
Abstract
The existent investigation deals with synthesis, characterization, computational analysis, and biological activities of some hydroxytriazene derivatives containing sulphonamide moiety. The compounds were screened for antidiabetic, antioxidant, and anti-inflammatory activities. The antidiabetic activity was assessed using α-glucosidase and α-amylase inhibition assays with IC50 values ranging from 32.0 to 759.13 μg/mL and 157.77 to 340.47 μg/mL while standard drug acarbose showed IC50 values 12.21 and 69.74 μg/mL, respectively. The antioxidant activity was evaluated using DPPH and ABTS radical scavenging assays with IC50 value ranging from 54.01 to 912.66 μg/mL and 33.22 to 128.11 μg/mL, and standard drug ascorbic acid showed IC50 values 29.12 μg/mL and 69.13 μg/mL, respectively. Anti-inflammatory activity was investigated using the carrageenan-induced paw edema method, where percentage inhibition was up to 93.0 and 98.57 for 2 h and 4 h, respectively, and all the compounds were found to exhibit excellent anti-inflammatory activity. Moreover, prediction of activity spectra for substance and molecular docking were also performed. The PASS prediction hypothesized the potential of the compounds for anti-inflammatory activity, and docking results suggested the best binding pose for compounds 1b and 2b with the least energy value from which compounds can be considered as potent COX-2 inhibitors. Furthermore, possible interactions between hydroxytriazene analogues and the targets of antioxidant NADPH oxidase and antidiabetic human maltase-glucoamylase enzyme have been identified. The HOMO and LUMO analysis revealed charge transfer within the compounds. These findings suggested that the synthesized compounds can be potential agents for the treatment of diabetes and inflammation.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM) is a chronic metabolic disease associated with a state of high plasma glucose levels known as hyperglycemia [1, 2]. DM is usually a chronic condition with multi-systemic complications. With the passage of time, alterations in protein and lipid metabolism often lead to the development of macrovascular (atherosclerotic-related vascular disease, coronary heart disease, cerebrovascular disease, and peripheral vascular disease) and microvascular (neuropathy, nephropathy, and retinopathy) complications [3]. DM can be classified into Type-1 (insulin-dependent Diabetes Mellitus) and Type-2 DM (insulin-independent Diabetes Mellitus). Type-1 DM includes about 5–10% of the diabetic population and is associated with complete insulin deficiency, primarily due to autoimmune destruction of pancreatic β-cells. Type-2 DM is characterized by dysfunction of β-cells, insulin resistance, and increased production of hepatic glucose. DM can be managed by inhibiting carbohydrate-hydrolyzing enzymes such as α-glucosidase and α-amylase. The method works by delaying the absorption of glucose and is becoming a promising and effective approach to control type-2 DM [4, 5].
As far the molecular mechanism behind DM is concerned, oxidative stress has been correlated with the disease pathogenesis. Indeed, a critical balance is needed between oxidants and antioxidants to maintain the structural integrity and to perform the normal function of cells and tissues. Antioxidants are known to prevent oxidative damage by scavenging free radicals in the biological system [6]. However, oxidative stress emerges when there is an imbalance between excessive production of reactive oxygen/ nitrogen species (ROS/RNS) like superoxide (O2), hydroxyl (HO), peroxyl (ROO), alkoxyl (RO), and nitric oxide (NO) radicals and the cell’s capacity to counteract them through antioxidant defence. Although these reactive species and redox reactions play an important role to fulfil the essential physiological functions at the basal level [7], the imbalance may damage cellular molecules such as nucleic acids, proteins, and lipids. Oxidative stress has been linked with many chronic and inflammatory health problems such as neurological toxicities, cardiovascular, sexual dysfunction, cancer, aging [8, 9]. Since the antioxidant activity of a compound is its capacity to react with free radicals and delays the oxidation process through free radical scavenging action, synthesis and design of compounds with potent antioxidant activity are a welcome asset.
Another aspect that has been associated with DM is inflammation. Inflammation is a multifactorial protective attempt of the immune system in response to the damaged cells or tissues caused by stimuli such as pathogens, injuries, or any toxic substance [10]. Inflammation involves an influx of the innate immune system, the secretion of pro-inflammatory cytokines, and tissue destruction [11]. There are four major signs of inflammation including pain, heat, swelling, and redness, which eventually lead to the loss of tissue’s function. These macroscopic characteristics show increased permeability of the vascular endothelium allowing secretion of serum ingredients and extravasations in immune cells. Consequently, an inflammatory response is terminated and the damaged cells or tissues are repaired [12,13,14]. Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the groups of compounds that are the most widely used drugs in the treatment of various inflammatory diseases and arthritic pain around the world [15,16,17].
Sulphonamide derivatives containing –SO2NH2 moiety have been abundantly employed as clinically and medicinally important molecules. Sulphonamide antibiotics contain an aromatic amine group which is considered as a trigger for drug reaction due to the formation of reactive hydroxylamine intermediates [18]. Numerous pharmacological activities such as antimicrobial, anti-HIV, anti-cancer, high ceiling diuretic, anti-thyroid, antitumor, analgesic, aldose reductase, and anti-inflammatory activities of these compounds have been reported [19,20,21,22,23,24,25,26,27]. Moreover, sulpha drugs are also known for their insulin-releasing antidiabetic property and are known insulin receptor tyrosine kinase activators, DPP-4, and α-glucosidase enzyme inhibitors [28,29,30]. Remarkably, these studies have suggested that sulphonamide-containing compounds may act as potential molecules for the management of diabetes.
Hydroxytriazenes (–N(OH)–N=N–) are bi-dentate chelating agents, which have been extensively studied by our group as analytical reagents [31, 32] and have been considered biologically important molecules possessing insecticidal [33], antimicrobial [34, 35], antidyslipidemic [36], anti-inflammatory properties [35, 37, 38]. In the last few years, we explored the synthesis and antidiabetic, antioxidant, and anti-inflammatory activity of sulpha drug-based hydroxytriazenes including sulphathiazole, sulphisoxazole, sulphamethoxazole, sulphanilamide sulphadiazine, sulphapyridine, and sulphamethazine drugs [39,40,41]. In these studies, all the compounds presented moderate activity. However, it seemed relevant to us, to further explore the antidiabetic, antioxidant, and anti-inflammatory potential of sulpha drug-based hydroxytriazines. Therefore, in continuation with our research interest, we hypothesized to explore sulphonamide drug-based hydroxytriaznes as new potential bioactive compounds. Herein, we prepared a new series of sulphanilamide- and sulphacetamide-based hydroxytriazenes and screened them for antidiabetic, antioxidant, and anti-inflammatory activities. Additionally, PASS (prediction of activity spectra for substance) prediction, density functional theory (DFT) and molecular docking studies were also performed to explore the structure–activity relationship.
Results and discussion
Chemistry
Hydroxytriazenes incorporated with sulpha drugs, namely sulphanilamide (1a–1d) and sulphacetamide (2a–2d), were synthesized using the reported method [37]. The synthetic pathway for hydroxytriazene derivatives is described in scheme 1. The general method involves reduction of nitro compounds using Zn dust in a neutral medium to obtain the corresponding hydroxylamines (Step I, Scheme 1) and diazotization of sulpha drugs (sulphanilamide and sulphacetamide) at 0–5 °C (Step II, Scheme 1). Further, the final product was obtained by the coupling reaction between the hydroxylamine and diazonium salt at 0–5 °C at a pH range of 5.5–6.5. The resultant compounds were recrystallized many times using methanol/ ethanol solvent and were characterized by spectral techniques such as FT-IR, 1H NMR, 13C NMR, and EI MS (SI). All the compounds yielded satisfactory data for the confirmation of the proposed structures. The formation of hydroxytriazenes was confirmed by the presence of signals around 3400 cm−1 and 1500 cm−1 in IR spectra for –N–OH and –N=N– groups, respectively. Moreover, appearance of a singlet peak for N–OH proton in 1H NMR spectra that appear around 12 ppm confirmed the structures.

Synthesis of hydroxytriazenes
Biology
Antidiabetic activity
In this study, all the synthesized hydroxytriazenes were examined for antidiabetic activity using two assays, namely α-glucosidase and α-amylase inhibition assay. The obtained results are presented in terms of IC50values as illustrated in Table 1 and SI. The results were compared with the standard drug acarbose having IC50 values 12.21 μg/mL (y = 0.226x + 47.24, R2 = 0.939) and 69.74 μg/mL (y = 0.435x + 19.66, R2 = 0.995) for α-glucosidase and α-amylase inhibition assays, respectively. The resultant IC50 value of test compounds varies from 32.0 to 594.28 μg/mL and 157.77 to 340.47 μg/mL for α-glucosidase and α-amylase inhibition assays, respectively. The compound 2d (32.0 μg/mL) presented an excellent α-glucosidase inhibition activity, which is comparable to the standard drug. The compounds 1c (226.80 μg/mL) and 2c (185.77 μg/mL) showed moderate activity, whereas the compounds 1a (594.28 μg/mL), 1b (448.46 μg/mL), 1d (759.13 μg/mL), 2a (577.02 μg/mL), and 2b (418 μg/mL) were found to be less active against the α-glucosidase enzyme. In α-amylase inhibition activity, the compounds 1d (157.77 μg/mL), 1b (211.60 μg/mL), 1c (232.08 μg/mL), 2a (166.50 μg/mL), 2b (220.95 μg/mL), and 2d (270.40 μg/mL) showed moderate activity, whereas the compounds 1a (340.47 μg/mL) and 2c (338.18 μg/mL) were found to be least active. It can be concluded that the synthesized compounds inhibit α-glucosidase enzyme better than the α-amylase enzyme.
