
Abstract
The development of new, more selective, environmental-friendly insecticide alternatives is in high demand for the control of Spodoptera frugiperda (S. frugiperda). The major objective of this work was to search for new potential S. frugiperda acetylcholinesterase (AChE) inhibitors. A ligand-based virtual screening was initially carried out considering six scaffolds derived from eugenol and the ZINC15, PubChem, and MolPort databases. Subsequently, molecular docking analysis of the selected compounds on the active site and a second region (determined by blind molecular docking) of the AChE of S. frugiperda was performed. Molecular dynamics and Molecular Mechanics Poisson–Boltzmann Surface Area analyses were also applied to improve the docking results. Finally, three new eugenol analogs were evaluated in vitro against S. frugiperda larvae. The virtual screening identified 1609 compounds from the chemical libraries. Control compounds were selected from the interaction fingerprint by molecular docking. Only three new eugenol analogs (1, 3, and 4) were stable at 50 ns by molecular dynamics. Compounds 1 and 4 had the best biological activity by diet (LC50 = 0.042 mg/mL) and by topical route (LC50 = 0.027 mg/mL), respectively. At least three new eugenol derivatives possessed good-to-excellent insecticidal activity against S. frugiperda.
Graphic abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Spodoptera frugiperda (S. frugiperda) is a worldwide polyphagous insect that affects more than 300 plant species [1]. In particular, S. frugiperda prefers corn, and it is considered corn’s primary pest. Worldwide production of maize in 2018 was 1,147,621,938 tons, of which 52.5% was from the American continent, 29% from Asia, 11.1% from Europe, 7.1% from Africa, and 0.1% from Oceania [2]. S. frugiperda during the larval stage is capable of causing a 45% reduction of total corn production due to the consumption of buds [3]. Current pest management systems use synthetic insecticides, such as organophosphates, carbamates, pyrethrins, pyrethroids, spinosyns, avermectins, nereistoxin, semicarbazones, oxadiazines, and diacylhydrazines to avoid large economic loss. These insecticides act on the nervous and/or muscular system [4] of S. frugiperda; however, extensive and long-term use (sometimes overuse) produces resistance [5]. Moreover, these synthetic insecticides cause soil and aquifer contamination and harm non-target organisms [6]. These adverse effects motivate an urgent need for developing environmentally benign insecticides against S. frugiperda. The major objective of this research is to develop new and novel eco-friendly insecticide(s) from secondary metabolites of plants. A few studies have identified acetylcholinesterase (AChE, EC 3.1.1.7) as a specific target. It is an essential enzyme for acetylcholine hydrolysis located in nerve cells and the neuromuscular synapse [7]. Therefore, its inhibition causes the insect’s death. Although three natural compounds (Fig. 1) have little or no AChE inhibitory and subsequent insecticidal activity against the genus Spodoptera [8], potent insecticidal AChE inhibitors of natural origin against the genus Spodoptera have been developed.

Natural secondary metabolites with AChE inhibition and biological activity against Spodoptera sp.
The natural compound, eugenol, and its derivatives (Fig. 2) have an inhibitory effect on AChE with lethal activity against S. frugiperda [9,10,11,12]. Based on this finding, a ligand-based virtual screening (LBVS) was carried out to find new eugenol analogs with more efficacy and eco-friendly insecticidal activity. Eugenol derivatives that demonstrated a half-maximal inhibitory concentration (IC50) on AChE (< 29.78 μg/mL) and a half-maximal lethal dose (LD50) against S. frugiperda (< 1 mg/g) were used to generate six scaffolds for a database search by similarity (TC, Tanimoto coefficient ≥ 0.8) and substructure in ZINC15, PubChem, and MolPort. Later, the Tice bioavailability rule for insecticides was applied to the compounds. The selected compounds were subjected to molecular docking and molecular dynamics analyses of the AChE of S. frugiperda. Finally, the top three hit compounds in this series were evaluated in an in vivo model (diet and topical) against S. frugiperda larvae to validate the in silico study.

Eugenol and its derivatives with inhibitory effects on AChE of S. frugiperda
Materials and methods
Scaffold selection
The scaffolds (Fig. 3) used in the LBVS of the ZINC15, PubChem, and MolPort databases were designed based on eugenol and its derivatives (Fig. 2) with lethal effects of less than 1 mg/g against S. frugiperda [9, 11,12,13].

The six scaffolds used in the LBVS in the ZINC15, PubChem, and MolPort chemical databases
LBVS
The search for compounds analogous to eugenol was carried out by substructure and similarity using scaffolds 1–6 (Fig. 3) in the three databases: ZINC15 (https://zinc15.docking.org/), PubChem (https://pubchem.ncbi.nlm.nih.gov/) and MolPort (https://www.molport.com/shop/index). Subsequently, using the OpenBabel program, the compounds with a TC value ≥ 0.8 regarding each scaffold were selected by similarity [14]. Duplicate compounds in the three chemical databases were removed. The Tice rule, which includes the following criteria, molecular weight ≤ 500 Da, partition coefficient ≤ 5, hydrogen bond donors ≤ 2, hydrogen bond acceptors ≤ 8, and rotatable bonds ≤ 12 [15], was applied. Subsequently, each compound was minimized, and polar hydrogens were added with prepare_ligand4.py from MGL-Tools; finally, they were changed to PDBQT format adding the charges and type of atom.
