
Abstract
Development of potential antitubercular molecules is a challenging task due to the rapidly emerging drug-resistant strains of Mycobacterium tuberculosis (M.tb). Structure-based approaches hold greater benefit in identifying compounds/drugs with desired polypharmacological profiles. These methods can be employed based on the knowledge of protein binding sites to identify the complementary ligands. In this study, polypharmacology guided computational drug repurposing approach was applied to identify potential antitubercular drugs. 20 important druggable protein targets in M.tb were considered from the target library of Molecular Property Diagnostic Suite–Tuberculosis (MPDSTB–http://mpds.neist.res.in:8084) for virtual screening. FDA approved drugs were collected, preprocessed and docked in the active sites of the 20 M.tb targets. The top 300 drug molecules from each target (20 × 300) were filtered-in and subsequently screened for possible antitubercular and antimycobacterial activity using PASS tool. Using this approach, 34 drugs with predicted antitubercular and anti-mycobacterial activity were identified along with good binding affinity against multiple M.tb targets. Interestingly, 21 out of the 34 identified drugs are antibiotics while 4 drug molecules (nitrofural, stavudine, quinine and quinidine) are non-antibiotics showing promising predicted antitubercular activity. Most of these molecules have the similar privileged antimycobacterial drugs scaffold. Further drug likeness properties were calculated to get deeper insights to M.tb lead molecules. Interestingly, it was also observed that the drugs identified from the study are under different stages of drug discovery (i.e., in vitro, clinical trials) for the effective treatment of various diseases including cancer, degenerative diseases, dengue virus infection, tuberculosis, etc. Krasavin et al., 2017 synthesized nitrofuran analogues with appreciable MICs (22–23 µM) against M.tb H37Rv. These experiments further add to the credibility of the drugs identified in this study (TB).
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, tuberculosis (TB) has taken a more dreadful shape as compared to any other time in human history. It has become one of the most prevalent diseases in the world, next to HIV-AIDS, and hence, it requires specific attention to cure the disease [1]. According to the recent WHO report, 10 million people are affected by TB and 1.4 million people have died due to TB. Amongst all the TB burdened countries, eight countries including India account for two-thirds of the total reported incidents [2]. M.tb drug resistance is the major challenge in antimycobacterial drug development due to the rapid mutations in genes, its complex cell wall, influx-efflux system, drug inactivity and modifying enzymes [3, 4]. The multidrug resistance and poor permeability of the drugs across the cell wall of membrane attributed to this unique arrangement of lipid-protein complex in the cell membrane [5,6,7]. The TB treatment includes the first line drugs rifampicin, isoniazid,ethambutol, streptomycin and pyrazinamide and second line of drugs (i.e., Fluoroquinolones, kanamycin and amikacin). Figure 1 showed the list of drugs under different stages to combat TB. In 2017, Sastry et al. [8], developed an indigenous disease specific web portal known as Molecular Property Diagnostic Suite for tuberculosis (MPDSTB). The portal contains different modules viz. data library, data processing and data analysis. The portal has extensive information about the M.tb genes, proteins, polypharmacological information, literature and it has the chemoinformatics, bioinformatics and computer aided drug design modules for lead finding, design and optimisation. The MPDSTB has a workflow management system where users can interconnect multiple modules for a user specific task (http://mpds.neist.res.in:8084). However, the emergence of novel M.tb strains has pushed us to develop novel alternative new treatment methods. Despite substantial efforts to arrive at more effective anti-TB therapeutic agents, the progress in this direction is poor in the last four decades. One possible reason may be because of its prevalence mainly in third world countries and also due to the high cost of testing and low-profit margins [9]. According to a recent report, U.S pharmaceutical companies spent about $1 billion to bring a new drug to the market [10]. Approximately 23 different antibiotics and non-antibiotics are under different clinical trials stages to find potential repurposable candidates as anti-TB drugs [11, 12]. The advancement in computational drug discovery has played a vital role in identifying potential lead compounds [13,14,15,16,17,18,19] and computational drug repurposing studies [20,21,22,23]. One-drug for one-target is the traditional drug discovery process, whereas, polypharmacology has emerged as an effective alternative paradigm in the discovery of potential lead/drug molecules that can simultaneously inhibit multiple targets [24]. It is well known that drug resistance is the major factor which need to be considered in designing effective anti-mycobacterial drugs. In this study, an attempt was made by combining polypharmacology which might be a promising approach to overcome drug resistance. Structure-based virtual screening has been employed to get the repurposable candidates against 20 M.tb targets. The drugs that has the potential activity against multiple M.tb targets were reported as possible anti-TB molecules.

Representation of first line, second line and new drugs under clinical trials used to combat TB
Materials and methods
Study design
20 M.tb targets have been selected after careful evaluation of their role in M.tb virulence, replication, cell wall synthesis, etc. Table S1 displayed the function of 20 mycobaterial proteins considered in this study and their functions. A systematic virtual screening was applied to identify ideal drug candidates that can be repurposed against M.tb. The drug molecules were collected from DrugBank. The drugs were carefully analysed by removing charged molecules, metals, Gadolinium-based contrast agents (due to associated toxicity), and other drugs which are unsuitable for docking and PASS calculations. The drug molecules were docked in the active site of the 20 selected M.tb targets, and virtual screening was carried out in three steps. In the first step, the top 300 molecules are screened-in for each target based on their high binding energies. In second step, the redundant drugs obtained from step 1 for the 20 targets were removed and finally screened the obtained hits using PASS tool to identify drugs showing high predicted antitubercular and/ antimycobacterial activity ≥ 0.5. After the three-step virtual screening, 34 drugs were identified which showed high binding energy in multiple targets with a concomitant high predicted probability of active (Pa ≥ 0.5) score for antitubercular and antimycobacterial activity. Interestingly, most of the hits identified hits are antibiotic drugs. In addition, a few non-antibiotic drugs were identified in the study. Among these we have reported 4 drug molecules as the possible repurposable candidates. Figure 2 displayed the workflow adopted in the study.