SAR studies: In both the series, only one compound 2d possesses excellent α-glucosidase inhibition activity with an IC50 value of 32.0 μg/mL. It is noteworthy that the presence of electron-donating group at the end of the compound enhances α-glucosidase inhibition activity. On the contrary, the presence of strong electron-withdrawing substituent such as -F made the molecules less active (1a; IC50 = 594.28 & 2a; IC50 = 577.02 μg/mL). Thus, the presence of electron-donating group can significantly improve the α-glucosidase and α-amylase inhibitory activity. The results reveal that both series showed moderate α-amylase inhibitory activity.
Carbohydrate metabolism provides a constant supply of energy to living cells and organisms. Sugar-hydrolyzing enzymes such as α-glucosidase and α-amylase are responsible for breaking dietary carbohydrates into absorbable molecules and play an important role in postprandial hyperglycemia [42]. In this regard, one of the therapeutic approaches to treat diabetes is the control of postprandial hyperglycemia by hindering the absorption of glucose through inhibition of α-glucosidase and α-amylase enzymes. The α-glucosidase and α-amylase inhibitors act by delaying the breakdown of carbohydrates in the digestive tract, thereby decreasing the postprandial blood glucose level.
Antioxidant activity
The antioxidant activity of hydroxytriazenes has been investigated using DPPH (2,2-diphenyl-1-picrylhydrazyl) and ABTS [2,2'-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)] scavenging assays, and the obtained results are expressed as IC50 values in Table 1 and SI. The results were compared with the standard drug ascorbic acid which has an IC50 value of 29.12 μg/mL (y = 0.1371 X + 46.017, R2 = 0.9609) and 69.13 μg/mL (y = 0.599X + 8.589, R2 = 0.991) for DPPH and ABTS assay, respectively. The IC50 values of hydroxytriazenes vary from 54.01 to 912.66 μg/mL for the DPPH assay. The DPPH scavenging activity of five hydroxytriazenes, viz. 1a (56.0 μg/mL), 1c (76.14 μg/mL), 2b (54.01 μg/mL), 2c (75.56 μg/mL), and 2d (84.42 μg/mL), displayed good results, while the compounds 1b (196.87 μg/mL) and 1d (285.85 μg/mL) showed moderate activity and the compound 2a (912.66 μg/mL) showed poor activity. In ABTS scavenging activity, the IC50 values vary from 33.22 to 128.11 μg/mL. The compounds 1a (48.75 μg/mL), 1b (40.58 μg/mL), 1d (33.22 μg/mL), 2a (64.64 μg/mL), 2b (50.94 μg/mL), and 2d (64.71 μg/mL) showed excellent ABTS radical scavenging activity while the compounds 1c (124.07 μg/mL) and 2c (128.11 μg/mL) exhibited moderate activity. The obtained results revealed that the synthesized compounds showed excellent ABTS radical scavenging activity as compared to the DPPH radical scavenging activity. These results are also consistent with our earlier reports [40, 41].
DPPH and ABTS methods have been used owing to their simple, rapid, and highly reproducible nature to investigate antioxidant activity of compounds. Both radical scavenging methods are based on the reduction of DPPH and ABTS radicals in the presence of antioxidants (X–H). Antioxidants can donate hydrogen radicals and form stable DPPH-H and ABTS-H diamagnetic molecular compounds. This study suggests that the synthesized hydroxytriazenes were able to reduce the DPPH and ABTS radicals and acted as radical scavengers. The degree of decolourization (Fig. 1) of the sample solution shows the radical scavenging capacity of the antioxidants in terms of H-donating ability. The intensity of change in colour for DPPH (Purple to yellow) and ABTS (Blue to colourless) assays could be measured spectrophotometrically at 517 nm and 734 nm, respectively.

Decolourization of DPPH and ABTS radicals
SAR studies: In this study, it has been reported that most of the compounds exhibited excellent IC50values in the range of 33.22 to 84.42 μg/mL. In DPPH and ABTS scavenging activity, compounds 1b (40.58 μg/mL; ABTS assay), 2b (54.01 μg/mL; DPPH assay & 50.94 μg/mL; ABTS assay), 1d (33.22 μg/mL; ABTS assay), 2d (84.42 μg/mL; DPPH assay & 64.71 μg/mL; ABTS assay) were found more potent due to the presence of electron-donating group at one of the ends of the phenyl ring attached with the hydroxytriazene moiety. Another reason for the potency could be lesser steric hindrance. In general, an electron-donating group significantly enhances the antioxidant activity of the compound by decreasing the dissociation energy of –OH group. In DPPH radical scavenging activity, the compounds 1a (56.0 μg/mL) and 1c (76.14 μg/mL) exhibited excellent activity, while the compounds 2a (912.66 μg/mL) and 1d (285.85 μg/mL) were found poorly active due to electron-withdrawing nature of the attached substituents. In ABTS radical scavenging activity, the compounds 1a (48.75 μg/mL) and 2a (64.64 μg/mL) were found to possess good activity while the compounds 1c (124.07 μg/mL) and 2c (128.11 μg/mL) were found to be moderately active.
Anti-inflammatory activity
Theoretical prediction for probable biological activity spectra has been done using PASS online web tool. The results of PASS (SI) revealed that all the synthesized hydroxytriazene derivatives could be good anti-inflammatory agents. Further, the theoretical prediction of anti-inflammatory activity has been validated experimentally using carrageenan-induced hind paw edema method. The results of anti-inflammatory activity have been reported as percentage inhibition of paw edema after 2 and 4 h of treatment as depicted in Table 1, Fig. 2, and SI. All the synthesized hydroxytriazenes showed excellent anti-inflammatory activity with 57.5–93.0% inhibition after 2 h of treatment and 80.0–98.57% inhibition after 4 h of treatment, which was better than the standard drug diclofenac (57.5% at 2 h and 94.28% at 4 h). The compounds 1b (2 h = 93%; 4 h = 97.14%) and 2b (2 h = 79.25%; 4 h = 94%) displayed highest percentage inhibition after 2 and 4 h which may be due to the presence of –CH2COOH group at the phenyl ring. Sulphanilamide drug-based compounds 1a (2 h = 79.5%; 4 h = 93.14%), 1c (2 h = 79.75%; 4 h = 93.42%), 1d (2 h = 90%; 4 h = 94.28%), and sulphacetamide drug-based compounds 2a (2 h = 57.5%; 4 h = 88.57%), 2c (2 h = 76.75%; 4 h = 98.57%), 2d (2 h = 77.5%; 4 h = 80%) were showing better activities as compared to the standard diclofenac drug. Comparing the results of anti-inflammatory activity for both series, it became evident that the sulphanilamide containing hydroxytriazenes (1a–1d) exhibited better results than sulphacetamide containing hydroxytriazenes. However, it is worth mentioning that both series showed excellent activities as compared to the standard drugs. The obtained results proved that the association of sulphonamide moiety with hydroxytriazene moiety enhanced the anti-inflammatory activity and the results are in good agreement with theoretical predictions.

Inhibition of inflammation by hydroxytriazenes
A few studies have been reported by our group exploring medicinal applications of sulpha drug-based hydroxytriazenes [39,40,41]. In 2020, Sharma et al. reported antidiabetic, antioxidant, and anti-inflammatory activity of hydroxytriazenes based on sulphanilamide, sulphadiazine, sulphapyridine, and sulphamethazine. They reported inhibition effect as IC50 values ranging from 148 to 401 μg/mL for α-glucosidase and 66 to 260 μg/mL for α-amylase enzyme. Besides, the authors obtained IC50 values for antioxidant activity ranging from 265.75 to 774 μg/mL and 24.18 to 488.25 μg/mL for DPPH and ABTS assays, respectively. In addition, the anti-inflammatory effect was achieved with 97.14% inhibition in paw edema, after 4 h of treatment [40]. In the same year, Dayma et al., studied the antidiabetic activity using α-glucosidase and α-amylase inhibition activities of sulphathiazole-, sulphisoxazole-, and sulphamethoxazole-based hydroxytriazenes and obtained IC50 values ranging from 160 to 341 μg/mL and 122 to 326 μg/mL, respectively [41]. Antioxidant activity by DPPH (367 to 858 μg/mL) and ABTS (48–401 μg/mL) radical scavenging assays was also studied. Moreover, the anti-inflammatory potential was evident from 89% inhibition of carrageenan-induced paw edema after 4 h of treatment with the synthesized compounds. Compared with the results presented by Sharma et al. and Dayma et al., the present study reports better results in α-glucosidase and α-amylase inhibition with IC50 values ranging from 32 to 759 μg/mL and 157 to 340 μg/mL, respectively. The compound 2d showed the best antidiabetic activity with α-glucosidase method among the synthesized as well as previously reported compounds. Among antioxidant activities, present compounds have displayed good results with the lowest IC50 values 54.01 and 33.22 μg/mL for DPPH and ABTS assays, respectively. Additionally, our compounds showed excellent reduction in edema by 98.57% after 4 h of treatment in the carrageenan-induced paw edema method. Thus, the present series of compounds have better results in comparison with our earlier reports.