AChE construction by homology modeling
The primary sequence of AChE I of S. frugiperda (AGK44160) has been partially determined; therefore, AChE I of S. litura was used and obtained from GenBank in FASTA format with access code AQQ79918 with a length of 694 amino acids (Fig. S1, Table S1). The three-dimensional structure of AChE I of S. frugiperda (AChEmSf) was generated with the Swiss-Model server in template search mode. The primary sequence of AChE I was entered in FASTA format. Multiple alignments were performed automatically in the SWISS-MODEL template library, selecting the templates with an identity greater than 40% and a resolution less than 2 Å, with a good global model quality estimate (GMQE) and quaternary structure quality estimate (QSQE) [16]. Dihedral angles were examined with PROCHECK [17].
AChEmSf preparation
The removal of additional molecules in the three-dimensional structure of the modeled AChE (AChEmSf) obtained from the Swiss-Model was done with the UCSF Chimera program. The polar hydrogens and the Gasteiger charges were added with Dock Prep [18]. The types of atoms were added with MGL-Tools 1.5.6 (http://mgltools.scripps.edu/) to convert to the PDBQT format [19].
Molecular docking of eugenol and derivatives on the AChEmSf
Eugenol and its derivatives with AChE inhibitory activity (Fig. 2) lack information related to enzyme–ligand interactions; therefore, blind molecular docking was performed on AChEmSf. The dimension of the box was 64 Å × 74 Å × 68 Å with a 1 Å spacing set with MGL-Tools. For AutoDock 4.2, a genetic algorithm was used with 100 evaluations, while in AutoDock Vina [20], the same dimensions of the box were maintained. Eugenol and its derivatives (Fig. 2) were drawn in MarvinSketch version 20.3.0 [21], saved in mol format, and then processed according to the ligand preparation section. The poses of the ligands were visualized in the PyMOL open-source version 2.4.0a0 program [22] to determine the regions where the highest number of dockings occurred.
Molecular docking of the new eugenol ligands on AChEmSf
The docking of the compounds obtained by LBVS was carried out with AutoDock Vina in two regions on the AChEmSf: (a) on the active site, where the center coordinates correspond to X = 3.442, Y = 2.478, Z = 27.206; and (b) on region 2, determined by blind molecular docking according to the highest number of poses docked in the protein with coordinates X = −4.332, Y = 6.078, Z = 38.027, both with a box dimension of 20 Å × 20 Å × 20 Å. Compounds with free binding energy lower than control, acetylcholine (CID 187), the natural substrate of AChE, malathion (CID 4004), a carbamate insecticide, methomyl (CID 5,353,758), an organophosphate insecticide, insecticides that inhibit AChE reversibly and irreversibly and are used to control S. frugiperda, and 9-(3-iodobenzylamino)-1,2,3,4-tetrahydroacridine (ZAI, PDB code: 1QON), a compound that inhibits the AChE of Drosophila melanogaster, were selected for further study. Interaction fingerprints of compounds with a lower free binding energy to malathion docked in AChEmSf were generated with the Open Drug Discovery Toolkit Version 0.7 program [23] using the Simple Interaction Fingerprint parameter [24], which considers eight types of interaction: hydrophobic contacts, face-to-face, edge-to-face π–π interaction, hydrogen donor, hydrogen acceptor, positive and negatively charged salt bridge, and an ionic bond with a metal ion. The interaction fingerprints of these new eugenol analogs were compared with the interaction fingerprints of the control compounds (malathion, acetylcholine, methomyl, and ZAI) and eugenol and its derivatives. The compounds with a TC > 7 were selected for further analysis [25]. Additionally, Protein–Ligand Interaction Profiler [26] was used to obtain the complex interaction profile.
Molecular dynamics of the AChEmSf–eugenol analog complex
Molecular dynamics analysis was performed with GROMACS version 2018.4 [27]. First, the topology of the eight compounds selected by LBVS, the ZAI control, and isoeugenol was generated in the LigParGen server [28] and the AChEmSf with GROMACS with the OPLS force field. Solvation was done with water molecules in a dodecahedron with a minimum distance from the wall of 10 Å, using the SPCE water model. Then, Na + and Cl− ions were added to neutralize the system with an energy minimization of 50,000 times. Later, the system was equilibrated with several particles, volume, and temperature (NVT), and several particles, pressure, and temperature (NPT), both at 100 ps. Finally, the simulation was performed at a temperature of 300 K for 50 ns [29, 30]. The root means square deviation (RMSD) of each complex was obtained to determine the stability of the complex. The binding energy was estimated with molecular mechanics Poisson–Boltzmann surface area (MMPBSA) with the g_mmpbsa [31] program, recovering 50 frames every 0.2 ns of 40–50 ns. Finally, the energy contribution of each amino acid was obtained with the MmPbSaDecomp.py script [32].
