
Abstract
Alzheimer’s disease (AD) is now ranked as the third leading cause of death after heart disease and cancer. There is no definite cure for AD due to the multi-factorial nature of the disease, hence, multi-target-directed ligands (MTDLs) have attracted lots of attention. In this work, focusing on the efficient cholinesterase inhibitory activity of tacrine, design and synthesis of novel arylisoxazole-tacrine analogues was developed. In vitro acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) inhibition assay confirmed high potency of the title compounds. Among them, compounds 7l and 7b demonstrated high activity toward AChE and BChE with IC50 values of 0.050 and 0.039 μM, respectively. Both compounds showed very good self-induced Aβ aggregation and AChE-induced inhibitory activity (79.4 and 71.4% for compound 7l and 61.8 and 58.6% for compound 7b, respectively). Also, 7l showed good anti-BACE1 activity with IC50 value of 1.65 µM. The metal chelation test indicated the ability of compounds 7l and 7b to chelate biometals (Zn2+, Cu2+, and Fe2+). However, they showed no significant neuroprotectivity against Aβ-induced damage in PC12 cells. Evaluation of in vitro hepatotoxicity revealed comparable toxicity of compounds 7l and 7b with tacrine. In vivo studies by Morris water maze (MWM) task demonstrated that compound 7l significantly reversed scopolamine-induced memory deficit in rats. Finally, molecular docking studies of compounds 7l and 7b confirmed establishment of desired interactions with the AChE, BChE, and BACE1 active sites.
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive and fatal neurodegenerative disorder, known as the main cause of dementia in elderly population. It is usually characterized by the loss of cognitive function and behavioral disturbances [1, 2]. Although some cases of AD have been observed before the age of 65, old age is still a major risk factor as the number of new cases of AD increases dramatically with age. According to the 2020 Alzheimer's disease facts and figures, the average annual incidence in people aged 65–74 is 0.4% and it increases to 3.2% in people aged 75–84. And as for the age demographic of 85 and older, the incidence is 7.6% [3]. Despite the tremendous efforts and financial expenditures aimed at finding an efficient treatment for AD, no definite cure has been found so far. Because AD is a multi-factorial disease and single-targeted drugs usually do not work efficiently [4, 5]. To date, autopsy studies and clinical diagnosis [6] have identified several factors responsible for the onset and progression of AD [7]. Intracellular formation of neurofibrillary tangles (NFTs) and neuropil threads (NTs) resulting from abnormal hyperphosphorylation of tau protein are of great importance [8]. In addition, the aggregation and extracellular deposition of amyloid beta (Aβ) protein is the main cause of plaque formation [9], which is usually catalyzed by β-secretase 1 (BACE-1) [10], leading to activation of cell death [11]. Also, metal-ion dysregulation has been known to interact with the Aβ peptide, trigging its aggregation and increasing toxicity [12]. Another important factor in the creation of AD which is described by the cholinergic hypothesis is associated with the reduction of acetylcholine (ACh) levels in the brain, up to 90 percent [13, 14].
ACh is the main neurotransmitter of the parasympathetic nervous system which is immediately hydrolyzed by cholinesterases (ChEs), acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). AChE and BChE are serine hydrolase, having similar three-dimensional structures but have shown diverse catalytic actions. For example, they have shown different substrate specificity and inhibitor sensitivity. In the case of hydrolysis of ACh, BChE usually plays a supportive role when AChE exists in low concentrations [15]. Despite the development of other theories, the cholinergic theory has played a significant role in the treatment of AD, since current FDA approved drugs such as donepezil, rivastigmine, and galantamine are ChE inhibitors (ChEIs) [16, 17] and development of both acetylcholinesterase and butyrylcholinesterase inhibitors (AChEIs and BChEIs) have been considered as a prominent strategy for the amelioration of AD symptoms [15, 18].