Correlation of antidiabetic, antioxidant, and anti-inflammatory activities
Many studies have evidenced that oxidative stress plays an important role in the pathogenesis of type 2 DM [43, 44]. The overproduction of free radicals/ROS may diminish the antioxidative defense system of the tissue. Oxidative stress in the diabetic patients might cause redox imbalance, auto-oxidation of glucose, decreased concentration of antioxidants, and metabolic abnormalities. This can lead to increase in lipid peroxidation, cellular and enzyme damage resulting in either enhanced insulin resistance or impaired secretion of insulin [45]. The antioxidant therapy may inhibit the formation of reactive oxygen species (ROS), thereby providing a therapeutic strategy to defend the βـcells against oxidative stress to prevent related diabetic vascular complications [46].
Oxidative stress is also closely associated with inflammation. Both oxidative stress and inflammation may cause cell injury and damage to cellular structure and functions [47]. The cellular vascular endothelium is sensitive to injury and hence is the site of inflammation. These cells release ROS, other oxidant radicals, and metabolites that cause damage to tissue and endothelial cell lining, eventually leading to endothelial dysfunction [48, 49]. Hyperglycemia-promoted oxidative stress directs the overproduction of pro-inflammatory proteins with cytokines and chemokines which leads to inflammation [50]. At the site of inflammation, accretion of ROS occurs because leucocytes and mast cells accumulate at the site which leads to a ‘respiratory burst’ due to an increased uptake of oxygen. This inflammatory environment can destroy healthy neighbouring epithelial and stromal cells. In general, long-term inflammation can cause the risk of pathologies such as cancer, atherosclerosis, and neurodegenerative diseases [51].
Prediction of biological activity spectra by PAAS
Computer-aided drug design (CADD) is an effective tool for the development of new lead generation. PASS web tool can be used to predict biological activity spectra of any chemical substance [52, 53]. This tool predicts over 6,400 kinds of biological activities including pharmacological effects, toxic effects, interaction with metabolic enzymes and transporters, mechanisms of action, influence on gene expression etc. PASS provides the basis for the selection of the most prospective leads based on the structure–activity relationships in a heterogeneous training set. PASS predicts activity with a mean accuracy of prediction of about 95%, based on the structural formula. Thus, the predicted activity spectrum may be defined as the “intrinsic” property depending on its physicochemical characteristics and structure. PASS uses MOL / SDF files as input and output are obtained as a list of biological activity names with Pa (the probability of compound being active) and Pi (the probability of compound being inactive) for the compound. The value of Pa and Pi varies from 0 to 1. If a compound has Pa > Pi, it would be considered as an active compound. If a compound has Pa > 0.7, there is a greater probability of activity in the experiment or clinical trial, and in many cases, the chemical compound may be a close analogue of a known pharmaceutical ingredient. If a compound has Pi value 0.5 < Pa < 0.7, there is a lesser probability of activity in the experiment or clinical trial and reveals that the chemical compound is not too similar with any known pharmaceutical ingredient. If a compound has Pa < 0.5, the chance of finding activity in the experiment or clinical trial is very less; however, if such a compound shows any activity, the chemical compound might be a new chemical entity. In the present investigation, theoretical predictions for all the synthesized compounds have been projected. All compounds showed a good probability for NADPH peroxidase inhibitor, anti-inflammatory activity, and cyclooxygenase-2 inhibitor (SI). The obtained Pa values were found to be 0.789–0.274 for NADPH peroxidase inhibitor, 0.432–0.268 for anti-inflammatory activity, and 0.555–0.282 for cyclooxygenase-2 inhibition property. Based on the results, anti-inflammatory and antioxidant activities were validated experimentally.
Anti-inflammatory molecular docking studies
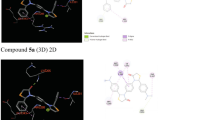
A molecular docking study was attempted to understand the nature of the interaction between the most active synthesized hydroxytriazenes and the COX-2 active site. The docking simulation was subjected to highly active 1b and 2b compounds. The analysis was performed on the X-ray crystal structure of the COX-2 complex with celecoxib PDB Id: 3LN1 active site obtained from the protein data bank using MOE 2015.1001 modelling software [54, 55]. Before predicting the binding pose for ligands, the docking method was validated for the replica of X-ray conformation of celecoxib at the active site of the COX-2 enzyme. For this purpose, the GBVI/WSA dG (Generalized-Born Volume Integral/Weighted Surface Area) scoring function was used to find out the binding energy values of the good poses [56]. The molecular modelling showed the reproducing experimental binding mode of the reference inhibitor celecoxib, with a binding energy of − 9.74 kcal/mol with a root mean square deviation of 0.42 Å (Fig. 3). The orientation and predicted binding mode of compounds 1b and 2b showed similar binding modes to the celecoxib conformation with a binding affinity of − 7.26 and − 8.38 kcal/mol, respectively. The obtained results of binding scores showed that the binding energy of the test compounds has lesser affinity for the COX-2 active site as compared to the binding score of the standard drug celecoxib. The binding mode of the compound 1b showed that the phenyl bearing sulphonamide group results in an arene–cation interaction with the phenyl moiety of Phe506 and a large number of hydrophobic residues such as Tyr371, Trp373, Phe367, Val335, Leu338, Ile503, Ala513, and Leu370 form extensive hydrophobic interactions with the phenyl ring of the 4-sulphamoylphenyl moiety. The interactions of the 4-sulphamoyl oxygen atoms create an electrostatic interaction with the Ser156 residue. The positively charged Arg106 residue exhibits an ionic interaction with the nitrogen and oxygen atoms of the hydroxytriaz-2-enyl fragment. The benzylic portion of the phenylacetic acid moiety interacts with Tyr341, Leu338, Phe504, Val509, and Ile503 through van der Waals and non-polar forces. The carboxylate polar functional group in the phenylacetic acid moiety forms polar contacts with the His75, Gln178 and Ser339 residues. The predicted binding energy of the compound 2b is higher than that of 1b owing to the formation of four strong H-bonding interactions between the N-acetylsulphamoyl and the backbone amide of the Phe504, Leu338, and Arg449 residues. Also, Thr79 and His75 residues form polar contacts with the N-acetylsulphamoyl fragment of the compound 2b. The lipophilic phenyl ring is attached between the N-acetylsulphamoyl and hydroxytriaz-2-enyl fragments, which make hydrophobic contacts with the Phe504, Val509, Tyr341, and Leu338 residues. The negatively charged fragment has a positive interaction with the Ser339, Ser516, and Arg106 residues. The phenylacetic acid fragment extensively forms hydrophobic interactions, and it is located in the hydrophobic tunnel which is formed by Phe367, Tyr371, Leu370, Tyr334, Trp373, Val334, Met508, and Leu338 as shown in Fig. 3. The two major dissimilar interactions such as H-bonding and lipophilic interactions between protein and ligands at the active site of the COX-2 enzyme will establish the anti-inflammatory potency of this chemical class of compounds. The other moderate and lower active compounds docking results are shown in SI.

Binding interactions of the compounds 1b (yellow colour) and 2b (pink colour) superimposed with celecoxib drug (red colour) at the active site of the COX-2 enzyme
Antidiabetic molecular docking studies
Maltase glucoamylase (MGAM) is one of the molecular targets of the diabetic disorder. This enzyme is responsible for the hydrolysis of polysaccharides into glucose, thereby lowering postprandial hyperglycemia (high blood glucose level after meals) and promoting glycaemic control in diabetic patients. Using MOE software, compounds 1a–2d were docked onto the crystal structure of the N-terminal catalytic domain of human intestinal maltase-glucoamylase (PDB Id: 3L4W). Compound 2d was the most active glucosidase inhibitor, with an IC50 of 32.00 g/mL and a better dock score (− 6.23 kcal/mol) than the other hydroxytriazene-based analogues (compound 1a-2c). In the compound 2d, the acetylphenyl moiety formed a strong Pi-Pi hydrophobic interaction with the Phe450 and Trp406 residues. The phenylsulphonylacetamide is surrounded by polar interaction residues such as Asp203, Asp504, Asp366, Asp443, Asp327, and Arg526. The Asp443 residue and the NH-sulphonyl group form a hydrogen bonding interaction among the polar residues. The methyl fragment in the phenylsulphonylacetamide moiety is exposed towards the residues of Phe575, Ile364, and Ile328 and forms hydrophobic interactions (Fig. 4). Moreover, the docking results of the other compounds (hydroxytriazene analogues) with the N-terminal MGAM are presented in SI.

Close-up view of the compound-2d at the active site of N-terminal domain of human maltase-glucoamylase enzyme
Antioxidant molecular docking studies
According to the DPPH and ABTS assays, compound 2b was selected for docking analysis onto the NADPH oxidase (PDB Id: 2CDU) due to its better DPPH and ABTS scavenging activities when compared with the standard ascorbic acid. The docking models predicted a binding affinity of − 7.96 kcal/mol, indicating that it has a higher binding affinity. Compound 2b forms two hydrogen bonding interactions with the enzyme active site residues Thr9 and Asp282 by means of polar functional groups present in the hydroxytriaz-2-enyl and phenylacetic acid moieties, respectively. Several polar residues, including Ser41, Cys133, Glu163, Lys134, Glu32, Gly281, Thr113, His10, Gly7, Gly114, Thr112, and Cys8, contribute to polar interactions with compound 2b, whereas other nearby residues, including Ile44, Ile160, Ala300, Ala303, Leu299, and Pro298, form hydrophobic interactions with the phenyl rings of compound 2b (Fig. 5). The docking outcomes for the other synthesized compounds are provided in SI.