S. frugiperda larvae
S. frugiperda larvae were collected from corn crops in H33V + JQ Celaya, Tamaulipas, Mexico. The broods were nurtured at a temperature of 26 °C, with a photoperiod of 12 h light: 12 h darkness and 60% relative humidity. The larvae were fed an artificial diet (Southland Products Inc., type diet: fall armyworm), and each larva was reared in a 3 cm diameter × 4 cm high plastic bottle. The pupae were transferred to a 20 cm diameter × 15 cm high plastic container lined with absorbent paper for moth oviposition. The adults were fed with 10% sucrose dissolved in sterile water. The moth eggs were collected every 24 h and transported to a hemispherical container with 30 mL of an artificial diet for the hatching of the larvae.
Insecticidal biological activity
Three compounds proposed by molecular dynamics were purchased from MolPort (Latvia) with the following codes: MolPort-011–119-237 (ZINC000034423376, 20 mg), MolPort-046–802-450 (20 mg) and MolPort-002–797-882 (ZINC000013941780, 20 mg). The evaluation of the insecticidal activity of the compounds was carried out with 25 neonatal larvae each time in triplicate (n = 75) by topical application and ingestion.
For topical evaluation, 1 μL was applied to the dorsum of the larva. Six concentrations were evaluated for each compound (1 mg/mL, 0.5 mg/mL, 0.250 mg/mL, 0.100 mg/mL, 0.05 mg/mL, and 0.01 mg/mL) dissolved in 2% DMSO in a 2 mL tube. The treated larvae were placed in flasks with pieces of an artificial diet with a 0.5 mm × 0.5 mm dimension. In the diet bioassay, the concentrations evaluated were 0.250 mg/mL, 0.100 mg/mL, 0.05 mg/mL, and 0.01 mg/mL dissolved in 2% DMSO. The compound was incorporated into 20 mL of artificial diet. Once solidified, the whole diet was divided into 75 small portions put in 1-oz jars where neonate larvae of S. frugiperda were added. Mortality (larvae that did not respond to a slight sting with a brush) was recorded for both topical application and ingestion and determined every 24 h until 168 h [12, 33].
The half-maximal lethal concentration (LC50) was determined with a Probit analysis in the SPSS statistical program with a significance level of 0.05.
Results and discussion
Plant-derived compounds have been associated with low toxicity and rapid reincorporation into the environment [8]. For this reason, six scaffolds of semisynthetic compounds (derived from eugenol) were generated in this study. Their main feature is a propyl group with and without the trans geometry [12]. This property is suggested to be related to the IC50 on AChE and the LC50 against S. frugiperda larvae.
AChE S. frugiperda construction by homology modeling
The structure of a T. californica AChE (PDB ID: 6G1U, resolution 1.79 Å) was selected as a template due to 44.82% identity and 77% coverage of the primary amino acid sequence of S. litura AChE, and the sequence AChE I of S. litura was used because it has 100% identity with the AChE I S. frugiperda (Fig. S1, Table S1). In the three-dimensional modeling of the AChE, I of S. frugiperda (AChEmSf), a QMEAN of −1.7 was obtained, indicating good quality since the acceptance range is 0 to −4 [34]. The dihedral angles of the amino acids were 87.8% in favored regions, 10.9% in additionally favored regions, whereas 1.3% are generally permitted regions (Fig. S2). A quality structure is considered when ≥ 90% of the residues are in the favored regions A, B, and L. Accordingly, the modeled AChEmSf possesses acceptable quality.
The structural comparison of 6G1U with AChEmSf in PDB-eFold [35] shows an RMSD of 0.56 Å, indicating that the overall folding AChEmSf is similar to the template (Fig. 4), which also allowed identification of homologous residues on the active site of AChEmSf, an important finding in the catalysis [7].

source Version 2.4
AChEmSf residues involved in acetylcholine hydrolysis. In T. californica (PDB: 6G1U) (a, b), in the AChEmSf (c, d). The residues for AChEmSf (d) in yellow correspond to the active site (S313, H553, and E439), in gray to the oxyanion cavity (G232, G233, and A314), in light blue, the anionic site (W198, Y244, Y442, and F443), in orange the acyl cavity (C400 and F402) and the peripheral anionic site (PAS) in aquamarine (I184, D186, Y235, W394, and Y446). The images were generated in PyMOL open-
The three-dimensional model of the AChEmSf shows an identity greater than 30%. This percentage is considered adequate for subsequent studies [36, 37]. Furthermore, the structural alignment of 6G1U and AChEmSf indicates that the amino acids that participate in the catalysis of acetylcholine were properly constructed, and all the dihedral angles of the amino acids were in the allowed regions; therefore, the structure of AChEmSf should be considered appropriate for molecular docking and dynamics studies.
Molecular docking of controls, eugenol, and its derivatives on the active site of AChEmSf
Initially, the molecular docking analysis of the control’s acetylcholine, malathion, methomyl, ZAI, eugenol, and its derivatives was carried out on the active site of AChEmSf. The binding energy values and the interaction profile are shown in Table 1. Considering the interaction profile, acetylcholine and methomyl interacted through hydrogen bonds with the catalytic residue (S313) and malathion with a salt bridge with H553. Only methomyl interacted with G233, which is part of the oxyanion cavity. The eugenol derivatives formed hydrogen bonds on the active site with S313, the oxyanion cavity G233, and eugenol itself with H553.