As previously mentioned, single-targeted drugs have not been completely successful in the treatment of AD and for this reason, design of multi-targeted ligands (MTDLs) has been in the center of attention in drug discovery research. One of the design strategies is based on a core, possessing potent ChEI activity which is hybridized with different functional groups, inducing different biological properties such as βA and BACE1 inhibitory activity as well as metal chelating ability and neuroprotectivity [19].
Tacrine (1,2,3,4-tetrahydroacridin-9-amine) (Fig. 1) has been removed from the market due to serious side effects such as hepatotoxicity [20]; however, its structure has remained as a versatile core in the design and synthesis of MTDLs [21] as it is a strong ChE inhibitor, possessing high affinity for desired interactions with enzymes active site. In this respect, tacrine-isatins have shown efficient ChEI activity in the nM range and among them, compound A (IC50 (AChE) = 0.42 nM, IC50 (BChE) = 0.57 nM, Fig. 1) showed significant capacity to chelate Fe2+ ions [22]. Tacrine-1,2,4-thiadiazoles have been designed and synthesized as potent multifunctional agents. In this series, compound B exhibited the highest ChE inhibitory activity (IC50 (AChE) = 0.431 µM, IC50 (BChE) = 0.004 µM, Fig. 1) along with the ability to scavenge free radicals [23]. Moreover, the efficacy of substituted tacrine analogues has attracted lots of attention since the presence of an appropriate substituent can lead to lower toxicity [24]. Compound C (Fig. 1), a tacrine-pyrazolo[3,4-b]pyridine hybrid has shown potent AChEI and BChEI activity with IC50 values of 0.12 and 0.045 µM, respectively [25].

The structure of tacrine based compounds
Tacrine-1,2,3-triazole hybrids D and E (Fig. 1) also depicted potent anti-ChE activity. The instructive point comes back to the effect of the presence of Cl on the tacrine moiety. The inhibitory activity of compound D was indicated by IC50 values of 0.52 and 0.05 μM toward AChE and BChE, respectively. Elimination of Cl (compound E) led to approximately a fourfold decrease in the AChEI activity; however, compound E showed stronger BChEI activity by 37 times. It should be noted that compound D showed remarkable neuroprotective activity caused by H2O2 in distinguished PC12 neuron cells [26].
Good results obtained by tacrine analogues possessing efficient anti-AD activity encouraged us to develop synthesis of novel tacrine-isoxazole hybrids as MTDLs. In this respect, all synthesized compounds were evaluated for their anti-ChE activity and the most potent compounds were candidate for complementary tests including in vitro self- and AChE-induced Aβ aggregation inhibitory activity, anti-BACE1 activity, metal chelating ability, neuroprotectivity against Aβ-induced damage in PC12 cells, and hepatotoxicity as well as in vivo studies by Morris water maze (MWM) task.
Methods and material
All chemicals and reagents were purchased from Merck and Aldrich. Melting points were determined on a Kofler hot stage apparatus and are uncorrected. The IR spectra were obtained on a Nicolet Magna FTIR 550 spectrometer (potassium bromide disks). NMR spectra were recorded on a Bruker 500 spectrometer and chemical shifts were expressed in δ (ppm) with respect to tetramethylsilane as the internal standard.
Chemistry
Synthesis of chlorotetrahydroacridines 3
A mixture of anthranilic acid derivative 1 (23 mmol) and cyclohexanone 2 (25 mmol) was heated in POCl3 (20 mL) for 3 h under reflux conditions. After completion of the reaction (TLC control), the reaction mixture was diluted with ethyl acetate, the excess POCl3 was neutralized using a 20% potassium carbonate solution, the organic phase was dried over anhydrous sodium sulfate, and the solvent was reduced under vacuum. The obtained product was purified through flash chromatography on silica gel using petroleum ether/ethyl acetate (9:1), yielding 70–80% of product [27, 28].