Close-up view of the compound-2b at the active site of NADPH oxidase enzyme
Density functional theory (DFT) calculations
We report the HOMO and LUMO energy values for the compounds 1b and 2b at − 0.325 and − 0.030 eV, respectively, using the DFT calculation by the PM3 method. Thus, we effectively produced a molecular system having a small HOMO–LUMO energy gap of 0.295 eV for both compounds. The obtained results revealed that both compounds 1b and 2b have slightly higher electron-accepting capability than the electron-donating capability. These results reveal energetically favourable interactions between the inflammatory target protein and the designed compounds. As shown in Fig. 6, deep red-coloured surfaces indicate an electron-rich spot around the molecule, whereas the deep blue-coloured surfaces indicate an electron-deficient spot around the molecule. The most electron-rich and deficient sites are distributed equally on the phenyltriaz-2-enyl scaffold while plotting the surfaces of HOMO and LUMO on the optimized structure of the compound 1b. In compound 2b, the surfaces of the HOMO plot are very similar to that of compound 1b, whereas the LUMO plot surfaces extend towards the phenyl ring which is attached to the acetic acid functional group. These DFT studies revealed that the key features of anti-inflammatory activities for compounds 1b and 2b are based upon the occurrence of the common phenyltriaz-2-enyl skeleton.

HOMO and LUMO electron density distribution maps of compounds 1b and 2b
Thus, theoretical predictions by PASS revealed a good probability for the compounds to have NADPH peroxidase inhibition potential, cyclooxygenase-2 inhibition potential, and anti-inflammatory activity. Similarly, a higher COX-2 inhibitory property was also predicted by molecular docking for compounds 1b (− 7.26 kcal/mol) and 2b (− 8.38 kcal/mol), which showed the best binding affinity score. The DFT calculation indicated a small HOMO and LUMO energy gap, i.e., 0.295 eV for compounds 1b and 2b, which revealed higher electron-accepting capability of both compounds. These energetically favourable interactions ensure feasibility of a chemical reaction with the inflammatory target protein. In the results of theoretical PASS predictions and docking studies, anti-inflammatory activities are not only in close agreement with each other, but also revealed excellent anti-inflammatory properties of the sulphonamide-based hydroxytriazenes.
Conclusions
In conclusion, we have investigated a new series of sulpha drug-based hydroxytriazenes for their antidiabetic, antioxidant, and anti-inflammatory activity. All the compounds displayed moderate to good α-glucosidase inhibition, DPPH, and ABTS radical scavenging activities. Moreover, all the compounds exhibited good to excellent inhibition of inflammation after 2 and 4 h, and interestingly, the findings are comparable with the results of the standard drug diclofenac. The PASS predictions, DFT and docking studies were also done to develop structure–activity relationships. Docking results showed a good binding pose of the tested compounds towards the COX-2 active site. Furthermore, significant interactions between hydroxytriazene analogues and antioxidant human maltase-glucoamylase and antidiabetic NADPH oxidase enzyme targets have been revealed. The active chemicals could be employed as essential components for more effective antioxidative and antidiabetic therapy. Thus, herein, we report sulpha drug-based hydroxytriazenes and their pharmacological properties guided by computer-aided drug designing and its experimental validation.
Experimental
General procedure for the synthesis of hydroxytriazenes
We adopted a general method involving three steps, for the synthesis of hydroxytriazenes. Step-I includes reduction of nitro compounds to the respective hydroxylamine. In a 100-mL beaker, 20 mmol of aryl nitro compound was taken and was dissolved in 30 mL of a water–ethanol mixture (1:1). A saturated solution of 1.06 gm (1 equiv) of ammonium chloride was added followed by the addition of 4.0 g of zinc dust in small lots with continuous stirring, maintaining temperature at 50–60 °C. After the addition of zinc dust, the mixture was stirred for another 20 min to complete the reaction and was then filtered. The obtained filtrate was used for the coupling reaction. In step-II, sulphanilamide and sulphacetamide were diazotized to form diazonium salts. For this, 13 mmol of the sulpha drug was dissolved in 7 mL of HCl-H2O mixture (1:1) and the temperature was maintained at 0 °C ± 5 °C in an ice bath. A saturated chilled solution of sodium nitrite (0.89 g) was added drop by drop with uninterrupted stirring. After completion of the reaction, the obtained diazonium salt was directly used for coupling with hydroxylamine. Step-III involves the coupling of the diazonium salt with the hydroxylamines as obtained in step-II and step-I, respectively. The diazonium salt was added to hydroxylamine with the temperature maintained at 0 °C ± 5 °C and pH maintained between 5.5 and 6.5 using sodium acetate. The final crude product was recrystallized with the appropriate solvent.
(E)-4-(3-(4-fluorophenyl)-3-hydroxytriaz-1-en-1-yl)benzenesulphonamide (1a): The title compound was synthesized according to general method using p–fluoronitrobenzene (2.82 gm, 20 mmol) and sulphanilamide (1.72 gm, 13 mmol) drug yielding a pure product as brown shining powder (Yield 3.70 g, 91.81%), m.p. 172 °C; FT–IR (KBr, cm−1): ѴO–H (3350 cm–1), ѴN–H (3256 cm–1, 3204 cm–1), ѴN=N (1409 cm–1), ѴC–N (1235 cm–1), ѴS=O (1159 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 7.27(s, 2H, Hn; –NH2), 7.39–7.43 (m, 2H, Hc,e; Ar–H), 7.62–7.64 (m, 2H, Hb,f; Ar–H), 7.79–7.81 (m, 2H, Hi,m; Ar–H), 8.15–8.19 (m, 2H, Hj,l; Ar–H), 12.28 (s, 1H, Hg; –OH); 13C NMR (100 MHz, DMSO–d6) ppm: δ 114.61 (2C; Cc–Ce), 116.24 (2C; Cb–Cf), δ122.34 (2C; Ci–Cm), 127.23 (2C; Cj–Cl), 137.48 (1C; Ck), 139.36 (1C; Ca), 143.14 (1C; Ch), 161.40 (1C; Cd); ESI–HRMS (m/z) calcd. for C12H11FN4O3S, 310.38; found 309.30.
(E)-2-(4-(1-hydroxy-3-(4-sulphamoylphenyl)triaz-2-en-1-yl)phenyl)acetic acid (1b): The title compound was synthesized according to general method using p–nitrophenyl acetic acid (3.62 gm, 20 mmol) and sulphanilamide (1.72 gm, 13 mmol) drug yielding a pure product as light cream yellow crystals (Yield 3.96 g, 87. 03%), m.p.148 °C; FT–IR (KBr, cm−1): ѴO–H (3340 cm–1), ѴN–H (3245 cm–1, 3187 cm–1), ѴN=N (1408 cm–1), ѴC–N (1233 cm–1), ѴS=O (1157 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 3.71(s, 2H, H; –CH2), 7.26 (s, 2H, Hn; –NH2), 7.46–7.48 (m, 2H, Hb,f; Ar–H), 7.62–7.65 (m, 2H, Hc,e; Ar–H), 7.79–7.82 (m, 2H, Hi,m; Ar–H), δ8.05–8.07 (m, 2H, Hj,l; Ar–H), 12.26 (s,1H, Hg; –OH), 12.51 (s, 1H, Hq; –OH); 13C NMR (100 MHz, DMSO–d6) ppm: δ 40.07 (1C; Co), 114.57 (2C; Cb–Cf), 119.78 (2C; Ci–Cm), 127.26 (1C; Cd), 130.27 (2C; Cj–Cl), 137.38 (2C; Cc–Ce), 137.40 (1C; Ck), 141.61 (1C; Ca), 143.23 (1C; C), 172.25 (1C; Cp); ESI–HRMS (m/z) calcd. for C14H14N4O5S, 350.34; found 351.04.
(E)-3-(1-hydroxy-3-(4-sulphamoylphenyl)triaz-2-en-1-yl)benzoic acid (1c): The title compound was synthesized according to general method using m–nitrobenzoic acid (3.34 gm, 20 mmol) and sulphanilamide (1.72 gm, 13 mmol) drug yielding a pure product as yellow powder (Yield 3.69 g, 84.43%), m.p. 168 °C; FT–IR (KBr, cm−1): ѴO–H (3366 cm–1), ѴN–H (3208 cm–1, 3094 cm–1), ѴN=N (1418 cm–1), ѴC–N (1232 cm–1), ѴS=O (1152 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 7.27 (s, 2H, Hn; –NH2), 7.63–7.66 (m, 2H, Hi, m; Ar–H), 7.69–7.73 (m, 1H, He; Ar–H), 7.80–7.83 (m, 2H, Hj, l; Ar–H), 8.09–8.11 (m, 1H, Hf; Ar–H), 8.37–8.40 (m, 1H, H d; Ar–H), 8.56 (m, 1H, H b; Ar–H), 12.40 (s,1H, Hg; –OH), 13.27 (s, 1H, Hp; –OH); 13C NMR (100 MHz, DMSO–d6) ppm: δ 114.76 (1C; Cb), 120.33 (1C; Cf), 123.95 (2C; Ci–Cm), 127.30 (1C; Ce), 129.78 (1C; Cc), 130.66 (2C; Cj–Cl), 132.03 (1C; Cd), 137.69 (1C; Ck), 143.01 (1C; Ca), 166.24 (1C; Co); ESI–HRMS (m/z) calcd. for C13H12N4O5S, 336.32; found 337.32.