LBVS databases
The identification of new eugenol analogs as potential inhibitors of AChEmSf from the ZINC15, PubChem, and MolPort databases was based on the search by similarity and substructure, using six scaffolds (Fig. 3, methodology). Initially, 22,137 compounds were selected by similarity from the three databases. This number was reduced to 1198 by applying a TC ≥ 0.8 [38] with respect to their scaffold. Similarly, 7200 were selected from the three databases by substructure. As a second criterion, the Tice rule was applied to select compounds with bioavailability; 582 and 1027 compounds obtained by similarity and substructure, respectively (Table S2), were selected for molecular docking analysis by AutoDock Vina, which is highly reliable [39] in predicting insecticidal activity [33] on the active site of AChEmSf.
Molecular docking of the compounds on the active site of AChEmSf
A total of 582 (similarity) and 1027 (substructure) compounds were simulated by molecular docking on the active site of AChEmSf. Considering the binding energy of methomyl (−5.1 kcal/mol, Table 1) as a cutoff point, 537 compounds by similarity and 998 compounds by substructure with a key substructure achieved this requirement. The interaction fingerprints of the compounds that reached the cutoff point were generated by the Open Drug Discovery Toolkit (ODDT), which compared the interaction fingerprints of the controls, eugenol, and its derivatives, selecting the compounds with a similarity (TC > 0.7) in the interaction, regarding 233 compounds by similarity and 331 compounds by substructure (Fig. S3), a criterion selected by Mojaddami et al. to obtain potentially active compounds. This method made it possible to obtain new compounds maintaining the eugenol structure with diversification in smaller functional groups since these were considered better in work carried out by Rosado-Solano et al.
The selected compounds were later ranked according to docking binding energy. The ten compounds (Fig. S4) with the best binding energy (from −10.8 to −11.6 kcal/mol) were selected. The binding energy could be attributed to the size of the compounds. Interactions such as hydrogen bonds on the active site and with an oxyanion cavity were observed. Also, most compounds have π–π stacking interactions since they have more than one aromatic ring. However, the compounds were discarded because they did not show the structure of some scaffolds, and these compounds are not commercially available.
A manual search of the 233 compounds (similarity) and 331 compounds (substructure) using the six scaffolds as a reference and their commercial availability was carried out. Therefore, five compounds belonging to scaffolds 1, 2, 4, and 6 with the best free binding energy (−6.6 kcal/mol to −7.8 kcal/mol) were selected for molecular dynamics analysis (Fig. 5).

Interactions of five new eugenol analogs on the active site of AChEmSf with the best free binding energy
All five compounds (Fig. 5) demonstrated interactions with AChEmSf residues that catalyze acetylcholine (Fig. 4); therefore, these compounds could compete with the natural substrate. Compounds 1–5 interacted on the active site by a hydrogen bond with S313. Only compound 5 showed interaction with H553 by a salt bridge. Compounds 2–5 on the oxyanion hole interacted with G232 and G233 through hydrogen bonds, while compound 1 only interacted with G232. Compound 2 on the anionic site interacted hydrophobically with W198, while compound 3 with π–π stacking; compounds 2–4 interacted hydrophobically with Y244, and compound 3 showed an additional interaction through a hydrogen bond. Compounds 1 and 4 interact hydrophobically with the residue F443, while compounds 1 and 5 with π–π stacking. Compounds 1 and 4 on the acyl pocket have hydrophobic interactions with F402, while compound 5 showed π–π stacking. Compounds 1, 4, and 5 on PAS interact with Y235 hydrophobically, and compound 4 showed an additional hydrophobic interaction with W394.
Blind molecular docking of controls, eugenol, and derivatives on AChEmSf
Since the binding mode of eugenol and derivatives on AChE has not been reported (Fig. 2), blind molecular docking (AutoDock 4.2 and AutoDock Vina) was carried out, counting the number of poses of each compound with the protein. The same analysis was also considered for the control compounds. The results showed six regions where the compounds had a higher affinity (Table S3). However, the sites with the highest number of docked poses were considered possible binding regions. Region 1, corresponding to the active site (previously analyzed), has 549 docked poses, and region 2 has 139 docked poses (Fig. 6). The remaining regions were discarded for further analysis due to the small number of poses counted (< 105, Table S3) on molecular docking, indicating that these regions have little affinity for eugenol and derivatives. Based on this information, the molecular docking of controls compounds, eugenol, and its derivatives in region 2 was considered for a new analysis.

Proposed regions as active sites for AChEmSf: The cavity in yellow is region 1 (the active site previously reported), and the cavity in green is region 2
The control compounds were docked on region 2, and a binding energy of −3.8 to −7.7 kcal/mol was observed, while for eugenol and its derivatives, the observed range was −5.2 to −5.6 kcal/mol (Table 2). In silico, acetylcholine demonstrated weak binding energy than the control compounds, and eugenol and derivatives had activity against the AChE of S. frugiperda.