Synthesis of compounds 5
A mixture of chlorotetrahydroacridine derivative 3 (2 mmol) and phenol (1 mmol) was heated under reflux conditions at 150° C for 30 min. Ethylene diamine 4 (2 mmol) was then added dropwise to the mixture and the reaction was continued at the same conditions for 3 h. After completion of the reaction (TLC control), it was cooled to room temperature, extracted by chloroform, and washed three times using 3% KOH solution. It was then dried over anhydrous sodium sulfate and the solvent was evaporated under vacuum. The oily product was obtained with a yield of 60–70% [28].
General procedure for the preparation of tacrine-arylisoxazoles hybrids 7a–l
A mixture of isoxazole-3-carboxylic acid derivative 6 (1 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) (1.1 mmol), and hydroxybenzotriazole (HOBT) (1 mmol) in dry acetonitrile (10 mL) was stirred at room temperature for 1 h. Then, the corresponding derivative 5 (1 mmol) was added and the reaction was continued for 24–48 h at room temperature. After completion of reaction (TLC control), the mixture was extracted using chloroform, and washed with a 10% NaHCO3 solution, 10% citric acid solution, and brine, respectively. The organic phase was then dried over anhydrous sodium sulfate and the solvent was removed under vacuum to obtain pure solid product [29].
5-(4-Fluorophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)isoxazole-3-carboxamide (7a)
Yield: 55%. Mp: 114–116 °C. IR (KBr): 3395, 3122, 2939, 2872, 1661, 1636, 1588, 1511, 1445 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.15 (t, J = 6.0 Hz, 1H, NH), 8.49 (d, J = 8.0 Hz, 1H, H8), 8.00–7.95 (m, 3H, H5, H6, NH), 7.86 (d, J = 8.0 Hz, 2H, H2', H6'), 7.52 (t, J = 8.0 Hz, 1H, H7), 7.40 (t, J = 8.0 Hz, 2H, H3', H5'), 7.35 (s, 1H, isoxazole), 4.09–4.06 (q, J = 6.0 Hz, 2H, CH2), 3.67–3.64 (q, J = 6.0 Hz, 2H, CH2), 3.00–2.99 (m, 2H, H4), 2.70–2.69 (m, 2H, H1), 1.82–1.81 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm):169.6, 164.4, 162.4, 160.2 (J C-F = 250.0 Hz), 159.3, 156.1, 150.7, 137.8, 132.5, 128.3, 125.1, 122.9, 119.1, 116.5 (J C-F = 12.5 Hz), 115.7, 111.6, 99.6, 46.7, 40.1, 27.9, 23.9, 21.4, 20.2. Anal. calcd. for C25H23FN4O2: C, 69.75; H, 5.39; N, 13.02. Found: C, 69.52; H, 5.26; N, 12.90.
5-(4-Chlorophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)isoxazole-3-carboxamide (7b)
Yield: 50%. Mp: 117–119 °C. IR (KBr): 3389, 3122, 2940, 2871, 1661, 1636, 1584, 1522, 1444 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.10 (bs, 1H, NH), 8.47 (d, J = 8.0 Hz, 1H, H8), 7.95 (d, J = 7.5 Hz, 2H, H2', H6'), 7.95–7.74 (m, 2H, H5, NH), 7.77–7.76 (m, 1H, H6), 7.63 (d, J = 7.5 Hz, 2H, H3', H5'), 7.59–7.58 (m, 1H, H7), 7.39 (s, 1H, isoxazole), 4.08–4.06 (m, 2H, CH2), 3.66–3.65 (m, 2H, CH2), 2.97–2.96 (m, 2H, H4), 2.71–2.70 (m, 2H, H1), 1.83–1.82 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm):169.9, 164.4, 162.1, 159.9, 155.4, 147.5, 145.8, 136.1, 132.2, 130.0, 127.4, 125.0, 122.5, 119.7, 115.1, 111.5, 99.8, 52.0, 40.0, 24.4, 21.8, 20.7, 20.3. Anal. calcd. for C25H23ClN4O2: C, 67.19; H, 5.19; N, 12.54. Found: C, 67.42; H, 4.83; N, 12.81.
