci can be incorporated into an existing marker panel without resynthesizing. Fourthly, it contains fixed markers targeting genomic regions, and is easy in data scoring, cross-lab comparison, accumulation, and management, compared to GBS that randomly captures genomic sequences and needs a huge amount of work in marker data imputation and treatment. Fifthly, it is suitable for polyploid species genotyping, heterozygote identification, and seed purity evaluation. In addition, GBTS can be implemented with different genotyping platforms by using currently available sequencing equipment.
SNP arrays/chips have been widely used in genetic studies for many plant and animal species including maize (Yan et al. 2010; Ganal et al. 2011; Unterseer et al. 2014; Rousselle et al. 2015; Xu et al. 2017b). However, the fixed marker design and high-genotyping cost restrict their applications in molecular breeding. Compared to chips, GBS can identify more variants across the genome and reduce the costs remarkably, but the unreliable imputation of missing data and the inevitable bias caused by restriction enzymes also make it unpractical to crop breeding (Schlötterer et al. 2014). The GBTS, as a new genotyping platform developed for maize in this study, combines the advantages of the fixed chips with the flexibility and low cost of GBS. GenoPlexs, as one of the GBTS option for small numbers of markers, is a multiplex-PCR target enrichment technique, which can be used to achieve single tube amplification of a few molecules to up to 5000 pairs of primers. GenoBaits, in-solution capture technique, as reported in this study, can simultaneously detect up to 90 K probes and 45 K markers in a sequencing reaction. With these two techniques, a series of marker panels can be developed to meet almost all the requirements of marker applications in the fields of genomics, genetics, and plant breeding (except GWAS using extremely high-density markers) (Table 2). The marker panel strategy and genotyping platform developed in this study for maize can be adopted and used in other plants and animals, which is supported by our ongoing efforts in other species including rice, wheat, cucumber, pig, and dog. As shown in this study, various marker numbers (i.e., 20 K, 10 K, 5 K, and 1 K) can be generated by using a single high-density marker panel but sequencing at different depths (Fig. 1), providing high flexibility and reduced genotyping cost for low-density marker panels (Table 1). With further cost reduction, one all-purpose marker panel containing 20 K or more markers (up to 45 K with in-solution capture technique) can be developed, allowing a more affordable platform that provides a foundation for data accumulating and sharing across laboratories and breeding programs.
GBTS-based genotyping provides a great advantage in cost reduction over other genotyping platforms. High-genotyping cost is one of the key issues that stop breeders from using molecular markers at a large scale due to limited research budgets, and the large population sizes required in breeding. When the genotyping cost using a high-density chip is 100 USD per sample, which has been the case in China for the chips containing 55 K or more markers, genotyping 500 samples, a reasonable population size required in plant breeding, will cost 50 K USD per population, which means that the total research budget for many small companies can be only enough for genotyping one population. Assuming that five laboratories/small companies would like to spend their research budget on genotyping one breeding population (n = 500) per year, the total market size per major crop for genotyping would be 500 × 5 = 2500 samples or 250 K USD. When the genotyping cost per sample is reduced to the half, i.e., 50 USD per sample, as many as up to 50 laboratories/companies would like to spend their research budget for marker application, each with at least two populations. As a result, the total genotyping market size for each major crop would be 500 × 50 × 2 = 50 K samples or 2.5 M USD. When the genotyping cost is further reduced to 20 USD per sample, approximately equivalent to or even lower than, the phenotypic selection cost, almost all laboratories/companies (as many as up to 200 each with at least five populations in China) would be happy to spend their research budget for routine marker application. As a result, the total genotyping market size for each major crop would be 500 × 200 × 5 = 500 K samples or 10 M USD, which is large enough for setting up a fully functional genotyping platform to support molecular breeding networks across crops and regions. Generally speaking, significant cost reduction in genotyping and associated activities will greatly stimulate marker application and associated market demand. The lower the genotyping cost is, the greater the market size will be.
Genotyping platform is not the sole requirement for molecular breeding. High-efficient breeding system requires seamless integration of genotyping with the platforms for precision phenotyping, envirotyping, breeding informatics, and decision support (Xu et al. 2017a). Multinational incorporations have established their highly efficient breeding platforms, supported by their large-scale breeding pipeline equipped with integrated breeding tools, which significantly reduces their operational and management costs. In developing countries, however, dispersed, small-scale breeding programs, each owned by small breeding companies or national organizations, have no way to set up fully functional breeding platforms for molecular breeding. Integration of the Excellence in Breeding platform established by CGIAR (EiB; http://excellenceinbreeding.org/) with open-source breeding network proposed (GOBii; http://gobiiproject.org/) could provide a highly efficient breeding pipeline for developing countries, by sharing all breeding related resources, including genotypic, phenotypic, and envirotypic information, marker effects, integrated pipeline services for DH production and genotyping, haplotype effects among environments and traits, genetic models and simulation results, and even breeding materials. By taking advantages of these sharable public platforms, developing countries can start to run their own molecular breeding programs. The GBTS, as a more affordable genotyping platform than ever, should be utilized to set up national or regional molecular breeding networks to support individual, small-scale breeding programs through resource-sharing, as being practiced by several molecular breeding initiatives/networks in China.