(E)-4-(3-(3-acetylphenyl)-3-hydroxytriaz-1-en-1-yl)benzenesulphonamide (1d): The title compound was synthesized according to general method using m–nitroacetophenone (3.30 gm, 20 mmol) and sulphanilamide (1.72 gm, 13 mmol) drug yielding a pure product as yellow shining crystals (Yield 3.91 g, 89.88%), m.p. 155 °C; FT–IR (KBr, cm−1): ѴO–H (3370 cm–1), ѴN–H (3266 cm–1, 3208 cm–1), ѴN=N (1419 cm–1), ѴC–N (1257 cm–1), ѴS=O (1155 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 2.67 (s, 3H, Hp; –CH3), 7.28 (s, 2H, Hn; –NH2), 7.64–7.67 (m, 2H, Hi, m; Ar–H), 7.70–7.74 (m, 1H, He; Ar–H), 7.81–7.83 (m, 2H, Hj,l; Ar–H), 8.11–8.13 (m, 1H, Hf; Ar–H), 8.36–8.38 (m, 1H, Hd; Ar–H), 8.51 (m, 1H, Hb; Ar–H), 12.39 (s,1H, Hg; –OH); 13C NMR (100 MHz, DMSO–d6) ppm: δ 26.89 (1C; Cp), 114.78 (1C; Cb), 118.87 (1C; Cf), 124.12 (2C; Ci–Cm), 127.27 (1C; Cd), 129.84 (1C; Cj–Cl), 137.68 (1C; Cc), 137.70 (1C; Ck), 143.01 (1C; Ca), 143.14 (1C; Ch), 196.99 (1C; Co); ESI–HRMS (m/z) calcd. for C14H14N4O4S, 334.35; found 335.35.
(E)-N-((4-(3-(4-fluorophenyl)-3-hydroxytriaz-1-en-1-yl)phenyl)sulphonyl)acetamide (2a): The title compound was synthesized according to general method using p–fluoronitrobenzene (2.82 gm, 20 mmol) and sulphacetamide (2.78 gm, 13 mmol) drug yielding a pure product as brown shining powder (Yield 3.67 g, 80.13%), m.p.149 °C; FT–IR (KBr, cm−1): ѴO–H (3438 cm–1), ѴN–H (3216 cm–1), ѴC=O (1711 cm–1), ѴN–O (1341 cm–1),ѴN=N (1449 cm–1), ѴC–N (1225 cm–1), ѴS=O (1154 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 1.92 (s, 3H, Hp, –CH3), 7.38–7.43 (m, 2H, Hc,e; Ar–H), 7.64–7.67 (m, 2H, Hb,f; Ar–H), 7.86–7.88 (m, 2H, Hi,m; Ar–H), 8.14–8.18 (m, 2H, Hj, l; Ar–H), 11.99 (s,1H, Hg; –OH), 12.37 (s, 1H, Hn; –NH–); 13C NMR (100 MHz, DMSO–d6) ppm: δ 114.58 (2C; Cc–Ce), 116.25 (2C; Cb–Cf), 122.35 (2C; Ci–Cm), 129.39 (2C; Cj–Cl), 131.94 (1C; Ck), 139.36 (1C; Ca), 144.67 (1C; Ch), 161.50 (1C; Cd), 168.65 (1C; Co); ESI–HRMS (m/z) calcd. for C14H13FN4O4S, 352.3; found 353.34.
(E)-2-(4-(3-(4-(N-acetylsulphamoyl)phenyl)-1-hydroxytriaz-2-en-1-yl)phenyl)acetic acid (2b): The title compound was synthesized according to general method using p–nitrophenylacetic acid (3.62 gm, 20 mmol) and sulphacetamide (2.78 gm, 13 mmol) drug yielding a pure product as yellow shining crystals (Yield 4.53 g, 88.82%), m.p.158 °C; FT–IR (KBr, cm−1): ѴO–H (3431 cm–1), ѴN–H (3129 cm–1), ѴC=O (1712 cm–1), ѴN–O (1596 cm–1),ѴN=N (1456 cm–1), ѴC–N (1223 cm–1), ѴS=O (1158 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 1.92 (s, 3H, Hp; –CH3), 3.71(s, 2H, Hq; –CH2), 7.46–7.49 (m, 2H, Hb, f; Ar–H), 7.64–7.67 (m, 2H, Hc, e; Ar–H), 7.85–7.88 (m, 2H, Hi, m; Ar–H), 8.04–8.07 (m, 2H, Hj, l; Ar–H), 12.03 (s, 1H, Hg; –OH), 12.36 (s, 1H, Hn; –NH–).13C NMR (100 MHz, DMSO–d6) ppm: δ 23.17 (1C; Cp), 40.06 (1C; Cq), 114.53 (2C; Cb–Cf), 119.87 (2C; Ci–Cm), 137.60 (1C; Ck), 141.62 (1C; Ca), 144.75 (1C; Ch), 168.65 (1C; Co), 172.21 (1C; Cr); ESI–HRMS (m/z) calcd. for C16H16N4O6S, 392.38; found 393.38.
(E)-3-(3-(4-(N-acetylsulphamoyl)phenyl)-1-hydroxytriaz-2-en-1-yl)benzoic acid (2c): The title compound was synthesized according to general method using m–nitrobenzoic acid (3.34 gm, 20 mmol) and sulphacetamide (2.78 gm, 13 mmol) drug yielding a pure product as yellow powder (Yield 4.02 g, 81.70%), m.p. 160 °C; FT–IR (KBr, cm−1): ѴO–H (3465 cm–1), ѴN–H (3153 cm–1), ѴC=O (1708 cm–1), ѴN–O (1332 cm–1),ѴN=N (1448 cm–1), ѴC–N (1222 cm–1), ѴS=O (1149 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 1.92 (s, 3H, Hp; –CH3), 7.66–7.68 (m, 2H, Hi, m; Ar–H), 7.69–7.73 (m, 1H, He; Ar–H), 7.88–7.90 (m, 2H, Hj,l; Ar–H), 8.09–8.11 (m, 1H, Hf; Ar–H), 8.35–8.38 (m, 1H, Hd; Ar–H), 8.55–8.56 (m, 1H, Hb; Ar–H), 11.94 (s,1H, Hg; –OH), 12.49 (s, 1H, Hn; –NH–); 13C NMR (100 MHz, DMSO–d6) ppm: δ23.16 (1C; Cp), 114.73 (1C; Cb), 120.43 (1C; Cf), 124.04 (2C; Ci–Cm), 129.44 (1C; Ce), 129.79 (1C; Cc), 130.82 (2C; Cj–Cl), 132.03 (1C; Cd), 132.16 (1C; Ck), 143.01 (1C; Ca), 144.53 (1C; Ch), 166.20 (1C; Co), 168.67 (1C; Cq); ESI–HRMS (m/z)calcd. for C15H14N4O6S, 378.35; found 379.35.
(E)-N-((4-(3-(3-acetylphenyl)-3-hydroxytriaz-1-en-1-yl)phenyl)sulphonyl)acetamide (2d): The title compound was synthesized according to general method using m–nitroacetophenone (3.30 gm, 20 mmol) and sulphacetamide (2.78 gm, 13 mmol) drug yielding a pure product as yellow shining crystals (Yield 4.12 g, 84.25%), m.p. 155 °C; FT–IR (KBr, cm−1): ѴO–H (3461 cm–1), ѴN–H (3178 cm–1), ѴC=O (1700 cm–1), ѴN–O (1342 cm–1),ѴN=N (1455 cm–1), ѴC–N (1245 cm–1), ѴS=O (1154 cm–1); 1H NMR (400 MHz, DMSO–d6) ppm: δ 1.93 (s, 3H, Hp; –CH3), 2.67 (s, 3H, Hr; –CH3), 7.67–7.69 (m, 2H, Hi, m; Ar–H), 7.70–7.74 (m, 1H, He; Ar–H), 7.88–7.90 (m, 2H, Hj, l; Ar–H), 8.11–8.13 (m, 1H, Hf; Ar–H), 8.35–8.37 (m, 1H, Hd; Ar–H), 8.51 (s, 1H, Hb; Ar–H), 11.99 (s,1H, Hg; –OH), 12.48 (s, 1H, Hn; –NH–); 13C NMR (100 MHz, DMSO–d6) ppm: δ 23.18 (1C; Cp), 26.88 (1C; Cr), 114.75 (1C; Cb), 119.01 (1C; Cf), 124.20 (2C; Ci–Cm), 129.41 (1C; Cd), 129.86 (1C; Ce), 129.97 (1C; Cj–Cl), 132.18 (1C; Ck), 137.69 (1C; Cc), 143.16 (1C; Ca), 144.54 (1C; Ch), 168.66 (1C; Co), 196.92 (1C; Cq); ESI–HRMS (m/z) calcd. for C16H16N4O5S, 376.38; found 377.38.