Virtual screening workflow for the identification of drug molecules with predicted antitubercular and antimycobacterial activity
Data collection
Three dimensional structures of 20 M.tb targets were collected from protein data bank (Table 1). The 20 targets are identified from various pathways that are critical for the survival of M.tb. These targets are chosen from a set of 450 crystal structures that are deposited at our in-house MPDSTB [8] target library. The 20 target proteins have been selected based on their role in M.tb (1) fatty acid biosynthesis, (2) cell wall biosynthesis, (3) DNA synthesis, (4) cell growth, (5) formation of DNA double helix, (6) DNA replication, (7) sterol biosynthesis. These protein structures are carefully evaluated and structures with (a) minimum or no missing residues, (b) high resolution, (c) no metals at the active site and (d) presence of co-crystal ligands at the active site were selected for the study. All the protein structures were prepared in MGLTools version 1.5.6 software. Hydrogen atoms were added, and unwanted water molecules were removed from the structure. The FDA approved drugs were collected from DrugBank [25]. All the drugs were prepared and converted to.pdbqt format using the raccon.py script.
Fixing missing residues and atoms in MtCA1, MtCA2 and loop modelling in MtDHPS
The crystal structures of MtCA1 (PDB ID: 1YLK), MtCA 2 (PDB ID: 2A5V) and MtDHPS (PDB ID: 1EYE) are selected from Protein Data Bank (PDB) [26]. However, in MtCA1 and 2, the considered proteins (PDB IDs: 1YLK and 2A5V) showed conformations with a very small active site volume (7 Å) which is unsuitable for inhibitor design at the active site. For MtDHPS, the available crystal structure 1EYE has missing residues in the catalytically important loop 2 region which makes it unsuitable for inhibitor design. Hence, the ligand bound conformations of MtCA1 and MtCA2 proteins are built using MODELLER v 9.10 [27]. From the BLAST analysis, ectodomain of human ADAM22 (PDB ID – 3G5C) and beta-carbonic anhydrase from V. Cholerae (PDB ID—5CXK) were obtained as templates for MtCA1 and MtCA2, respectively. However, both these structures are apo-proteins, with small active site volume and lacking a co-crystal at the active site making them unsuitable templates for MtCA homology modelling. Nonetheless, the activity of MtCA with acetazolamide has been widely studied [28,29,30]. Coccomyxa beta-carbonic anhydrase (PDB ID—3UCJ), which is the only acetazolamide bound form of MtCA, has been chosen as a template for building homology models of MtCA 1 and 2. DHPS from E.coli (PDB ID—1AJ0) has been selected as a template for building the missing residues of loop 2 in MtDHPS followed by refinement of the loop residues. The MtCA1 and MtCA2 models are minimised using steepest-descent method for 500 steps in GROMACS [31]. All three models are further validated using Ramachandran (RC) plot and other tools available at the Structure Analysis and Verification Server (SAVES) server [32].
Virtual screening
The virtual screening was performed using AutoDockVina [33] against the 20 targets. The grids for each protein were constructed with the spacing of 1.00 Å on the centroid of the existing co-crystal ligands. The gird was set around the co-crystal ligand.
Biological activity prediction
The unique drugs from each target protein were subsequently filtered through PASS analysis. Different pharmacological effects, toxic and adverse effects, mechanisms of actions, the influence of gene expression, etc., can be predicted through PASS online web server [34, 35]. The PASS developers have formed a structure activity relationships equation based on a training set of > 300,000 biologically active substances (i.e., drugs, drug like molecules, lead compounds, toxins, etc.). There are two terms namely probability of active (Pa) and probability of inactive (Pi) for each category (i.e., anticancer, antibacterial, antiviral, antimycobacterial, etc.). Probability of active value close to 1 indicates that the compounds may be active for the particular property whereas the Probability of inactive value close to 1 indicates that the compound is inactive to the specific property.
Calculation of drug likeness properties
Drug likeness properties of 34 compounds were calculated to get a deeper understanding of the screened drug molecules. DruLiTo tool was used to calculate these properties. Drug likeness rules are a set of guidelines for the structural properties of compounds, used for the fast calculation of drug like properties. Various drug like properties such as Lipinski’s rule, MDDR-like rule, Veber rule, Ghose filter, BBB rule, CMC-50 like rule can be calculated by DruLiTo tool [36].
Results and discussion
Homology modelling of ligand bound form of MtCA1 and MtCA2
BLAST Sequence similarity search was performed against PDB to identify the templates for building homology models of MtCA1 and MtCA2. The BLASTp generated a list of templates based on their sequence similarity to the query sequence. 3QY1 (34% identity) and 1I6O (35% identity) for MtCA2 and 1G5C (33% identity) for MtCA1 are identified as templates based on their identity and query coverage. However, both the proteins are apo-proteins without co-crystals at the active site making them unsuitable for modelling ligand bound form of MtCA proteins. Nonetheless, experimental inhibition data of MtCA1 and MtCA2 with AZM has been widely studied and reported in the literature [28,29,30]. Therefore, PDB ID: 3UCJ, which is the only available ligand bound form of β-carbonic anhydrase, is chosen as a template for homology modelling. The primary sequences of 3UCJ, 1YLK and 2A5V are obtained from the SWISS-PROT database, and pairwise sequence similarity is done between these two sequences in CLUSTAL—Omega.
Validation of MtCA1 and MtCA2 homology models
Once the models are generated, they are further subjected to short energy minimisation using steepest-descent method for 1000 steps. This is followed by model validation using RC plot and various validation tools available in the SAVES server. The RC plot of MtCA1 and MtCA2 is shown in Fig. 3. The RC plot of MtCA1 and MtCA2 shows more than 80% of the non-glycine and non-proline residues in the favoured region and less than 10% of the residues are lying in the allowed region and less than 2% of the residues are outliers which depict the quality of the model. In addition to the RC plot, the models are also validated using ERRAT and Verify 3D scores which are available at the SAVES server. In both MtCA1 and MtCA2, 70% of the residues have 3D to 1D score of more than 0.2 as shown in their Verify3d score. A similar trend is observed with the ERRAT score where both MtCA1 and 2 have a score of more than 70% which depicts that the protein is modelled fairly well.

The homology modelled structure and their Ramachandran plot for the proteins A, D) MtDHPS; B, E) MtCA1 C, F) MtCA2
Homology modelling of missing residues of loop 2 of MtDHPS
1EYE which is the only available crystal structure of MtDHPS is taken from RCSB-PDB. 1EYE has missing loop 2 region (Glu51-Val64), along with the residues 1–4 and 275–280 at the N and the C–terminal, respectively. While the terminal missing residues are not catalytically important, loop 2 harbour residues which account for the sulphonamide resistance as observed in M. leprae which correspond to residues Ser53 and Pro55 in loop 2 of M.tb. Since the loop 2 is clearly seen in E. coli DHPS, 1AJ0 is chosen as template for loop modelling which also coincided with the BLASTp analysis. The missing loop 2 residues are built as reported in previous study [37] followed by refinement of the loop residues as implemented in MODELLER. The obtained model is thereafter validated using RC plot and other validation tools of SAVES server. The modelled loop 2 is shown in Fig. 3a.