References
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376
Bernardo R (2008) Molecular marker and selection for complex traits in plants: learning from the last 20 years. Crop Sci 48:1649–1664
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am J Hum Genet 32:314–331
Collard BC, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc London Ser B 363:557–572
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Dreher K, Khairallah M, Ribaut JM, Morris M (2003) Money matters (I): costs of field and laboratory procedures associated with conventional and marker-assisted maize breeding at CIMMYT. Mol Breed 11:221–234
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L, Hunt S, McBride C, Bibikova M, Rubano T, Chen J, Wickham E, Doucet D, Chang W, Campbell D, Zhang B, Kruglyak S, Bentley D, Haas J, Rigault P, Zhou L, Stuelpnagel J, Chee MS (2003) Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 68:69–78
Ganal MW, Durstewitz G, Polley A, Bérard A, Buckler ES, Charcosset A, Clarke JD, Graner EM, Joets J, Le Paslier MC, McMullen MD, Montalent P, Rose M, Schön CC, Sun Q, Walter H, Martin OC, Falque M (2011) A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One 6:e28334
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler ES, Qian Q, Zhang Q, Li JY, Han B (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
Huang YF, Poland JA, Wight CP, Jackson EW, Tinker NA (2014) Using genotyping-by-sequencing (GBS) for genomic discovery in cultivated oat. PLoS One 9:e102448
Hyten DL, Song Q, Choi IY, Yoon MS, Specht JE, Matukumalli LK, Nelson RL, Shoemaker RC, Young ND, Cregan PB (2008) High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor Appl Genet 116:945–952
Jiang L, Liu X, Yang J, Wang H, Jiang J, Liu L, He S, Ding X, Liu J, Zhang Q (2014) Targeted resequencing of GWAS loci reveals novel genetic variants for milk production traits. BMC Genomics 15:1105
Krasileva KV, Vasquez-Gross HA, Howell T, Bailey P, Paraiso F, Clissold L, Simmonds J, Ramirez-Gonzalez RH, Wang X, Borrill P, Fosker C, Ayling S, Phillips AL, Uauy C, Dubcovsky J (2017) Uncovering hidden variation in polyploid wheat. Proc Natl Acad Sci U S A 114:E913–E921
Kuchel H, Ye GY, Fox R, Jefferies S (2005) Genetic and economic analysis of a targeted marker-assisted wheat breeding strategy. Mol Breed 16:67–78
Li L, Fang Z, Zhou J, Chen H, Hu Z, Gao L, Chen L, Ren S, Ma H, Lu L, Zhang W, Peng H (2017) An accurate and efficient method for large-scale SSR genotyping and applications. Nucleic Acids Res 45:e88
Liu K, Goodman M, Muse S, Smith JS, Buckler E, Doebley J (2003) Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 165:2117–2128
Lu Y, Yan J, Guimarães CT, Taba S, Hao Z, Gao S, Chen S, Li J, Zhang S, Vivek BS, Magorokosho C, Mugo S, Makumbi D, Parentoni SN, Shah T, Rong T, Crouch JH, Xu Y (2009) Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet 120:93–115
Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ (2010) Target-enrichment strategies for next-generation sequencing. Nat Methods 7:111–118
Prasanna BM, Pixley K, Warburton ML, Xie CX (2010) Molecular marker-assisted breeding options for maize improvement in Asia. Mol Breed 26:339–356
Rasheed A, Hao YF, Xia XC, Khan A, Xu YB, RK HZH (2017) Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol Plant 10:1047–1064
Rousselle Y, Jones E, Charcosset A, Moreau P, Robbins K, Stich B, Knaak C, Flament P, Karaman Z, Martinant JP, Fourneau M, Taillardat A, Romestant M, Tabel C, Bertran J, Ranc N, Lespinasse D, Blanchard P, Kahler A, Chen J, Kahler J, Dobrin S, Warner T, Ferris R, Smith S (2015) Study on essential derivation in maize: III. Selection and evaluation of a panel of single nucleotide polymorphism loci for use in European and North American germplasm. Crop Sci 55:1170–1180
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci U S A 81:8014–8018
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetics trees. Mol Biol Evol 4:406–425
Samorodnitsky E, Datta J, Jewell BM, Hagopian R, Miya J, Wing MR, Damodaran S, Lippus JM, Reeser JW, Bhatt D, Timmers CD, Roychowdhury S (2015) Comparison of custom capture for targeted next-generation DNA sequencing. J Mol Diagn 17:64–75
Schlötterer C, Tobler R, Kofler R, Nolte V (2014) Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nucleic Acids Res 15:749–763