Biological assays
α-Glucosidase inhibition assay
In this research, α-glucosidase inhibition activity was evaluated by using the reported method [57]. In this assay, 1.0 mg of rat-intestinal acetone powder was dissolved in 100 mL of saline water and sonicated at 4 °C. After sonication, the suspension was centrifuged (3000 rpm, 4 °C, 30 min), and obtained supernatant was used for the assay. A reaction mixture containing 50 µL of phosphate buffer (50 mM, pH 6.8), 50 µL of rat α-glucosidase, and 50 µL of samples of varying concentrations (100–800 µg/mL) was pre-incubated for 5 min at 37 °C, and then, 50 µL of p-nitrophenyl-α-D-glucopyranoside, PNPG (3 mM), was added to the mixture as a substrate. After incubation at 37 °C for 30 min, enzymatic activity was quantified by measuring the absorbance at 405 nm on a multimode microplate reader (Synergy H4, Bio-TEK, USA). Acarbose was used as positive control and experiments were done in triplicates. The following equation has been used for the calculation of percentage inhibition (SI).
IC50 values were calculated graphically using a linear regression algorithm. The obtained results are expressed as IC50 values and given in Table 1.
α-Amylase inhibition assay
The α-amylase inhibition activity was determined by the previously reported method [40, 58]. In this assay, 50 µL of various concentrations of test samples and 50 µL of α-amylase solution (0.5 mg/ mL) containing 0.02 M sodium phosphate buffer (pH 6.9) were incubated at 25 °C for 10 min. After pre-incubation, 50 µL of 1% starch solution prepared in 0.02 M sodium phosphate buffer (pH 6.9) was added to each tube, and the reaction mixture was again incubated at 25 °C for 10 min. The reaction was stopped with the addition of 100 µL of DNS solution, and microplates were again incubated for 10 min at 85–90 °C to develop colour and left to cool at RT. Then, the reaction mixture was diluted with 100 µL of distilled water and enzymatic activity was quantified by measuring the absorbance at 540 nm on a multimode microplate reader (Synergy H4, Bio-TEK, USA). Acarbose was used as positive control, and experiments were done in triplicates. The following equation has been used for the calculation of percentage inhibition (given in SI).
IC50 values were calculated graphically using a linear regression algorithm. The obtained results are expressed as IC50 values and illustrated in Table 1.
DPPH scavenging assay
DPPH radical scavenging activity was determined using the reported method [58]. In this assay, antiradical activity was screened by taking 125 µL of test samples of various concentrations (10–100 μg/mL), 125 µL of DPPH solution (0.004% w/v in methanol), 50 µL of 0.1 M Tris–HCl buffer (pH 7.4) in 96-well microplate. The reaction mixture was incubated for half an hour at RT in dark, and then, absorbance was recorded at 517 nm on BioTek Synergy H4 hybrid multimode microplate reader. Radical quenching efficiency of test samples was determined by comparison with 125 μL methanol, 50 μL of tris–HCl buffer, and 125 μL of DPPH solution treated as control absorbance. In this assay, ascorbic acid was used as a positive control. The following equation has been used for the calculation of percentage inhibition (SI).
IC50 values were calculated graphically using a linear regression algorithm. The obtained results are expressed as IC50 values and given in Table 1.
ABTS scavenging assay
The ABTS radical scavenging activity was estimated using the reported method [59]. The working ABTS·+ solution was obtained by reacting 7 mM ABTS solution with 2.45 mM potassium persulphate solution in an equal ratio, and the mixture was left to stand in the dark at room temperature for 18–24 h before use. The Sample solution of various concentrations (20–300 μg/mL) was added to ABTS·+ solution. The absorbance of sample solutions along with standard drug was recorded at 734 nm on BioTek Synergy H4 hybrid multimode microplate reader. In this assay, ascorbic acid was used as a positive control. The following equation has been used for the calculation of percentage inhibition (SI).
IC50 values were calculated graphically using a linear regression algorithm. The obtained results are expressed as IC50 values and given in Table 1.
Anti-inflammatory activity
The anti-inflammatory activity of hydroxytriazenes was evaluated by using the carrageenan-induced paw edema method as described in the literature [60]. Wister albino rats either male or female (150–200 g) were obtained from the animal house, B.N. College of Pharmacy, Udaipur (Raj). The experimental protocol was accepted and reviewed by the Institutional Animal Ethics Committee (IAEC), Protocol No. is 52/ACR/BNCP-10/IAEC and Registration No. is 870/ac/08/ CPCSEA. In this method, rats were divided into eleven groups. Each group contains six rats. The rats were starved overnight with free access of water. Group I were given only water as control. Group II rats were given carrageenan (0.1 mL of 1% mg/kg, bw, p.o.) while group III rats were given carrageenan (0.1 mL of 1% mg/kg) and diclofenac drug (12.5 mg/kg, bw, p.o.). In groups IV-XI, rats were given carrageenan (0.1 mL of 1% mg/kg, bw, p.o.) and synthesized hydroxytriazenes (200 mg/kg, bw, p.o.) orally. After injection into the plantar side of the right hind paw, the paw volume was recorded plethysmographically at different time intervals, i.e., 1 h, 2 h, 3 h and 4 h, and the percent inhibition of hydroxytriazenes on carrageenan-induced paw edema in 2 h and 4 h shown in Fig. 2. The obtained results were compared with control and % inhibition was calculated by using the following formula.
V0 and Vt indicate the volume of hind paw edema of at 0, 2, and 4 h. The obtained results are given in Table 1.
The anti-inflammatory activity of NSAIDs is associated with inhibition of the activity of cyclooxygenases (COXs) enzymes, key enzymes in the biosynthesis of prostaglandins (PGs), which causes inflammatory pain. COXs exist as COX-1 and COX-2; both are regulated differently. Constitutive COX-1 provides cytoprotection in the gastrointestinal (GI) tract, whereas inducible COX-2 mediates pro-inflammatory conditions [61, 62]. It is recognized that some selective COX-2 inhibitors lead to adverse cardiovascular side effects [63, 64]. Chronic NSAID reduces the symptoms of many arthritic pain and gastroduodenal damage but also invites adverse GI complications [65, 66]. Therefore, existing COX-2 are not able to eliminate NSAID-induced complications, and their long-term use is associated with cardiovascular diseases [67, 68] and the search for anti-inflammatory drugs with promising results is still continuing. The marketed selective COX-2 inhibitor (celecoxib) having a suphonamide moiety shows high anti-inflammatory activity with reduced GI toxicity.
Molecular docking protocol
In silico docking studies were performed on all of the compounds to investigate the orientations and binding affinities against the anti-inflammatory, antidiabetic, and antioxidant targets of cyclooxygenase-2, maltase-glucoamylase, and NADPH oxidase, respectively. The X-ray 3D crystal structure enzymes were obtained from the protein data bank website (PDB entry 3LN1, 3L4W & 2CDU; www.rcsb.org). Using the MMFF94 force field Molecular Operating Environment software, the co-crystallized ligands were flexibly re-docked to validate the docking protocol (MOE 2015.1001). Water molecules were removed from the proteins, and structural issues with the proteins were resolved by adding polar hydrogen and optimizing the hydrogen bond network. The protein was fixed with the MMFF94x forcefield setup and partial charges. The ligands were prepared, optimized, and docked. To search for and generate good fit conformers in the active site of the enzymes, the triangle matcher placement technique was used, followed by a rigid receptor protocol. The docked poses were scored and rescored using the London dG and GBVI/WSA dG functions. The target molecules' and receptors' configurations were evaluated and chosen based on their scores and root mean square deviation values.
DFT protocol
The 3-D chemical structure of the most promising anti-inflammatory compounds (1b and 2b) by using the "builder" module available in the Molecular Operating Environment (MOE 2015.1001) software. Further, geometry optimizations were accomplished with the Parameterized Method 3 (PM3) parameterization of the Modified Neglect of Diatomic Overlap (MNDO) method. This is based on the Neglect of Diatomic Differential Overlap (NDDO) approximate hamiltonian for the DFT calculations by using Argus Lab Software (V: 4.0.1) [69, 70]. Quantum chemical properties of frontier molecular orbitals such as HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) energy, and the energy level difference between HOMO and LUMO were calculated, and the electron density cloud surface plotted around the molecules using the Quickplot module in Argus lab software [71].
PASS predication protocol
PASS predication tool is an online freely accessible web resource which can predict biological activity spectrum of a compound. The biological activity spectrum of compound involves various pharmacological activities, mechanisms of action, toxicity. PASS predication has been made freely accessible web tool by logging in to http://www.way2drug.com/passonline.
Data availability
Materials and methods and all the instrumentation spectra like IR, 1H NMR, 13C NMR, and mass spectra are provided in the SI.