Subsequently, the 537 compounds obtained by similarity and the 997 by substructure, complied with the Tice rule, were analyzed by molecular docking on region 2 of AChEmSf to obtain their binding energy and interaction fingerprint. The compounds that showed an interaction similarity > 0.7 with respect to controls, eugenol, and derivatives were selected (Table 2), resulting in 172 compounds by similarity and 235 compounds by substructure in region 2 (Fig. S5). The ten compounds (Fig. S6) with the best binding energy (−8.1 to −10.1 kcal/mol) were selected. The compounds have more than one aromatic ring responsible for hydrophobic interactions. However, the compounds were discarded because they did not have the structure of some scaffolds, and they were not commercially available.
Therefore, based on the six scaffolds (Fig. 3) and their commercial availability, a manual search of the 172 (similarity) and 235 (substructure) compounds was carried out. Compounds 1, 2, and 3 (Fig. 7), respectively, derived from scaffolds 2, 3, and 5, showed the best binding energy (−5.6 kcal/mol to −6.3 kcal/mol) over region 2 of AChEmSf. These compounds were selected for molecular dynamics analysis (Fig. 7).

Interactions of eugenol analogs on region 2 of AChEmSf with the best free binding energy
In region 2, due to the lack of information on the proposed cavity and its ability to bind inhibitors, the residues observed in interaction with the four compounds were emphasized. Hydrophobic interactions with P345, Y511, W640, Q641, L644, P645, and hydrogen bonds with T343 were considered.
Molecular dynamics analysis of the new eugenol analogs on AChEmSf
Many molecules were selected from the interaction fingerprint. Molecular dynamics were carried out based on the commercial availability and binding affinity on the active site and region 2 of AChEmSf. Initially, the molecular dynamics analysis of AChEmSf was carried out, showing an RMSD of 0.01 to 3.06 Å. The complex with the reference compound ZAI varied from 0.79 to 4.63 Å, showing a difference between the oscillations of 3.83 Å, suggesting a complex with good stability. Furthermore, isoeugenol showed an oscillation of 0.85 to 6.72 Å in the first 10 ns and then stabilized with a fluctuation of 4.17 to 6.52 Å. Subsequently, the eight selected new eugenol analogs were analyzed (5 compounds that interact on the active site and 3 that interact on region 2 of the AChEmSf). This analysis allowed determining the stability of the complex formed in the molecular docking. Compounds with an RMSD lower than ZAI (4.63 Å) were selected: compound 1 (ZIN000034423376) with a fluctuation from 0.60 to 3.55 Å, compound 3 (Molport-046–802-450) from 0.67 to 3.94 Å, and compound 4 (ZIN000013941780) from 0.54 to 3.55 Å (Fig. 8), with a difference between the oscillations of 2.95 Å, 3.27 Å, and 3.01 Å, respectively. These compounds appeared to be the most stable. In most molecular dynamics simulations, the trajectory generally has a difference of 0.7–9 Å [40,41,42,43] depending on the receptor–ligand complex analyzed. However, according to Zhang et al. [44], the compound with the lowest RMSD and minimal differences between the oscillations is the most stable. Based on the above, these three compounds were purchased from MolPort for in vivo evaluation against S. frugiperda larvae. The result of molecular dynamics for the five remaining compounds presented a very large fluctuation of RMSD. This finding was interpreted as an unstable complex over time; thus, these compounds were discarded for subsequent biological activity studies (see RMSD Fig. S7).

RMSD of ZAI, isoeugenol, and three new eugenol analogs with good stability in the molecular dynamic’s simulation
Per residue energy decomposition
The energy contribution analysis in molecular dynamics is an important step in determining residues of key amino acids in their interaction with ligands.
The energy contribution of amino acids in the binding energy of isoeugenol and new eugenol analogs was analyzed by molecular dynamics in AChEmSf (Fig. 9). Isoeugenol presented six amino acids with an energy contribution less than −0.5 kcal/mol: S313 (−0.6 kcal/mol), W346 (−1.14 kcal/mol), E401 (−0.57 kcal/mol), F402 (−0.63 kcal/ mol), F443 (−0.8 kcal/mol), and F513 (−0.73 kcal/mol), except E312 (1.27 kcal/mol). Compound 1 presented nine residues that contribute to the free binding energy less than −0.5 kcal/mol: G233 (−0.68 kcal/mol), Y235 (−0.56 kcal/mol), W346 (−1.74 kcal/mol), E393 (−0.51 kcal/mol), F402 (−0.91 kcal/mol), E439 (−0.58 kcal/mol), F443 (−2.01 kcal/mol), H512 (−1.0 kcal/mol), and F513 (−0.89 kcal/mol), and a residue contributes more than 1 kcal/mol, while for compound 3 four residues are involved with less than −0.5 kcal/mol in binding energy: W198 (−1.99 kcal/mol), E401 (−0.53 kcal/mol), Y442 (−0.79 kcal/mol), and F443 (−0.64 kcal/mol), except E312 (1.25 kcal/mol). Eleven AChEmSf residues aid binding with less than −0.5 kcal/mol in the compound 4: W198 (−0.62 kcal/mol), G233 (−0.78 kcal/mol), Y235 (−0.74 kcal/mol), S313 (−0.84 kcal/mol), A314 (−1.07 kcal/mol), F402 (−1.4 kcal/mol), E439 (−0.58 kcal/mol), F443 (−2.48 kcal/mol), Y447 (−1.78 kcal/mol), F513 (−1.07 kcal/mol), H553 (−0.52 kcal/mol), except E312 (3.41 kcal/mol).