Validation of the MtDHPS model
The MtDHPS model obtained from loop modelling is validated using RC plot and other validation tools of the SAVES server as in the MtCA1 and MtCA2 models. The RC plot shows that more than 94.55 of the residues are within favourable region and less than 1% of the residues are outliers. The RC plot of MtDHPS is shown in Fig. 3. The verified score shows that 90% of the residues are having 3D-1D score which shows that the secondary structures of the protein are built well. The ERRAT shows an overall quality of 97.78%. The Ramachandran plot results and ERRAT scores have been given in Table 2.
Analysis of 20 selected M.tb targets
20 different druggable targets were chosen from M.tb H37Rv genome family for this study. These 20 targets belong to different families such as short-chain dehydrogenase, dihydrofolate reductase, alanine racemase, NDA-dependent DNA ligase, protein kinase, carbonic anhydrase, cytochrome P450 family, etc. The 20 targets are involved in different biological functions such as cell wall synthesis (2), DNA precursor synthesis (1), catalytic reaction (12), virulence factor (1), cell growth (1) and amino acid synthesis (1). In order to understand the sequence similarity and identify, multiple sequence alignment (MSA) was carried out using T-Coffee alignment among 20 targets [38]. The MSA analysis revealed that the 20 target sequences shared less than 20% sequence identity, and it signifies that these sequences are independent of each other. Further, BLAST analysis was performed for 20 target proteins with Homo sapiens. It was also observed that 11 proteins have no sequence identity, two proteins have 25–28% sequence identity, six proteins have 30–39% sequence identity, and one protein have the maximum of 71% sequence identity. Analysis of the 20 selected target structures indicated that the selection of these 20 structures can be a good initiation to perform the poly-pharmacological approach to get structural insights for designing novel potent inhibitors against multiple M.tb targets.
Identification of 34 polypharmacological hits
A set of 1428 approved drugs have been used to carry out a high throughput virtual screening (HTVS) on 20 M.tb druggable targets. The collected 1428 drugs are docked into the active site of 20 targets considered in the study. Based on their docking score and binding conformation, the top 300 drugs in each protein (6000 drugs) have been screened. The redundant molecules are removed, and 982 unique drugs are identified for further analysis. These 982 drugs are further analysed in the PASS 2017 tool to identify the drugs with high predicted antitubercular (AT)/Antimycobacterial (AM) activity. We have considered the Probable active value of 0.5 for antitubercular (AT)/Antimycobacterial (AM) activity from PASS analysis. The obtained results are further processed by removing eleven drugs that are already known to show anti-tuberculosis activity. This has led to the identification of 34 non-antitubercular drugs. Tables 3 and 4 showed the 34 drugs have interaction with 20 targets and their mechanism of action and PASS predicted activity, respectively.
Physiochemical properties of selected hits
Different filters such as Lipinski’s rule, MDDR-like rule, Veber rule, Ghose filter, BBB rule and CMC-5 were calculated to understand the different properties of the screened drug molecules. As shown in Table 5, from Lipinski’s filter out of 34 compounds, 22 compounds were observed to have molecular weight ≤ 500, 34 compounds have fulfilled AlogP criteria (≤ 5), and 23 compounds have ≤ 5 hydrogen bond donor, and 20 compounds have ≤ 10 hydrogen bond acceptors count which satisfies the property of the rule. In total, 20 compounds followed all four properties of the Lipinski filter. From Ghose’s filter, it can be observed that 19 compounds have a molecular weight in the range of 160 to 480, 8 compounds have AlogP value ranging between -0.4 to 5.6, 24 compounds have nAtom numbering between 20 to 70 atoms, and 23 compounds have molar refractivity in the range of 40 to 130. In total, 8 compounds followed all four property criteria for fulfilling Ghose filter. Further, from Veber’s, it is observed that 34 compounds have ≤ 10 rotatable bond counts, and 15 compounds have polar surface area ≤ 140 which indicates a total of 15 compounds satisfying the criteria and followed Veber filter. Table S2 has shown the drug like properties of the selected 34 compounds. This analysis helpful in understanding the different physiochemical properties among identified antitubercular drugs.
In addition, biopharmaceutics classification system (BCS) analysis was carried out for predicting the intestinal drug absorption of the 34 polypharmacological hits. Based on the predictive partition coefficient (XlogP) and intrinsic solubility (logS) values, BCS classifies the compounds into four classes [8, 36]. Out of 34 compounds, the maximum number of compounds (23 compounds) was found in the class 3 which indicated the compounds with high solubility and low permeability. This was followed by the class 2 with 7 compounds having low solubility and high permeability and 3 compounds in the class 1 category with high solubility and high permeability. The least number of compounds (1 compound) were found in the class 4 with low solubility and low permeability. Table S3 has shown the logS and XlogP values along with BCS classification.
Identification of privileged scaffolds and their analysis
To identify the privileged scaffolds, the 34 compounds/hits were taken to generate three levels of scaffolds. As the scaffold tree level increases, the size of the scaffold was observed to increase with the reduction of common scaffolds between the compound groups. From the scaffold tree analysis, level 1, 2 and 3 consists of 24, 25 and 26 scaffolds, respectively, where the compounds were arranged based on the number of times the compounds occur in the level tree. In addition, the identified scaffolds of each level were compared with existing 14 M.tb drugs scaffolds. The scaffold comparison between the two trees showed similarities with 3 scaffolds (Benzene, Tetrahydro-2H-pyran-2-olate,4-Hydroxypyridine), 3 scaffolds (2H-Pyran, 2-(cyclohexyloxy)tetrahydro-, 2-(Oxolan-3-yloxy)oxane, 4-Hydroxyquinoline) and 4 scaffolds (7-Piperazin-1-yl-1H-quinolin-4-one,2-[3-(Oxan-2-yloxy)cyclohexyl]oxyoxane etc.) similarities in level 1, 2 and 3, respectively. Hence, the compounds present in level 2 and 3 can be considered as potential candidates for designing effective drugs against M.tb. The scaffolds presented in level 2 and level 3 has been shown in Fig. 4.