Semagn K, Babu R, Hearne S, Olsen M (2013) Single nucleotide polymorphism genotyping using Kompetitive allele specific PCR (KASP): overview of the technology and its application in crop improvement. Mol Breed 33:1–14
Tanksley SD, Young ND, Paterson AH, Bonierbale MW (1989) RFLP mapping in plant breeding: new tools for an old science. Bio/Technology 7:257–264
Unterseer S, Bauer E, Haberer G, Seidel M, Knaak C, Ouzunova M, Meitinger T, Strom TM, Fries R, Pausch H, Bertani C, Davassi A, Mayer KF, Schön CC (2014) A powerful tool for genome analysis in maize: development and evaluation of the high density 600 k SNP genotyping array. BMC Genomics 15:823
Wu X, Li Y, Shi Y, Song Y, Wang T, Huang Y, Li Y (2014) Fine genetic characterization of elite maize germplasm using high-throughput SNP genotyping. Theor Appl Genet 127:621–631
Xu C, Ren Y, Jian Y, Guo Z, Zhang Y, Xie C, Fu J, Wang H, Wang G, Xu Y, Li P, Zou C (2017b) Development of a maize 55K SNP array with improved genome coverage for molecular breeding. Mol Breed 37:20
Xu Y (2003) Developing marker-assisted selection in plant breeding hybrid rice. Plant Breed Rev 23:73–174
Xu Y (2010) Molecular plant breeding. CABI Publishing, Wallingford
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48:391–407
Xu Y, Li P, Zou C, Lu YL, Xie C, Zhang X, Prasanna BM, Olsen MS (2017a) Enhancing genetic gain in the era of molecular breeding. J Exp Bot 68:2641–2666
Yan J, Yang X, Shah T, Sánchez H, Li J, Warburton M, Zhou Y, Crouch JH, Xu Y (2010) High-throughput SNP genotyping with the GoldenGate assay in maize. Mol Breed 25:441–451
Yang L, Yin X, Wu L, Chen N, Zhang H, Li G, Ma Z (2013) Targeted exome capture and sequencing identifies novel PRPF31 mutations in autosomal dominant retinitis pigmentosa in Chinese families. BMJ Open 3:e004030
Funding
This research is supported by the National Key Research and Development Program of China (2016YFD0101201 and 2017YFD0101201) and the Agricultural Science and Technology Innovation Program (ASTIP) of Chinese Academy of Agricultural Sciences (CAAS) (CAAS-XTCX2016009), and Research activities of CIMMYT staff were supported by the Bill and Melinda Gates Foundation and the CGIAR Research Program MAIZE.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Fig. 1
Distribution of minor allele frequency (MAF) for maize 20 K SNP marker panel as revealed by 96 representative inbred lines (96-genotype panel) (PDF 263 kb)
Supplementary Fig. 2
Distribution of polymorphic information contents (PICs) for four marker panels using 96 representative inbred lines (96-genotype panel) (PDF 284 kb)
Supplementary Fig. 3
Distribution of gene diversity (GD) as revealed by 96 maize inbred lines (96-genotype panel) and four marker panels (PDF 295 kb)
Supplementary Fig. 4
Distribution of polymorphic rates as revealed by 96 maize inbred lines (96-genotype panel) and four marker panels (PDF 284 kb)
Supplementary Fig. 5
Phylogenetic trees constructed using 387 maize breeding lines and 96 representative maize inbred lines (483 genotypes) (PDF 1883 kb)
Supplementary Table 1
The 96 diverse maize inbred lines (96-genotype panel) were used in this study (XLSX 14 kb)
Supplementary Table 2
The 387 breeding lines (387-genotype panel) selected from the ongoing breeding programs at CAAS (XLSX 24 kb)
Supplementary Table 3
Pairwise differences among eight heterotic groups of maize inbred lines as revealed by four marker panels. The lower diagonal are the average genetic distances between groups. The diagonal cells are pairwise nucleotide diversities within groups (XLSX 13 kb)
Supplementary Table 4
The field cost involved in phenotypic selection across three experimental stations in China (XLSX 14 kb)
Supplementary Table 5
Savings that can be achieved by marker-assisted selection for Fusarium verticillioides ear rot across three experimental stations in China (XLSX 13 kb)
Rights and permissions
About this article

Cite this article
Guo, Z., Wang, H., Tao, J. et al. Development of multiple SNP marker panels affordable to breeders through genotyping by target sequencing (GBTS) in maize. Mol Breeding 39, 37 (2019). https://doi.org/10.1007/s11032-019-0940-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0940-4