5-(4-Bromophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)isoxazole-3-carboxamide (7c)
Yield: 50%. Mp: 115–118 °C. IR (KBr): 3397, 3121, 2940, 2870, 1663, 1635, 1582, 1538, 1445 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.14 (bs, 1H, NH), 8.48 (d, J = 8.0 Hz, 1H, H8), 7.92 (d, J = 8.0 Hz, 1H, H5), 7.88–7.74 (m, 4H, H6, H2', H6', NH), 7.77 (d, J = 8.0 Hz, 2H, H3', H5'), 7.58 (t, J = 8.0 Hz, 1H, H7), 7.41 (s, 1H, isoxazole), 4.10–4.08 (m, 2H, CH2), 3.67–3.65 (m, 2H, CH2), 3.01–2.99 (m, 2H, H4), 2.70–2.69 (m, 2H, H1), 1.82–1.81 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 170.0, 163.2, 162.1, 159.5, 154.5, 148.5, 145.1, 135.9, 133.0, 130.5, 127.7, 125.0, 123.5, 119.6, 116.2, 111.2, 99.9, 51.8, 40.3, 24.4, 21.7, 20.5, 20.0. Anal. calcd. for C25H23BrN4O2: C, 61.11; H, 4.72; N, 11.40. Found: C, 60.89; H, 4.78; N, 11.24.
5-Phenyl-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)isoxazole-3-carboxamide (7d)
Yield: 55%. Mp: 100–103 °C. IR (KBr): 3399, 3123, 2933, 2863, 1661, 1635, 1576, 1542, 1443 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.80 (bs, 1H, NH), 9.15 (d, J = 7.5 Hz, 1H, H8), 8.59–8.52 (m, 5H, H5, H6, H2', H6', NH), 8.23–8.21 (m, 4H, H7, H3', H4', H5'), 8.02 (s, 1H, isoxazole), 4.74–4.73 (m, 2H, CH2), 4.34–4.33 (m, 2H, CH2), 3.66–3.65 (m, 2H, H4), 3.38–3.37 (m, 2H, H1), 2.52–2.50 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 170.5, 164.4, 159.2, 156.0, 150.9, 147.5, 136.1, 132.5, 131.0, 129.3, 126.2, 125.7, 125.0, 119.3, 115.8, 111.7, 99.8, 46.8, 40.0, 28.0, 23.9, 21.4, 20.3. Anal. calcd. for C25H24N4O2: C, 72.80; H, 5.86; N, 13.58. Found: C, 72.56; H, 5.49; N, 13.28.
N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-5-(p-tolyl)isoxazole-3-carboxamide (7e)
Yield: 55%. Mp: 102–104 °C. IR (KBr): 3379, 3130, 2941, 2870, 1660, 1634, 1586, 1518, 1446 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.10 (t, J = 6.0 Hz, 1H, NH), 8.47 (d, J = 8.0 Hz, 1H, H8), 7.91–7.85 (m, 2H, H5, NH), 7.83–7.80 (m, 3H, H6, H2', H6',), 7.58–7.57 (m, 1H, H7), 7.36 (d, J = 8.0 Hz, 2H, H3', H5'), 7.25 (s, 1H, isoxazole), 4.07–4.05 (m, 2H, CH2), 3.66–3.63 (m, 2H, CH2), 2.98–2.96 (m, 2H, H4), 2.71–2.68 (m, 2H, H1), 2.38–2.37 (m, 4H, H2, H3), 1.83 (s, 3H, CH3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 169.8, 161.5, 159.9, 154.1, 148.0, 144.7, 143.1, 140.2, 138.4, 134.0, 131.8, 130.2, 129.1, 127.7, 125.0, 118.4, 116.1, 48.8, 40.3, 29.1, 24.0, 21.7, 20.5, 20.0. Anal. calcd. for C26H26N4O2: C, 73.22; H, 6.14; N, 13.14. Found: C, 73.05; H, 5.89; N, 12.93.