Scaffold similarities between 34 novel compounds and 14 M.tb drugs present in level 2 and 3, respectively
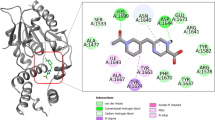
Binding mode analysis of 4 four drugs in the active site of 20 druggable targets
In this section, the binding mode of the four drugs has been analysed in detail along with the co-crystal ligands. Most of the selected targets in this study are promising druggable targets. Thus, to get a deep atomic level understanding, the binding mode of the top four compounds with the co-crystallised ligands are analysed in detail. Figure S1 displayed the binding mode of four drug molecules in the 20 M.tb targets. Table 6 displayed the docking score of 4 drug molecules in different M.tb protein targets. A comparison chart of drug molecules interaction with 20 different targets including co-crystal ligands has been displayed in Table S4.
Enoyl–ACP reductase (InhA)
The InhA protein involves in the fatty acid biosynthesis of M.tb that has two different systems such as FAS-I and FAS-II for the production of fatty acid in M.tb. Both the enzymes are responsible for the production of mycolic acids which are a major component for M.tb cell wall synthesis [39, 40]. The solved InhA enzyme from Rozwarski et al., (1999) was considered for the virtual screening [41]. The active site of InhA is majorly surrounded by hydrophobic residues. A198, M199, A201, I202, L207, I202, L207 I215, L218, M103, F149, Met155, Y158 and M161 are the major amino acids that reside in this region. The F149 is an important amino acid that may help in the turn of the fatty acyl substrate U-shaped conformation. The nitrofural and stavudine compounds have this major interaction with InhA. Another amino acid Y158 is conserved in bacteria and plants, and this key interaction is essential for fatty acyl substrate. The stavudine has another interaction with Y158.
Dihydrofolate reductase
Dihydrofolate reductase is an important enzyme catalysed the NDPH-dependent reduction of dihydrofolate to tetrahydrofolate. The reduction reaction is essential for the synthesis of several amino acids and thus inhibiting this protein leads to arrest DNA synthesis and cell death. Li et al., [42] solved the crystal structure of M.tb dihydrofolate reductase with the presence of inhibitor methotrexate. The study explained that there are two major avenues involved in the binding of new selective inhibitors [42]. Among these, second avenue is a pocket filled with glycerol and essentially absent in human enzyme. I20, R23, Q28, R146 are the major binding site residues at this pocket. The three identified drugs quinidine, stavudine and quinine have interaction with these amino acids.
Dihydrodipicolinatereductase
Dihydrodipicolinate reductase acts as catalysing enzyme to produce tetrahydropicolinate. This enzyme is essential to catalyse bacterial biosynthetic pathway which is used to M.tb cell wall synthesis. Thus, inhibiting this enzyme will be helpful to rupture the bacterial cell wall. The crystal structure of dihydro dipicolinate reductase has been solved by Cirilli et al., (2003) [43] in presence of NADPH. The active site analysis revealed the binding site of DHPR located in the C-terminal domain. The pyridine nitrogen atom from the K136 amino group is proposed to function in catalysis [43]. Interestingly, nitrofural drug have this major interaction. Other drugs (quinidine, stavudine, quinine) are also binding in the same sites. However, no interaction was observed with K136.
Lipoprotein ligase B
The overexpression of Lipoprotein ligase B protein is identified from patients with multidrug-resistant M.tb. This makes LipB enzyme an attractive target for drug development. Ma et al., (2006) [44] solved this crystal structure with the presence of co-crystallised ligand. The atomistic study revealed that R76, K79, H83, I146, G147 are the major active sites [44]. Among the four drugs, nitrofural was binding at the same active site in LipB enzyme.
Alanine racemase
Alanine racemase is one of the promising targets for the development for anti-microbacterial agents. This enzyme is essential since it contains pyridoxal 5’-phosphate (PLP) which is essential for the growth of the bacteria. Analysis of known alanine recemases binding site properties revealed some of the conserved amino acids K42, W88, H172, Y271 and Y364 which are important active sites from a study carried out by LeMagueres et al., Most known inhibitors bound to this region to arrest alanine racemase activity [45]. The four drug molecules are also having major interactions with the above mentioned amino acids.
Tyrosine phosphatase
Phosphatase and kinases are the major families involved in signal transduction. The main function of these enzymes is involved in the production or regulation of exopolysaccharides and capsular polysaccharides. Protein phosphorylation and dephosphorylation are controlled by these enzymes. Madhurantakam et al., (2005) [45] solved the crystal structure of protein tyrosine phosphatase from M.tb. The major active sites are L12, R17, W48, C11 and E56. Among these L12 plays a major role in PTP loop stabilisation [46]. The stavudine molecule has an interaction with L12. Other than that R17 and W48 are the important amino acids. The four molecules have interactions with R17 or W48.
Carbonic anhydrase-1
Reversible hydration of CO2 to form bicarbonate is a universal reaction require for fatty acid synthesis. Carbonic anhydrases catalyse this reaction. Carbonic anhydrase-1 structure was modelled due to its small active site. The residues involved in the metal chelation were considered as active sites. R39, H104, R55 amino acids involved in metal chelation [47]. The nitrofural drug has interactions with G92, R103 and I105.
Decaprenylphosphoryl-beta-D-ribose oxidase (DprE1)
The DprE1 enzyme is a key precursor involved in catalyses the first step of epimerisation. Thus DprE1 is an attractive drug target for M.tb. Crystal structure from Batt et al., (2012) revealed Y60, K134, K367 are the key interactions for substrate binding [48]. Among the four drugs, quinidine has interaction with DprE1. It has the interactions with Y60, K118 of the DprE1 active site.
NAD+-dependent DNA ligase
Phosphodiester bonds are essential for the formation of DNA double helix. The phosphodiester bonds are catalysed by DNA ligases utilising either ATP or NAD+ as a cofactor. Thus DNA ligase is an attractive drug target since it needs NAD+ for ligase activity. Structural analysis of DNA ligase by Srivastava et al., (2005) revealed that N94, E121, L122, I124 are the major interactions essential for AMP binding [49]. In the docking analysis, four drug molecules have occupied the same binding site where AMP binds in NAD+ -dependent DNA ligase.
Protein Kinase B (PknB)
Protein Kinase B (PknB) is a trans-membrane Ser/Thr protein kinase (STPK) present in Mycobacterium tuberculosis which is involved in cell growth control. The crystal structure of the kinase domain of PknB is reported with ATP analogue by Wehenkel et al., (2006) [50]. The regulation of PknB is carried out through autophosphorylation and dephosphorylation by the Ser/Thr protein phosphatase PstP. It is known that the PknB enzyme is predominantly expressed during exponential growth whereas if it is overexpressed it leads to defects in cell wall synthesis and cell division. A compound used in cancer treatment namely mitoxantrone is reported to be a PknB inhibitor preventing mycobacterial cell growth which indicates that bacterial kinases can be a potential target for drug design [49]. The major active sites of PknB interacting with inhibitor include L17, G18, V25, A38, M92, E93, Y94, V95, M145, M155, V95, N143, D76, R35, A64, V74, N67 and E93. Among the four drug molecules, nitrofural interacts with V95, quinidine and quinine bind to L17.