References
Mukherjee N, Lin L, Contreras CJ, Templin AT (2021) Β-cell death in diabetes: past discoveries, present understanding, and potential future advances. Metabolites 11:796
Esser N, Utzschneider KM, Kahn SE (2020) Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 63:2007–2021
Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X, Song X, Ren Y, Shan PF (2020) Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci Rep 8:1–1
Min SH, Yoon JH, Hahn S, Cho YM (2018) Efficacy and safety of combination therapy with an α-glucosidase inhibitor and a dipeptidyl peptidase-4 inhibitor in patients with type 2 diabetes mellitus: a systematic review with meta-analysis. J Diabetes Investig 9:893–902
Zhang L, Chen Q, Li L, Kwong JS, Jia P, Zhao P, Wang W, Zhou X, Zhang M, Sun X (2016) Alpha-glucosidase inhibitors and hepatotoxicity in type 2 diabetes: a systematic review and meta-analysis. Sci Rep 6:1–8
Dahlen A, Dashi G, Maslov I, Attwood MM, Jonsson J, Trukhan V, Schiöth HB (2022) Trends in antidiabetic drug discovery: FDA approved drugs, new drugs in clinical trials and global sales. Front Pharmacol 19:807548
Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, Qin X, Ji Y, Yang X, Wang H (2019) Biochemical basis and metabolic interplay of redox regulation. Redox Biol 26:101284. https://doi.org/10.1016/j.redox.2019.101284
Sharma GN, Gupta G, Sharma P (2018) A comprehensive review of free radicals, antioxidants, and their relationship with human ailments. Crit Rev Eukaryot Gene Expr 28:139–154
Karam HM, Radwan RR (2019) Metformin modulates cardiac endothelial dysfunction, oxidative stress and inflammation in irradiated rats: A new perspective of an antidiabetic drug. Clin Exp Pharmacol Physiol 46:1124–1132. https://doi.org/10.1111/1440-1681.13148
Greten FR, Grivennikov SI (2019) Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 16:27–41
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737. https://doi.org/10.1038/nri3073
Li D, Wu M (2021) Pattern recognition receptors in health and diseases. Sig Transduct Target Ther 6:291
Tourki B, Halade GV (2021) Heart failure syndrome with preserved ejection fraction is a metabolic cluster of non-resolving inflammation in obesity. Front Cardiovasc Med 8:695952
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454:428–435. https://doi.org/10.1038/nature07201
Geronikaki AA, Lagunin AA, Hadjipavlou-Litina DI, Eleftheriou PT, Filimonov DA, Poroikov VV, Alam I, Saxena AK (2008) Computer-aided discovery of anti-inflammatory thiazolidinones with dual cyclooxygenase/lipoxygenase inhibition. J Med Chem 51:1601–1609. https://doi.org/10.1021/jm701496h
Amatruda JG, Katz R, Peralta CA, Estrella MM, Sarathy H, Fried LF, Newman AB, Parikh CR, Ix JH, Sarnak MJ, Shlipak MG (2021) Association of non-steroidal anti-inflammatory drugs with kidney health in ambulatory older adults. J Am Geriatrics Soc 69:726–34
Akgul O, Mannelli CDL, Vullo D, Angeli A, Ghelardini C, Bartolucci G, Altamimi ASA, Scozzafava A, Supuran TC, Carta F (2018) Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs–CAIs) for the management of rheumatoid. J Med Chem 61:4961–4977. https://doi.org/10.1021/acs.jmedchem.8b00420
Apaydın S, Torok M (2019) Sulfonamide derivatives as multi-target agents for complex diseases. Bioorg Med Chem Lett 15:2042–2050
Zhang J, Tan Y, Li G, Chen L, Nie M, Wang Z, Ji H (2021) Coumarin sulfonamides and amides derivatives: design, synthesis, and antitumor activity in vitro. Molecules 26:786
Badgujar JR, More DH, Meshram JS (2018) Synthesis, antimicrobial and antioxidant activity of pyrazole based sulfonamide derivatives. Indian J Microbiol 58:93–99. https://doi.org/10.1007/s12088-017-0689-6
Shafique M, Hameed S, Naseer MM, Al-Masoudi NA (2018) Synthesis of new chiral 1,3,4-thiadiazole-based di- and tri-arylsulfonamide residues and evaluation of in vitro anti-HIV activity and cytotoxicity. Mol Divers 22:957–968. https://doi.org/10.1007/s11030-018-9851-2
Berredjem M, Bouzina A, Bahadi R, Bouacida S, Rastija V, Djouad SE, Sothea TO, Almalki FA, Hadda TB, Aissaoui M (2022) Antitumor activity, X-Ray crystallography, in silico study of some-sulfamido-phosphonates. Identification of pharmacophore sites. J Mol Struc 1250:131886
Demir Y, Koksal Z (2020) Some sulfonamides as aldose reductase inhibitors: therapeutic approach in diabetes. Arch Physiol Biochem 23:1–6. https://doi.org/10.1080/13813455.2020.1742166
Poudapally S, Battu S, Velatooru LR, Bethu MS, Rao JV, Sharma S, Sen S, Pottabathini N, Iska VBR, Katangoor V (2017) Synthesis and biological evaluation of novel quinazoline-sulfonamides as anti-cancer agents. Bioorg Med Chem Lett 27:1923–1928. https://doi.org/10.1016/j.bmcl.2017.03.042
Pervaiz M, Riaz A, Munir A, Saeed Z, Hussain S, Rashid A, Younas U, Adnan A (2020) Synthesis and characterization of sulfonamide metal complexes as antimicrobial agents. J Mol Struct 1202:127284. https://doi.org/10.1016/j.molstruc.2019.127284
Irfan A, Ahmad S, Hussain S, Batool F, Riaz H, Zafar R, Kotwica-Mojzych K, Mojzych M (2021) Recent updates on the synthesis of bioactive quinoxaline-containing sulfonamides. Appl Sci 11:5702. https://doi.org/10.3390/app11125702
Ovung A, Bhattacharyya J (2021) Sulfonamide drugs: structure, antibacterial property, toxicity, and biophysical interactions. Biophys Rev 13:259–272. https://doi.org/10.1007/s12551-021-00795-9
Taha M, Imran S, Salahuddin M, Iqbal N, Rahim F, Uddin N, Shehzad A, Farooq RK, Alomari M, Khan KM (2021) Evaluation and docking of indole sulfonamide as a potent inhibitor of α-glucosidase enzyme in streptozotocin induced diabetic albino wistar rats. Bioorg Chem 110:104808. https://doi.org/10.1016/j.bioorg.2021.104808
Alyar S, Şen T, Ozdemir UO, Alyar H, Adem S, Şen C (2019) Synthesis, spectroscopic characterizations, enzyme inhibition, molecular docking study and DFT calculations of new Schiff bases of sulfa drugs. J Mol Struc 1185:416–424. https://doi.org/10.1016/j.molstruc.2019.03.002
Mendes CP, Postal BG, Oliveira GT, Castro AJ, Frederico MJ, Moraes AL, Neuenfeldt PD, Nunes RJ, Menegaz D, Silva FR (2019) Insulin stimulus-secretion coupling is triggered by a novel thiazolidinedione/sulfonylurea hybrid in rat pancreatic islets. J Cell Physiol 234:509–520
Purohit DN (1967) Hydroxytriazenes-a review of a new class of chelating agents. Talanta 14:353–359. https://doi.org/10.1016/0039-9140(67)80009-2
Purohit DN, Tyagi MP, Bhatnagar R, Bishnoi IR (1992) Hydroxytriazenes as chelating agents: a review. Revs Anal Chem 11:269–303. https://doi.org/10.1515/REVAC.1992.11.3-4.269
Kumar S, Garg M, Jodha JS, Singh RP, Pareek N, Chauhan RS, Goswami AK (2009) Studies on insecticidal activity of some hydroxytriazenes derivatives. E-J Chem 6:466–468. https://doi.org/10.1155/2009/943576
Ombaka AO, Muguna AT, Gichumbi JM (2012) Antibacterial and antifungal activities of novel hydroxytriazenes. J Environ Chem Ecotoxicol 4:133–136. https://doi.org/10.5897/JECE12.006
Jain S, Dayma V, Sharma P, Bhargava A, Baroliya PK, Goswami AK (2019) Synthesis of some new hydroxytriazenes and their antimicrobial and anti-inflammatory activities. Anti-Inflamm Anti-Allergy Agents Med Chem 19:50–60. https://doi.org/10.2174/1871523018666190301151826
Regar M, Baroliya PK, Patidar A, Dashora R, Mehta A, Chauhan RS, Goswami AK (2016) Antidyslipidemic and antioxidant effects of novel hydroxytriazenes. Pharm Chem J 50:310–314. https://doi.org/10.1007/s11094-016-1442-x
Goswami AK, Ameta KL, Khan S (2020) Hydroxytriazenes and triazenes: the versatile framework and medicinal applications, 1st edn. CRC Press, Boca Raton
Singh K, Patel P, Goswami AK (2008) Anti-inflammatory activity of hydroxytriazenes and their Vanadium complexes. E-J Chem 5:1144–1148. https://doi.org/10.1155/2008/830737
Agarwal S, Baroliya PK, Bhargava A, Tripathi IP, Goswami AK (2016) Synthesis, characterization, theoretical prediction of activities and evaluation of biological activities of some sulfacetamide based hydroxytriazenes. Bioorg Med Chem Lett 26:2870–2873. https://doi.org/10.1016/j.bmcl.2016.04.051