The energy contribution of amino acids in AChEmSf calculated with MMPBSA. Derivative of eugenol (a), compound 1 (b), compound 3 (c), and compound 4 (d). In the X-axis, the number of residues is shown; in the Y-axis, the energy contribution in kcal/mol; the dotted blue lines are equivalent to ± 1 kcal/mol; and the residues that contribute less than ± 0.5 kcal/mol in the interaction are in blue
Analysis by MMPBSA showed that in compounds 1, 3, and 4, the residue F443 of the anionic site largely contributed to the binding energy of −0.64 to −2.48 kcal/mol and the polar part E312 with 1.25 at 3.41 kcal/mol. Additionally, the residues greater than 1 kcal/mol in compound 1 were W346 and H51; the latter close to the active site; in compound 3, only W198 (anionic site); in compound 4, it was A314 (oxyanion cavity), F402 (acyl cavity), and Y447. Finally, the number of residues contributing less than −0.5 kcal/mol in compounds 4 > 1 > 3 could explain why compound 4 had the best in vivo activity.
Biological activity of the new eugenol analogs against S. frugiperda larvae
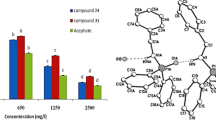
Compounds 1, 3, and 4 were evaluated against G3 neonate larvae of S. frugiperda for 7 days in two ways: diet and topical. Through a Probit analysis in IBM SPSS with a significance level of 0.05, the LC50 for the three compounds was determined. In the evaluation of the three tested compounds by diet, four concentrations were used.
In the diet bioassay against S. frugiperda larvae after 168 h, compound 1 presented the best LC50 (0.042 mg/mL) (Table 3). However, it did not have the lowest binding free energy (−6.6 kcal/mol) in the analysis of molecular docking of the three selected compounds. However, compound 1 is a derivative of scaffold 1 (based on trans-anethole, LD50 = 0.027 mg/g), presenting two primary amine groups without the trans geometry in the propyl moiety, which could favor biological activity through a free rotation and hydrogen bond. Compounds 3 and 4 presented LC50 values (0.060 mg/mL and 0.062 mg/mL), both derived from reference scaffold 4, which could explain the similar LC50 values. The first compound with the imidazolidine-2,4-dione group bond to the trans geometry could restrict rotation; therefore, it could influence the LC50 value, while compound 4, with an improvement of 0.002 at the LC50 with regard to compound 3, has a more bulky group N-(2-hydroxypyridin-3-yl)prop-2-enamide than the other two compounds. Precisely, this group is the one that forms the hydrogen bonds with the active site and the cavity oxyanion.
The LC50 of compound 3 could not be determined in the topical evaluation due to similar mortality in all the concentrations evaluated. The best LC50 was shown by compound 4 (0.027 mg/mL) (Table 3). It was the second best in the diet test, followed by compound 1 (0.037 mg/mL). In both bioassay application routes, the LC50 was calculated, except for compound 3. These findings indicate that the compounds could be effective in the field through these two application routes. Comparing the compounds with the best LC50 values, compound 4 (topical) is 1.55 times better than compound 1 (diet). With this, it was observed that the route of application affects the effectiveness of each compound, for which it can be inferred that compound 4 is better overcoming the barrier of the insects in the tegument; however, in the diet, it presented an LC50 of 0.060 mg/mL, which suggests that it would not have a good inhibitory effect on AChE.
The LC50 of the three compounds is consistent with the study of Torres et al. [45], where they showed the extract (secondary metabolites) of Yucca periculosa with LC50 values of 5.4 ppm (0.0054 mg/mL), 6.4 ppm (0.0064 mg/mL), and 27.6 ppm (0.0276 mg/mL) with a structure similar to the compounds evaluated in this work, supporting that the LC50 of the three compounds is comparable to the synthesized compounds, LD50 = 0.65 mg/g and LD50 = 27.6 mg/g [33]. The compounds evaluated in this work were more effective.
Conclusions
The use of six scaffolds from eugenol and its derivatives with inhibitory activity on the AChE of S. frugiperda for the LBVS of compounds by substructure and similarity in the databases used led to the identification of five new eugenol analogs with excellent affinity to AChEmSf. The blind molecular docking of eugenol and its derivatives with activity on S. frugiperda AChE elucidated an additional region to the active site with a higher affinity. Additionally, the combination of LBVS and subsequent molecular docking resulted in the selection of eight new eugenol analogs that mimicked the interaction of the control compounds, eugenol, and their derivatives on AChEmSf. However, the molecular dynamics analysis optimized compound selection by evaluating the binding stability of the eight compounds in an aqueous system to obtain three new eugenol analogs (1, 3, and 4) with an RMSD with an oscillation between 1 and 3.9 Å. The determination of the insecticidal activity of the three compounds in neonate S. frugiperda larvae allowed validation of the in silico study by obtaining compounds with an LC50 lower than 0.062 mg/mL. This study may help in developing new and novel eco-friendly insecticides through further research.
Data availability
Data are available from the authors upon reasonable request.