Lysine ε-aminotransferase (LAT)
Lysine ε-aminotransferase (LAT) is a PLP-dependent enzyme which is involved in catalysing the reactions involved in transferring the ε-amino group of L-lysine to α-ketoglutarate to yield L-glutamate and α-aminoadipate-δ-semialdehyde. The internal aldimine form of the enzyme is reported to exhibit the characteristic Schiff base linkage with the active site residue K300. Residues G128, A129, F167, H168, E238, D271, V273, Q274, K300, S329 and T330 are in close contact within 4 Å of PLP in LAT [51]. The docking study revealed that all four compounds are binding to the major active site. Quinidine has interaction with F167 and V273 residue and nitrofural with Q274 and quinine having an interaction with one of the most important residue E243. This interaction can be considered to be essential to prevent unwanted transamination reactions at the α-amino group to occur for further resections.
b-ketoacyl ACP synthase (KasA)
KasA (b-ketoacyl ACP synthase) is an enzyme involved in catalysing the condensation between malonyl-AcpM and acyl chain in fatty acid elongation cycle. It is one of the important enzymes within the FAS-II system of mycobacteria. The depletion of KasA is reported to induce cell lysis and transposon-site hybridisation which is essential for cell growth [52]. It is also essential in the biosynthesis of long-chain fatty acids which binds to natural product inhibitor thiolactomycin (TLM). The TLM binding active sites of KasA are P280, G318, F402, F237, A279, P280, G403, F404, D273, F392 and G406. The four drugs are also found to bind to the major actives sites of KasA protein among which nitrofural interacts with G403, stavudine interacts with P280 and quinidine interacts with A279.
Pantothenate (vitamin B5)
Pantothenate (vitamin B5) is a precursor for biosynthesising coenzyme A and acyl-carrier proteins which are involved in cellular processes, energy metabolism and fatty acid metabolism. The study conducted by Wang et al., (2006) [53] reported the dimer nature of the protein crystal structure and the active sites of these dimers are independent of each other in terms of their catalytic functions. The major active sites reported in the binding pockets are M40, G158, S196, K160 and R132 [53]. Among the four compounds, nitrofural forms major interaction with S196, quinidine at K160 and stavudine at G164. These interactions can play an important role in inhibiting the activity of pantothenate (vitamin B5) in the mycobacterial cellular processes.
Isopentenyl diphosphate
Isopentenyl diphosphate is an enzyme that is produced by the mevalonate pathway by an alternate route starting from acetyl-CoA in plants, protozoa, and many bacteria. As reported by Henriksson et al., (2007) [54], it is a precursor of vital isoprenoids. In the second step of non-mevalonate pathway, NADPH is catalysed by 1-Deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) and reduction of 1-deoxy-D-xylulose 5-phosphate (DXP) takes place for the formation of 2-C-methyl-D-erythritol 4-phosphate. For the formation of isopentenyl diphosphate, many enzymes are present within the non-mevalonate pathway in the bacteria which makes the enzymes as potential targets. The major active sites of 2JCV include S152, E153, W203, G206, N209, K219 and S23. Among the active sites, D151, E153, E222 are reported to interact with M.tb inhibitor fosmidomycin which also interacts with residues S152, S177, H200, N218 and K219 [54]. In the docking studies, we observed that quinine interacts with 2JCV at binding residue at S23. Nitrofural binds at S152 and E153 residue where S152 is reported to be a binding site of the inhibitor fosmidomycin which suggests that these interactions are necessary for inhibition.
Carbonic anhydrase-2
Carbonic anhydrase-2 is working as a catalysing agent as carbonic anhydrase-1 which is required for many functions including pH homeostasis and assimilation [47]. In case of this enzyme, major interaction was observed only with nitrofural compared to other three drugs.
3-oxoacyl-[acyl-carrier-protein] synthatase-3 (FabH)
M.tb FabH involves acyl-coenzyme A (CoA) precursors of mycolic acids, which are mainly involved in bacterial cell wall synthesis. Sachdeva et al., (2008) solved the crystal structure of FabH with alkyl-CoA sulphide inhibitors in the active site [55]. Binding site analysis revealed that C112, W195, V205, A306 are the important amino acids in the active sites. The four drug molecules have major interactions with these amino acids suggested that they have the potential to inhibit FabH enzyme.
N‐Acetylglucosamine‐1‐phosphate uridyltransferase (GlmU)
N‐Acetylglucosamine‐1‐phosphate uridyltransferase (GlmU) is an acetyltransferase/uridyltransferase enzyme that is involved in catalysing the formation of UDP-GlcNAc from GlcN-1-P which is a substrate for lipopolysaccharide and peptidoglycan synthesis pathways. GlmU is an essential gene for the proper growth of M.tb. The mechanism of cell wall synthesis has been reported by Zhang et al., in 2009, and this enzyme is a target for several antibiotics vancomycin, fosfomycin, nisin and bacitracin. The major actives sites of GlmU consist of R323, TW460, V461, A470, A473, R463, T89, E166, N181, G151, D114, S112, Q83, G88, N239, R19 [56]. Among the major active sites, it was observed that nitrofural has interaction with G88, D114 and S112. Quinidine has interaction with T89, stavudine with N181 and quinine with T89 and S112.
Cytochrome P450 14α-sterol demethylase (CYP51)
In eukaryotes, CYP51 is an important enzyme for sterol biosynthesis. CYP51 inhibitors (i.e., fluconazole and itraconazole) are used as antifungal agents. Podust et al., (2001) [57] solved Ecoli expressed MTCYP51 in the presence of azole inhibitors. MTCYP51 has a unique substrate access channel in the active site. Mutation analysis revealed that F78, A256, H259, T260 are the major hotspots in the MTCYP51 active site. Though the identified drug molecules have less interaction, it properly bound in the MTCYP51 active site.
Thymidylate kinase
Thymidylate kinase enzyme is essential for DNA replication. Thus, this enzyme is an attractive drug target for M.tb infection. Fioravanti et al., (2005) [58] solved M.tb thymidylate kinase in the presence of competitive inhibitor (AZTMP). The binding analysis of AZTMP in thymidylate kinase in the active site revealed that some of the amino acid residues D9, R95, D163 are crucial for inhibitory activity. Among all the four drugs, nitrofural and quinidine have D9 interaction, and stavudine and quinine have R95 interaction.