Sharma P, Dayma V, Dwivedi A, Baroliya PK, Tripathi IP, Vanangamudi M, Chauhan RS, Goswami AK (2020) Synthesis of sulpha drug-based hydroxytriazene derivatives: anti-diabetic, antioxidant, anti-inflammatory activity and their molecular docking studies. Biorg Chem 96:103642. https://doi.org/10.1016/j.bioorg.2020.103642
Dayma V, Chopra J, Sharma P, Dwivedi A, Tripathi IP, Bhargava A, Murugesan V, Goswami AK, Baroliya PK (2020) Synthesis, antidiabetic, antioxidant and anti-inflammatory activities of novel hydroxytriazenes based on sulphadrugs. Heliyon 6:e04787. https://doi.org/10.1016/j.heliyon.2020.e04787
Hasaninezhad F, Tavaf Z, Panahi F, Nourisefat M, Khalafi-Nezhad A, Yousefi R (2020) The assessment of antidiabetic properties of novel synthetic curcumin analogues: α-amylase and α-glucosidase as the target enzymes. J Diabetes Metab Disord 19:1505–1515. https://doi.org/10.1007/s40200-020-00685-z
Zhang P, Li T, Wu X, Nice EC, Huang C, Zhang Y (2020) Oxidative stress and diabetes: antioxidative strategies. Front Med 14:583–600. https://doi.org/10.1007/s11684-019-0729-1
Ceriello A, Testa R, Genovese S (2016) Clinical implications of oxidative stress and potential role of natural antioxidants in diabetic vascular complications. Nutr Metab Cardiovasc Dis 26:285–292. https://doi.org/10.1016/j.numecd.2016.01.006
Balbi ME, Tonin FS, Mendes AM, Borba HH, Wiens A, Llimos FF, Pontarolo R (2018) Antioxidant effects of vitamins in type 2 diabetes: a meta-analysis of randomized controlled trials. Diabetol Metab Syndr 10:1–12. https://doi.org/10.1186/s13098-018-0318-5
Oguntibeju OO (2019) Type 2 diabetes mellitus, oxidative stress and inflammation: examining the links. Int J Physiol Pathophysiol Pharmacol 11:45–63
Ng CY, Kamisah Y, Faizah O, Jaarin K (2012) The role of repeatedly heated soybean oil in the development of hypertension in rats: association with vascular inflammation. Int J Exp Pathol 93:377–387. https://doi.org/10.1111/j.1365-2613.2012.00839.x
Pacurari M, Kafoury R, Tchounwou BP, Ndebele K (2014) The Renin-angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflamm 2014:1–13. https://doi.org/10.1155/2014/689360
Horio E, Kadomatsu T, Miyata K, Arai Y, Hosokawa K, Doi Y, Ninomiya T, Horiguchi H, Endo M, Tabata M, Tazume H, Tian Z, Takahashi O, Terada K, Takeya M, Hao H, Hirose N, Minami T, Suda T, KiyoharaY OH, Kaikita K, Oike Y (2014) Role of endothelial cell-derived Angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arterioscler Thromb Vasc Biol 34:790–800. https://doi.org/10.1161/ATVBAHA.113.303116
Ito F, Sono Y, Ito T (2019) Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants 8:72
De Lavor EM, Fernandes AW, de Andrade Teles RB, Leal AE, de Oliveira Júnior RG, Gama e Silva M, De Oliveira AP, Silva JC, de Moura Fontes Araujo MT, Coutinho HD, De Menezes IR (2018) Essential oils and their major compounds in the treatment of chronic inflammation: a review of antioxidant potential in preclinical studies and molecular mechanisms. Oxid Med Cell Longev 23:6468593
Geronikaki A, Lagunin A, Poroikov V, Filimonov D, Hadjipavlou-litina D, Vicini P (2002) Computer aided prediction of biological activity spectra: Evaluating versus known and predicting of new activities for thiazole derivatives. SAR QSAR Environ Res 13:457–471. https://doi.org/10.1080/10629360290014322
Filimonov DA, Lagunin AA, Gloriozova TA, Rudik AV, Druzhilovskii DS, Pogodin PV, Poroikov VV (2014) Prediction of the biological activity spectra of organic compounds using the PASS online web resources. Chem Heterocycl Compd 50:444–457. https://doi.org/10.1007/s10593-014-1496-1
Wang JL, Limburg D, Granato MJ, Springer J, Joseph HRB, Liao S, Pawlitz JL, Kurumbail RG, Maziasz T, Talley JJ, Kiefer JR, Carter J (2010) The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg Med Chem Lett 20:7159–7163. https://doi.org/10.1016/j.bmcl.2010.07.054
Molecular Operating Environment (MOE) (2016) 2015.10; Chemical Computing Group Inc., 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, https://www.chemcomp.com/index.htm
Galli CL, Sensi C, Fumagalli A, Parravicini C, Marinovich M, Eberini I (2014) A computational approach to evaluate the androgenic affinity of iprodione, procymidone, Vinclozolin and their metabolites. PLoS one 9:e104822. https://doi.org/10.1371/journal.pone.0104822
Tripathi IP, Dwivedi A (2016) Synthesis, characterization and α-glucosidase inhibition of some copper, cobalt, nickel and zinc complexes with N-Methylethylenediamine. Br J Med Med Res 16:1–11. https://doi.org/10.9734/BJMMR/2016/26100
Ilyasov IR, Beloborodov VL, Selivanova IA, Terekhov RP (2020) ABTS/PP decolorization assay of antioxidant capacity reaction pathways. Int J Mol Sci 21:1131. https://doi.org/10.3390/ijms21031131
Re R, Pellegrini N, Proteggente A, Pannala A, Yan M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26:1231–1237. https://doi.org/10.1016/S0891-5849(98)00315-3
Winter CA, Risley EA, Nuss GW (1962) Carrageenan-induced edema in hind paw of the rat as an assay for anti-inflammatory drugs. Exp Biol Med 111:544–547
Blobaum AL, Marnett LJ (2007) Structural and functional basis of Cyclooxygenase inhibition. J Med Chem 50:1425–1441. https://doi.org/10.1021/jm0613166
Li X, Mazaleuskaya LL, Ballantyne LL, Meng H, FitzGerald GA, Funk CD (2018) Genomic and lipidomic analyses differentiate the compensatory roles of two COX isoforms during systemic inflammation in mice1,2[S]. J Lipid Res 59:102–112. https://doi.org/10.1194/jlr.M080028
Simone RD, Chini MG, Bruno I, Riccio R, Mueller D, Werz O, Bifulco G (2011) Structure-based discovery of inhibitors of microsomal prostaglandin E2 Synthase-1,5-lipoxygenase and 5-lipoxygenase-activating protein: promising hits for the development of new anti-inflammatory agents. J Med Chem 54:1565–1575. https://doi.org/10.1021/jm101238d
Khalil NA, Ahmed EM, Mohamed KO, Nissan YM, Zaitone Abo-Bakr S (2014) Synthesis and biological evaluation of new pyrazolone–pyridazine conjugates as anti-inflammatory and analgesic agents. Bioorg Med Chem 22:2080–2089. https://doi.org/10.1016/j.bmc.2014.02.042
Cash JM, Klippel JH (1994) Second-line drug therapy for rheumatoid arthritis. New Eng J Med 330:1368–1375. https://doi.org/10.1056/NEJM199405123301908
Lanas A, Baron JA, Sandler RS, Horgan K, Bolognese J, Oxenius B, Quan H, Watson D, Cook TJ, Schoen R, Burke C, Loftus S, Niv Y, Ridell R, Morton D, Bresalier R (2007) Peptic ulcer and bleeding events associated with rofecoxib in a 3-year colorectal adenoma chemoprevention trial. Gastroenterology 132:490–497. https://doi.org/10.1053/j.gastro.2006.11.012
Harirforoosh S, Asghar W, Jamali F (2013) Trial Adverse effects of nonsteroidal anti-inflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J Pharm Pharmaceut Sci 16:821–847
Wu KKW, Sung JJY, Lee CW, Yu J, Cho CH (2010) Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett 295:7–16. https://doi.org/10.1016/j.canlet.2010.03.015
Noureddine O, Issaoui N, Al-Dossary O (2021) DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19. J King Saud Univ Sci 33:101248
Dehkordi MM, Asgarshamsi MH, Fassihi A, Zborowski KK (2022) A comparative DFT study on the antioxidant activity of some novel 3-hydroxypyridine-4-one derivatives. Chem Biodivers. 8:e202100703
Thompson M A (2004) Arguslab computational chemistry software: a molecular modeling, graphics and drug design program. http://www.arguslab.com
Acknowledgements
Authors are thankful to SAIF, Chandigarh, India and MNIT, Jaipur, India for providing spectral analysis.
Funding
P.K. Baroliya acknowledges the financial support from the Science and Engineering Research Board, Government of India, for the award of the TARE grant (TAR/2018/000282).
Author information
Authors and Affiliations
Contributions
The manuscript was written through the contributions of all authors. All authors have approved to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that there are no conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chauhan, L.K., Chopra, J., Vanangamudi, M. et al. Hydroxytriazenes incorporating sulphonamide derivatives: evaluation of antidiabetic, antioxidant, anti-inflammatory activities, and computational study. Mol Divers 27, 223–237 (2023). https://doi.org/10.1007/s11030-022-10420-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10420-w