5-(3-Methoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)isoxazole-3-carboxamide (7f)
Yield: 65%. Mp: 105–107 °C. IR (KBr): 3382, 3127, 2928, 2863, 1661, 1635, 1574, 1525, 1460 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.07 (bs, 1H, NH), 8.46 (d, J = 8.0 Hz, 1H, H8), 7.84–7.80 (m, 3H, H5, H6, H7), 7.58 (bs, 1H, NH), 7.50–7.45 (m, 3H, H4', H5', H6'), 7.37 (s, 1H, isoxazole), 7.10 (s, 1H, H2'), 4.10–4.08 (m, 2H, CH2), 3.84 (s, 3H, OCH3), 3.65–3.63 (m, 2H, CH2), 2.99–2.98 (m, 2H, H4), 2.71–2.69 (m, 2H, H1), 1.87–1.85 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 171.0, 165.5, 161.8, 160.2, 159.4, 155.7, 150.1, 139.2, 137.0, 131.8, 127.7, 125.9, 124.1, 122.6, 119.1, 116.3, 114.7, 111.4, 99.7, 54.5, 46.3, 40.0, 28.0, 24.1, 21.5, 20.3. Anal. calcd. for C26H26N4O3: C, 70.57; H, 5.92; N, 12.66. Found: C, 70.73; H, 6.02; N, 13.01.
5-(4-Nitrophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)isoxazole-3-carboxamide (7g)
Yield: 40%. Mp: 121–123 °C. IR (KBr): 3237, 3079, 2936, 1673, 1543, 1522, 1449, 1425, 1345, 1313 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.13 (bs, 1H, NH), 8.38 (d, J = 8.0 Hz, 2H, H3', H5'), 8.21 (d, J = 8.0 Hz, 2H, H2', H6'), 7.66 (s, 1H, isoxazole), 7.62–7.58 (m, 2H, H8, NH), 7.48–7.34 (m, 3H, H5, H6, H7), 4.27–4.26 (m, 2H, CH2), 4.10–4.03 (m, 2H, CH2), 3.11–3.07 (m, 2H, H4), 2.89–2.75 (m, 2H, H1), 2.06–1.97 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 168.1, 162.8, 159.7, 157.8, 150.9, 145.5, 143.7, 137.5, 135.4, 131.1, 128.8, 127.0, 125.5, 117.6, 112.5, 111.0, 124.5, 50.5, 40.1, 29.1, 26.1, 22.5, 21.1. Anal. calcd. for C25H23N5O4: C, 65.64; H, 5.07; N, 15.31. Found: C, 65.37; H, 4.87; N, 15.04.
N-(2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)-5-(4-fluorophenyl)isoxazole-3-carboxamide (7h)
Yield: 50%. Mp: 125–127 °C. IR (KBr): 3333, 3050, 2940, 1673, 1632, 1578, 1506, 1446 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.1 (bs, 1H, NH), 8.51 (d, J = 8.5 Hz, 1H, H8), 8.04 (s, 1H, H5), 8.02–7.94 (m, 3H, H2', H6', NH), 8.57 (d, J = 8.5 Hz, 1H, H7), 7.42–7.38 (m, 2H, H3', H5'), 7.35 (s, 1H, isoxazole), 4.10–4.06 (m, 2H, CH2), 3.66–3.65 (m, 2H, CH2), 2.99–2.97 (m, 2H, H4), 2.68–2.67 (m, 2H, H1), 1.83–1.81 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 169.4, 164.5, 162.2, 160.0 (J C-F = 250.2 Hz), 159.3, 156.4, 150.2, 143.1, 137.5, 134.0, 132.5, 128.7, 125.5, 122.8, 118.9, 116.0 (J C-F = 12.5 Hz), 99.6, 46.75, 40.10, 27.9, 23.9, 21.4, 20.2..Anal. calcd. for C25H22ClFN4O2: C, 64.59; H, 4.77; N, 12.05. Found: C, 64.23; H, 4.69; N, 11.89.