6-Hydroxymethyl-7, 8-dihydropteroate Synthase
6-Hydroxymethyl-7, 8-dihydropteroate synthase enzyme is essential for the condensation reaction. The activity of this enzyme is essential for de novo synthesis of folate in prokaryotes and eukaryotes. Inhibition of this enzyme leads to cell death, and thus, it is an important enzyme in M.tb inhibition [59]. The interaction analysis of binding pocket revealed that amino acids D86, N105, V107, R253 and H255 are important amino acids to inhibit the activity. Among the four compounds, nitrofural has the interaction with this enzyme. The drug is placed in the active site of the enzyme.
In this section, 20 different M.tb targets function and active sites were extensively studied. Binding mode analysis of 4 drugs in 20 targets revealed that these drugs can bind and inhibit the enzymes with major amino acids interaction.
Repurposable opportunities for the selected drugs
Though the identified four drugs have shown promising antitubercular activity against the M.tb targets and other drug likeness properties, it is important to analyse their repurposable feasibility. An extensive literature survey has been made to identify an alternative therapeutic indication of the identified molecules. The top four compounds original and possible repurposable indication has been reported in Table 7. The original indication of nitrofural is an anti-infective agent. However, it has been reported that nitrofural derivatives can be repurposed for various disease conditions from cancer to urinary tract infections [3, 60]. Stavudine is an HIV reverse transcriptase inhibitor and experiments suggested that it can be repurposed against degenerative diseases [61]. Similarly, antimalarial drug quinine and sodium channel blocker quinidine are experimentally proved for treating dengue virus infection and as an anti-depressant, respectively [62, 63]. Interestingly, among these four drugs, nitrofural is under clinical trials for repurposing against M.tb [60]. These studies have enormously supported our computational approach that 1. Nitrofural can be a potential repurposable candidate for M.tb infection, 2. The other three molecules can also be validated against M.tb infection, and it can be the repurposable candidates against different diseases.
Conclusions
Drug repurposing of existing drugs for the new indications is becoming an important approach for drug discovery [64]. The majority of antimycobacterial drugs are ineffective due to the emergence of drug-resistant strains of M.tb. Drug resistance is the major bottleneck in the discovery of potential anti-TB molecules. Although several studies have been carried out to understand the molecular mechanism underlying the TB disease, no effective agents are available to treat the disease. Most of the M.tb drugs have adverse side effects, and the drugs are very expensive. Thus, screening existing FDA approved drugs can be an effective way to identify potential lead molecules against drug resistance M.tb strains.
In the present study, an in silico guided polypharmacology has been applied to identify potential drug molecules against various M.tb targets. 982 potential polypharmacological hits were identified against 20 M.tb targets. The 982 hits were prioritised based on their docking scores against 20 M.tb targets and PASS predicted antitubercular activity. 34 drug molecules were filtered, and 11 among them are antibiotics. The four molecules identified in the study using polypharmacology approaches will be potential interesting candidates to study the activity on drug resistance strains of M.tb.
References
Huber FD, Sánchez A, Gomes HM, Vasconcellos S, Massari V, Barreto A, Cesconi V, de Almeida Machado SM, Gomgnimbou MK, Sola C, Larouzé B, Suffys PN, Saad MH (2014) Insights into the population structure of Mycobacterium tuberculosis using spoligotyping and RDRio in a southeastern Brazilian prison unit. Infect Genet Evol 26:194–202. https://doi.org/10.1016/j.meegid.2014.05.031
WHO (2019) global tuberculosis report
Krasavin M, Parchinsky V, Kantin G, Manicheva O, Dogonadze M, Vinogradova T, Brönstrup M (2017) New nitrofurans amenable by isocyanide multicomponent chemistry are active against multidrug-resistant and poly-resistant Mycobacterium tuberculosis. Bioorg Med Chem 25(6):1867–1874. https://doi.org/10.1016/j.bmc.2017.02.003
Seung KJ, Keshavjee S, Rich ML (2015) Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis. Cold Spring Harb Perspect Med 5(9):a017863. https://doi.org/10.1101/cshperspect.a017863
Groenewald W, Baird MS, Verschoor JA, Minnikin DE, Croft AK (2014) Differential spontaneous folding of mycolic acids from Mycobacterium tuberculosis. Chem Phys Lipids 180:15–22. https://doi.org/10.1016/j.chemphyslip.2013.12.004
Scordo JM, Arcos J, Kelley HV, Diangelo L, Sasindran SJ, Youngmin E, Wewers MD, Wang SH, Balada-Llasat JM, Torrelles JB (2017) Mycobacterium tuberculosis cell wall fragments released upon bacterial contact with the human lung mucosa alter the neutrophil response to infection. Front Immunol 8:307. https://doi.org/10.3389/fimmu.2017.00307
Nagamani S, Sastry GN (2021) Mycobacterium tuberculosis (M.tb) cell wall permeability model generation using chemoinformatics and machine learning approaches. ACS Omega 6:17472–17482
Gaur AS, Bhardwaj A, Sharma A, John L, Vivek MR, Tripathi N, Bharatam PV, Kumar R, Janardhan S, Mori A, Banerji A (2017) Assessing therapeutic potential of molecules: molecular property diagnostic suite for tuberculosis (MPDSTB). J Chem Sci 129(5):515–531. https://doi.org/10.1007/s12039-017-1268-4
Tomioka H, Namba K (2006) Development of antituberculosis drugs: current status and future prospects. Kekkaku 81(12):753–74
Wouters OJ, McKee M, Luyten J (2020) Estimated research and development investment needed to bring a new medicine to market 2009–2018. JAMA 323(9):844–853. https://doi.org/10.1001/jama.2020.1166
An Q, Li C, Chen Y, Deng Y, Yang T, Luo Y (2020) Repurposed drug candidates for antituberculosis therapy. Eur J Med Chem 192:112175. https://doi.org/10.1016/j.ejmech.2020.112175