References
EPPO (2020) European and Mediterranean Plant Protection Organization. Spodoptera frugiperda. https://gd.eppo.int/taxon/LAPHFR. Accessed 15 Mar 2020
FAO (2020) Food and Agriculture Organization of the United Nations. FAOSTAT, Data, Production, Crops. http://www.fao.org/faostat/en/#compare. Accessed 08 Nov 2020
Blanco CA, Pellegaud JG, Nava-Camberos U, Lugo-Barrera D, Vega-Aquino P, Coello J et al (2014) Maize pests in Mexico and challenges for the adoption of integrated pest management programs. J Integr Pest Manag 5(4):E1–E9. https://doi.org/10.1603/IPM14006
IRAC (2020) Lepidoptera Insecticide Mode of Action Classification: A key to effective insecticide resistance management. https://irac-online.org/lepidoptera/. Accessed 03 Sept 2020
Sparks TC, Nauen R (2015) IRAC: mode of action classification and insecticide resistance management. Pest Biochem Physiol 121:122–128. https://doi.org/10.1016/j.pestbp.2014.11.014
Prasanna B, Huesing J, Eddy R, Peschke V (2018) Fall armyworm in Africa: a guide for integrated pest management, 1st edn. CIMMYT, Mexico, CDMX
Dvir H, Silman I, Harel M, Rosenberry TL, Sussman JL (2010) Acetylcholinesterase: from 3D structure to function. Chem Biol Interact 187(1–3):10–22. https://doi.org/10.1016/j.cbi.2010.01.042
Hernández-Carlos B, Gamboa-Angulo M (2019) Insecticidal and nematicidal contributions of Mexican Flora in the search for safer biopesticides. Molecules 24(5):897. https://doi.org/10.3390/molecules24050897
Cruz GS, Wanderley-Teixeira V, Oliveira JV, D’assunção CG, Cunha FM, Teixeira ÁA et al (2017) Effect of trans-anethole, limonene and your combination in nutritional components and their reflection on reproductive parameters and testicular apoptosis in Spodoptera frugiperda (Lepidoptera: Noctuidae). Chem Biol Interact 263:74–80. https://doi.org/10.1016/j.cbi.2016.12.013
Melani D, Himawan T, Afandhi A (2016) Bioactivity of sweet flag (Acorus calamus Linnaeus) essential oils against Spodoptera litura Fabricius (Lepidoptera: Noctuidae). J Trop Life Sci 6(2):86–90. https://doi.org/10.11594/jtls.06.02.04
Menichini F, Tundis R, Loizzo MR, Bonesi M, Marrelli M, Statti GA et al (2009) Acetylcholinesterase and butyrylcholinesterase inhibition of ethanolic extract and monoterpenes from Pimpinella anisoides V Brig. (Apiaceae). Fitoterapia 80(5):297–300. https://doi.org/10.1016/j.fitote.2009.03.008
Vargas-Méndez LY, Sanabria-Flórez PL, Saavedra-Reyes LM, Merchan-Arenas DR, Kouznetsov VV (2018) Bioactivity of semisynthetic eugenol derivatives against Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae infesting maize in Colombia. Saudi J Biol Sci. https://doi.org/10.1016/j.sjbs.2018.09.010
Mukherjee PK, Kumar V, Mal M, Houghton PJ (2007) In vitro acetylcholinesterase inhibitory activity of the essential oil from Acorus calamus and its main constituents. Planta Med 73(03):283–285. https://doi.org/10.1055/s-2007-967114
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminform 3(1):33. https://doi.org/10.1186/1758-2946-3-33
Tice CM (2001) Selecting the right compounds for screening: does Lipinski’s Rule of 5 for pharmaceuticals apply to agrochemicals? Pest Manag Sci Former Pest Sci 57(1):3–16. https://doi.org/10.1002/1526-4998(200101)57:1%3c3::AID-PS269%3e3.0.CO;2-6
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303. https://doi.org/10.1093/nar/gky427
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291. https://doi.org/10.1107/S0021889892009944
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ (2016) Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11(5):905–919. https://doi.org/10.1038/nprot.2016.051
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
ChemAxon (2020) Marvin. https://chemaxon.com/products/marvin. Accessed 03 Feb 2020
Seeliger D, de Groot BL (2010) Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 24(5):417–422. https://doi.org/10.1007/s10822-010-9352-6
Wójcikowski M, Zielenkiewicz P, Siedlecki P (2015) Open drug discovery toolkit (ODDT): a new open-source player in the drug discovery field. J Cheminform 7(1):1–6. https://doi.org/10.1186/s13321-015-0078-2
Chupakhin V, Marcou G, Gaspar H, Varnek A (2014) Simple ligand-receptor interaction descriptor (SILIRID) for alignment-free binding site comparison. Comput Struct Biotechnol J 10(16):33–37. https://doi.org/10.1016/j.csbj.2014.05.004
Baptista LPR, Sinatti VV, Da Silva JH, Dardenne LE, Guimarães AC (2019) Computational evaluation of natural compounds as potential inhibitors of human PEPCK-M: an alternative for lung cancer therapy. Adv Appl Bioinform Chem 12:15. https://doi.org/10.2147/AABC.S197119
Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M (2015) PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res 43(W1):W443–W447. https://doi.org/10.1093/nar/gkv315