N-(2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)-5-(4-chlorophenyl)isoxazole-3-carboxamide (7i)
Yield: 55%. Mp: 120–122 °C. IR (KBr): 3338, 3050, 2941, 1673, 1632, 1577, 1519, 1444 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.12 (t, J = 6.0 Hz, 1H, NH), 8.50 (d, J = 9.5 Hz, 1H, H8), 7.98–7.96 (m, 2H, H5, NH), 7.94 (d, J = 8.0 Hz, 2H, H2', H6'), 7.62 (d, J = 8.0 Hz, 2H, H3', H5'), 7.58 (d, J = 9.5 Hz, 1H, H7), 7.39 (s, 1H, isoxazole), 4.08–4.04 (m, 2H, CH2), 3.66–3.62 (m, 2H, CH2), 2.98–2.96 (m, 2H, H4), 2.67–2.65 (m, 2H, H1), 1.84–1.82 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 169.5, 164.5, 159.9, 156.8, 150.7, 147.2, 144.4, 143.1, 140.6, 138.9, 135.1, 132.1, 130.3, 129.1, 125.5, 118.6, 116.1, 46.5, 40.3, 28.6, 23.0, 21.7, 20.2. Anal. calcd. for C25H22Cl2N4O2: C, 62.38; H, 4.61; N, 11.64. Found: C, 62.12; H, 4.28; N, 11.55.
N-(2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)-5-phenylisoxazole-3-carboxamide (7j)
Yield: 65%. Mp: 114–117 °C. IR (KBr): 3381, 3064, 2927, 1661, 1635, 1574, 1524, 1459 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.11 (bs, 1H, NH), 8.51 (d, J = 9.0 Hz, 1H, H8), 8.03–7.99 (m, 2H, H5, NH),7.92–7.90 (m, 2H, H2', H6'), 7.59–7.54 (m, 4H, H7, H3', H4', H5'), 7.33 (s, 1H, isoxazole), 4.07–4.06 (m, 2H, CH2), 3.65–3.64 (m, 2H, CH2), 2.97–2.96 (m, 2H, H4), 2.67–2.66 (m, 2H, H1), 1.82–1.80 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 169.0, 162.3, 162.0, 159.9, 153.5, 147.7, 144.5, 136.5, 134.2, 131.1, 130.2, 125.1, 124.9, 118.2, 116.2, 115.8, 99.5, 46.2, 40.1, 28.6, 23.9, 21.4, 20.2.Anal. calcd. for C25H23ClN4O2: C, 67.19; H, 5.19; N, 12.54. Found: C, 66.95; H, 4.89; N, 12.68.
N-(2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)-5-(p-tolyl)isoxazole-3-carboxamide (7k)
Yield: 45%. Mp: 110–112 °C. IR (KBr): 3270, 2941, 1664, 1577, 1508, 1445 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 9.08 (bs, 1H, NH), 8.50 (d, J = 9.5 Hz, 1H, H8), 8.05 (bs, H, NH), 7.93 (s, 1H, H5), 7.94 (d, J = 7.5 Hz, 2H, H2', H6'), 7.60 (d, J = 9.5 Hz, 1H, H7), 7.35 (d, J = 7.5 Hz, 2H, H3', H5'), 7.26 (s, 1H, isoxazole), 4.07–4.05 (m, 2H, CH2), 3.98–3.95 (m, 2H, CH2), 2.82–2.81 (m, 2H, H4), 2.67–2.66 (m, 2H, H1), 2.37 (s, 3H, CH3), 1.80–1.77 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 169.7, 161.9, 159.9, 153.8, 147.8, 144.4, 143.1, 140.6, 138.9, 134.3, 131.1, 130.2, 129.6, 128.7, 125.1, 118.6, 116.1, 46.8, 40.1, 28.0, 23.9, 21.4, 20.6, 20.0.Anal. calcd. for C26H25ClN4O2: C, 67.75; H, 5.47; N, 12.15. Found: C, 67.52; H, 5.38; N, 11.81.