Adeniji AA, Knoll KE, Loots DT (2020) Potential anti-TB investigational compounds and drugs with repurposing potential in TB therapy: a conspectus. Appl Microbiol Biotechnol 104(13):5633–5662. https://doi.org/10.1007/s00253-020-10606-y
Kumar N, Sarma H, Sastry GN (2021) Repurposing of approved drug molecules for viral infectious diseases: a molecular modelling approach. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.1905558
Srivastava HK, Sastry GN (2012) Molecular dynamics investigation on a series of HIV protease inhibitors: assessing the performance of MM-PBSA and MM-GBSA approaches. J Chem Inf Model 52(11):3088–3098. https://doi.org/10.1021/ci300385h
Badrinarayan P, Sastry GN (2011) Virtual high throughput screening in new lead identification. Comb Chem High Throughput Screen 14(10):840–860. https://doi.org/10.2174/138620711797537102
Ravindra GK, Achaiah G, Sastry GN (2008) Molecular modeling studies of phenoxypyrimidinyl imidazoles as p38 kinase inhibitors using QSAR and docking. Eur J Med Chem 43(4):830–838. https://doi.org/10.1016/j.ejmech.2007.06.009
Badrinarayan P, Sastry GN (2012) Virtual screening filters for the design of type II p38 MAP kinase inhibitors: a fragment based library generation approach. J Mol Graph Model 34:89–100. https://doi.org/10.1016/j.jmgm.2011.12.009
Choudhury C, Priyakumar UD, Sastry GN (2015) Dynamics based pharmacophore models for screening potential inhibitors of mycobacterial cyclopropane synthase. J Chem Inf Model 55(4):848–860. https://doi.org/10.1021/ci500737b
Jha V, Rameshwaram NR, Janardhan S, Raman R, Sastry GN, Sharma V, Rao JS, Kumar D, Mukhopadhyay S (2019) Uncovering structural and molecular dynamics of ESAT-6: β2M interaction: Asp53 of human β2-microglobulin is critical for the ESAT-6: β2M complexation. J Immunol 203(7):1918–1929. https://doi.org/10.4049/jimmunol.1700525
Reddy AS, Pati SP, Kumar PP, Pradeep HN, Sastry GN (2007) Virtual screening in drug discovery - a computational perspective. Curr Protein Pep Sci 8(4):329–351
Nagamani S, Sahoo R, Muneeswaran G, Sastry GN (2019) Data science driven drug repurposing for metabolic disorders. In silico Drug Design 191–227.
Gaur AS, Nagamani S, Tanneeru K, Druzhilovskiy D, Rudik A, Poroikov V, Sastry GN (2018) molecular property diagnostic suite for diabetes mellitus (MPDSDM): an integrated web portal for drug discovery and drug repurposing. J Biomed Inform 85:114–125
Bohari MH, Sastry GN (2012) FDA approved drugs complexed to their targets: evaluating pose prediction accuracy of docking protocols. J Mol Model 18(9):4263–4274
Goel RK, Gawande DY, Lagunin AA, Poroikov VV (2018) Pharmacological repositioning of Achyranthes aspera as an antidepressant using pharmacoinformatic tools PASS and PharmaExpert: a case study with wet lab validation. SAR QSAR Environ Res 29(1):69–81. https://doi.org/10.1080/1062936X.2017.1408683
Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 36(Database issue):D901–D906. https://doi.org/10.1093/nar/gkm958
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242. https://doi.org/10.1093/nar/28.1.235
Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform 54:5.6.1-5.6.37. https://doi.org/10.1002/cpbi.3
Güzel O, Maresca A, Scozzafava A, Salman A, Balaban AT, Supuran CT (2009) Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial beta-carbonic anhydrases Rv1284 and Rv3273. J Med Chem 52(13):4063–4067. https://doi.org/10.1021/jm9004016
Nishimori I, Minakuchi T, Vullo D, Scozzafava A, Innocenti A, Supuran CT (2009) Carbonic anhydrase inhibitors Cloning, characterization, and inhibition studies of a new beta-carbonic anhydrase from Mycobacterium tuberculosis. J Med Chem 52(9):3116–20. https://doi.org/10.1021/jm9003126
Nishimori I, Minakuchi T, Maresca A, Carta F, Scozzafava A, Supuran CT (2010) The β-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr Pharm Des 16(29):3300–3309. https://doi.org/10.2174/138161210793429814
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50(3):437–450. https://doi.org/10.1002/prot.10286
Trott O, Olson AJ (2010) AutoDock Vina: improve ng the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Lagunin A, Stepanchikova A, Filimonov D, Poroikov V (2000) PASS: prediction of activity spectra for biologically active substances. Bioinformatics 16(8):747–748. https://doi.org/10.1093/bioinformatics/16.8.747
Filimonov DA, Lagunin AA, Gloriozova TA, Rudik AV, Druzhilovskiy DS, Pogodin PV, Poroikov VV (2014) Prediction of the biological activity spectra of organic compounds using the pass online web resource. Chem Heterocycl Compd 50:444–457. https://doi.org/10.1007/s10593-014-1496-1
http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_Reference.html
Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Houl WG (2000) Crystal structure of Mycobacterium tuberculosis 7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol 302(5):1193–1212
Janardhan S, John L, Prasanthi M, Poroikov V, Narahari Sastry G (2017) A QSAR and molecular modelling study towards new lead finding: polypharmacological approach to Mycobacterium tuberculosis. SAR QSAR Environ Res 28(10):815–832. https://doi.org/10.1080/1062936X.2017
Zumla AI, Gillespie SH, Hoelscher M, Philips PP, Cole ST, Abubakar I, McHugh TD, Schito M, Maeurer M, Nunn AJ (2014) New ant tuberculosis drugs, regimens, and adjunct therapies: needs, advances, and future prospects. Lancet Infect Dis 14(4):327–340. https://doi.org/10.1016/S1473-3099(13)70328-1
Davis CE, Carpenter JL, McAllister CK, Matthews J, Bush BA, Ognibene AJ (1985) Tuberculosis. Cause of death in antibiotic era. Chest 88(5):726–729. https://doi.org/10.1378/chest.88.5.726
Rozwarski DA, Vilchèze C, Sugantino M, Bittman R, Sacchettini JC (1999) Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 274(22):15582–9. https://doi.org/10.1074/jbc.274.22.15582
Li R, Sirawaraporn R, Chitnumsub P, Sirawaraporn W, Wooden J, Athappilly F, Turley S, Hol WG (2000) Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J Mol Biol 295(2):307–23. https://doi.org/10.1006/jmbi.1999.3328