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B et al (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Dodda LS, Cabeza de Vaca I, Tirado-Rives J, Jorgensen WL (2017) LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res 45(W1):W331–W336. https://doi.org/10.1093/nar/gkx312
Polishchuk P, Kutlushina A, Bashirova D, Mokshyna O, Madzhidov T (2019) Virtual screening using pharmacophore models retrieved from molecular dynamic simulations. Int J Mol Sci 20(23):5834. https://doi.org/10.3390/ijms20235834
Lemkul J (2018) From proteins to perturbed Hamiltonians: a suite of tutorials for the GROMACS-2018 molecular simulation package [article v1. 0]. Living J Comput Mole Sci 1(1):5068. https://doi.org/10.33011/livecoms.1.1.5068
Kumari R, Kumar R, Lynn A (2014) g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–62. https://doi.org/10.1021/ci500020m
Liu J-Y, Chen X-E, Zhang Y-L (2015) Insights into the key interactions between human protein phosphatase 5 and cantharidin using molecular dynamics and site-directed mutagenesis bioassays. Sci Rep 5(1):1–11. https://doi.org/10.1038/srep12359
Rosado-Solano DN, Sanabria-Florez PL, Barón-Rodríguez MA, Luna-Parada LK, Puerto Galvis CE, Zorro-González AF et al (2019) Synthesis, biological evaluation and in silico computational studies of 7-Chloro-4-(1H–1, 2, 3-triazol-1-yl) quinoline derivatives. Search for new controlling agents against Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae. J Agric Food Chem. https://doi.org/10.1021/acs.jafc.9b01067
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27(3):343–350. https://doi.org/10.1093/bioinformatics/btq662
Velankar S, Alhroub Y, Best C, Caboche S, Conroy MJ, Dana JM, et al (2012) PDBe: Protein Data Bank in Europe. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr998
Haddad Y, Adam V, Heger Z (2020) Ten quick tips for homology modeling of high-resolution protein 3D structures. PLoS Comput Biol 16(4):e1007449. https://doi.org/10.1371/journal.pcbi.1007449
Xiang Z (2006) Advances in homology protein structure modeling. Current Protein Peptide Sci 7(3):217–227. https://doi.org/10.2174/138920306777452312
Fortney K, Griesman J, Kotlyar M, Pastrello C, Angeli M, Sound-Tsao M et al (2015) Prioritizing therapeutics for lung cancer: an integrative meta-analysis of cancer gene signatures and chemogenomic data. PLoS Comput Biol 11(3):e1004068. https://doi.org/10.1371/journal.pcbi.1004068
Nguyen NT, Nguyen TH, Pham TNH, Huy NT, Bay MV, Pham MQ et al (2019) Autodock vina adopts more accurate binding poses but autodock4 forms better binding affinity. J Chem Inf Model 60(1):204–211. https://doi.org/10.1021/acs.jcim.9b00778
Bhowmik D, Jagadeesan R, Rai P, Nandi R, Gugan K, Kumar D (2020) Evaluation of potential drugs against leishmaniasis targeting catalytic subunit of Leishmania donovani nuclear DNA primase using ligand based virtual screening, docking and molecular dynamics approaches. J Biomole Struct Dyn. https://doi.org/10.1080/07391102.2020.1739557
Kumari M, Subbarao N (2020) Virtual screening to identify novel potential inhibitors for Glutamine synthetase of Mycobacterium tuberculosis. J Biomole Struct Dyn 38(17):5062–5080. https://doi.org/10.1080/07391102.2019.1695670
Liao KH, Chen K-B, Lee W-Y, Sun M-F, Lee C-C, Chen CY-C (2014) Ligand-based and structure-based investigation for Alzheimer’s disease from traditional Chinese medicine. Evidence-Based Complement Altern Med. https://doi.org/10.1155/2014/364819
Rampogu S, Son M, Park C, Kim H-H, Suh J-K, Lee KW (2017) Sulfonanilide derivatives in identifying novel aromatase inhibitors by applying docking, virtual screening, and MD simulations studies. Biomed Res Int. https://doi.org/10.1155/2017/2105610
Zhang X, Yan J, Wang H, Wang Y, Wang J, Zhao D (2020) Molecular docking, 3D-QSAR, and molecular dynamics simulations of thieno [3, 2-b] pyrrole derivatives against anticancer targets of KDM1A/LSD1. J Biomole Struct Dyn. https://doi.org/10.1080/07391102.2020.1726819
Torres P, Avila JG, de Vivar AR, Garcia AM, Marin JC, Aranda E et al (2003) Antioxidant and insect growth regulatory activities of stilbenes and extracts from Yucca periculosa. Phytochemistry 64(2):463–73. https://doi.org/10.1016/S0031-9422(03)00348-0
Funding
The authors have no relevant financial or non-financial interests to disclose.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Méndez-Álvarez, D., Herrera-Mayorga, V., Juárez-Saldivar, A. et al. Ligand-based virtual screening, molecular docking, and molecular dynamics of eugenol analogs as potential acetylcholinesterase inhibitors with biological activity against Spodoptera frugiperda. Mol Divers 26, 2025–2037 (2022). https://doi.org/10.1007/s11030-021-10312-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10312-5