Cirilli M, Zheng R, Scapin G, Blanchard JS (2003) The three-dimensional structures of the Mycobacterium tuberculosis dihydrodipicolinate reductase-NADH-2,6-PDC and -NADPH-2,6-PDC complexes. Structural and mutagenic analysis of relaxed nucleotide specificity. Biochemistry 42(36):10644–50. https://doi.org/10.1021/bi030044v
Ma Q, Zhao X, Nasser Eddine A, Geerlof A, Li X, Cronan JE, Kaufmann SH, Wilmanns M (2006) The Mycobacterium tuberculosis LipB enzyme functions as a cysteine/lysine dyad acyltransferase. Proc Natl Acad Sci U S A 103(23):8662–8667. https://doi.org/10.1073/pnas.0510436103
LeMagueres P, Im H, Ebalunode J, Strych U, Benedik MJ, Briggs JM, Kohn H, Krause KL (2005) The 1.9 A crystal structure of alanine racemase from Mycobacterium tuberculosis contains a conserved entryway into the active site. Biochemistry 44(5):1471–81. https://doi.org/10.1021/bi0486583
Madhurantakam C, Rajakumara E, Mazumdar PA, Saha B, Mitra D, Wiker HG, Sankaranarayanan R, Das AK (2005) Crystal structure of low-molecular-weight protein tyrosine phosphatase from Mycobacterium tuberculosis at 1.9-A resolution. J Bacteriol 187(6):2175–81. https://doi.org/10.1128/JB.187.6.2175-2181
Covarrubias AS, Larsson AM, Högbom M, Lindberg J, Bergfors T, Björkelid C, Mowbray SL, Unge T, Jones TA (2005) Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J Biol Chem 280(19):18782–18789. https://doi.org/10.1074/jbc.M414348200
Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, Alderwick LJ, Fütterer K, Besra GS (2012) Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci U S A 109(28):11354–11359. https://doi.org/10.1073/pnas.1205735109
Srivastava SK, Tripathi RP, Ramachandran R (2005) NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis. Crystal structure of the adenylation domain and identification of novel inhibitors. J Biol Chem 280(34):30273–81. https://doi.org/10.1074/jbc.M503780200
Wehenkel A, Fernandez P, Bellinzoni M, Catherinot V, Barilone N, Labesse G, Jackson M, Alzari PM (2006) The structure of PknB in complex with mitoxantrone, an ATP-competitive inhibitor, suggests a mode of protein kinase regulation in mycobacteria. FEBS Lett 580(13):3018–3022. https://doi.org/10.1016/j.febslet.2006.04.046
Tripathi SM, Ramachandran R (2006) Direct evidence for a glutamate switch necessary for substrate recognition: crystal structures of lysine epsilon-aminotransferase (Rv3290c) from Mycobacterium tuberculosis H37Rv. J Mol Biol 362(5):877–886. https://doi.org/10.1016/j.jmb.2006.08.019
Luckner SR, Machutta CA, Tonge PJ, Kisker C (2009) Crystal structures of Mycobacterium tuberculosis KasA show mode of action within cell wall biosynthesis and its inhibition by thiolactomycin. Structure 17(7):1004–1013. https://doi.org/10.1016/j.str.2009.04.012
Wang S, Eisenberg D (2006) Crystal structure of the pantothenate synthetase from Mycobacterium tuberculosis, snapshots of the enzyme in action. Biochemistry 45(6):1554–1561. https://doi.org/10.1021/bi051873e (PMID: 16460002)
Henriksson LM, Unge T, Carlsson J, Aqvist J, Mowbray SL, Jones TA (2007) Structures of Mycobacterium tuberculosis 1-deoxy-D-xylulose-5-phosphate reductoisomerase provide new insights into catalysis. J Biol Chem 282(27):19905–19916. https://doi.org/10.1074/jbc.M701935200
Sachdeva S, Musayev FN, Alhamadsheh MM, Scarsdale JN, Wright HT, Reynolds KA (2008) Separate entrance and exit portals for ligand traffic in Mycobacterium tuberculosis FabH. Chem Biol 15(4):402–412. https://doi.org/10.1016/j.chembiol.2008.03.007
Zhang Z, Bulloch EM, Bunker RD, Baker EN, Squire CJ (2009) Structure and function of GlmU from Mycobacterium tuberculosis. Acta Crystallogr D Biol Crystallogr 65(Pt 3):275–283. https://doi.org/10.1107/S0907444909001036
Podust LM, Poulos TL, Waterman MR (2001) Crystal structure of cytochrome P450 14alpha -sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci U S A 98(6):3068–3073. https://doi.org/10.1073/pnas.061562898
Fioravanti E, Adam V, Munier-Lehmann H, Bourgeois D (2005) The crystal structure of Mycobacterium tuberculosis thymidylate kinase in complex with 3’-azidodeoxythymidine monophosphate suggests a mechanism for competitive inhibition. Biochemistry 44(1):130–137. https://doi.org/10.1021/bi0484163
Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WG (2000) Crystal structure of Mycobacterium tuberculosis 7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol 302(5):1193–1212. https://doi.org/10.1006/jmbi.2000.4094
Zuma NH, Aucamp J, N’Da DD (2019) An update on derivatisation and repurposing of clinical nitrofuran drugs. Eur J Pharm Sci 1(140):1050902. https://doi.org/10.1016/j.ejps.2019.105092
La Rosa F, Clerici M, Ratto D, Occhinegro A, Licito A, Romeo M, Rossi P (2018) The gut-brain axis in Alzheimer’s disease and Omega-3 A critical overview of clinical trials. Nutrients 10(9):1267. https://doi.org/10.3390/nu10091267
Malakar S, Sreelatha L, Dechtawewat T, Noisakran S, Yenchitsomanus PT, Chu JJH, Limjindaporn T (2018) Drug repurposing of quinine as antiviral against dengue virus infection. Virus Res 15(255):171–178. https://doi.org/10.1016/j.virusres.2018.07.018
Murrough JW, Abdallah CG, Mathew SJ (2017) Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov 16(7):472–486. https://doi.org/10.1038/nrd.2017
Rani J, Silla Y, Borah K, Ramachandran S, Bajpai U (2020) Repurposing of FDA-approved drugs to target MurB and MurE enzymes in Mycobacterium tuberculosis. J Biomol Struct Dyn 38(9):2521–2532. https://doi.org/10.1080/07391102.2019.1637280
Acknowledgements
GNS thanks DST for the award of J C Bose National fellowship. MSS thanks DST for the Grant (Project No: SR/WOS-A/CS-1091/2014). EJ thanks DST-INSPIRE fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Madugula, S.S., Nagamani, S., Jamir, E. et al. Drug repositioning for anti-tuberculosis drugs: an in silico polypharmacology approach. Mol Divers 26, 1675–1695 (2022). https://doi.org/10.1007/s11030-021-10296-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10296-2