N-(2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)-5-(3-methoxyphenyl)isoxazole-3-carboxamide (7l)
Yield: 50%. Mp: 118–120 °C. IR (KBr): 3255, 3064, 2938, 1665, 1633, 1576, 1437 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ (ppm):): 9.10 (t, J = 6.0 Hz, 1H, NH), 8.49 (d, J = 9.0 Hz, 1H, H8), 7.98 (s, 1H, H5), 7.94–7.92 (bs 1H, NH), 7.56 (d, J = 9.0 Hz, 1H, H7), 7.47–7.44 (m, 2H, H5', H6'), 7.39–7.38 (m, 2H, H2', isoxazole), 7.09 (d, J = 7.0 Hz, 1H, H4'), 4.05–4.04 (m, 2H, CH2), 3.83 (s, 3H, OCH3), 3.64–3.63 (m, 2H, CH2), 2.96–2.95 (m, 2H, H4), 2.66–2.65 (m, 2H, H1), 1.81–1.79 (m, 4H, H2, H3). 13C NMR (DMSO-d6, 125 MHz)δ (ppm): 170.4, 164.2, 162.9, 159.7, 159.2, 156.0, 150.6, 139.0, 132.4, 127.4, 125.4, 124.1, 122.2, 121.0, 117.9, 116.8, 115.4, 110.8, 100.4, 55.4, 46.8, 39.8, 27.1, 24.4, 21.3, 20.2.Anal. calcd. for C26H25ClN4O3: C, 65.47; H, 5.28; N, 11.75. Found: C, 65.33; H, 5.03; N, 12.01.
Biological activity
Cholinesterase inhibitory activity
Acetylcholinesterase (AChE, E.C. 3.1.1.7, Type V-S, lyophilized powder, from electric eel, 1000 unit), butyrylcholinesterase (BChE, E.C. 3.1.1.8, from equine serum), acetylthiocholine iodide (ATCI), and 5,5-dithiobis-(2-nitrobenzoic acid) (DTNB) were purchased from Sigma-Aldrich. Potassium dihydrogen phosphate, dipotassium hydrogen phosphate, potassium hydroxide, and sodium hydrogen carbonate were obtained from Fluka.
The in vitro cholinesterase inhibitory activity of all synthesized compounds 7a–l was studied using the modified Ellman's method, exactly according to our previous studies [30,31,32]. For this purpose, synthesized compounds 7a–l were dissolved in DMSO (1 mL) and diluted with a mixture of DMSO and methanol to achieve desired concentrations of the test compounds and obtain final ratio of 50/50 DMSO/methanol. Each well contained 50 μL potassium phosphate buffer (KH2PO4/ K2HPO4, 0.1 M, pH 8), prepared sample as described above (25 μL), AChE (25 μL) with final concentration of 0.22 U/mL in buffer. They were preincubated for 15 min at room temperature, and then DTNB (3 mM in buffer) (125 μL) was added. Characterization of hydrolysis of ATCI catalyzed by the AChE, was spectrometrically performed at 405 nm following with the addition of substrate (ATCI, 3 mM in water). After 15 min, the change in the absorbance was measured at 405 nm. A control experiment was performed under the same conditions without inhibitor, containing buffer, water, DTNB, and substrate.The IC50 values were graphically determined using inhibition curves. BChE inhibition assay was performed in the same described method. For all synthesized compounds, four different concentrations were tested in triplicate, to obtain an appropriate range of 20–80% inhibition for both enzymes.
Kinetic studies
Kinetic studies were carried out for the inhibition of AChE and BChE, respectively by compounds 7l and 7b according to the Ellman's method used for the inhibition assay, using various concentrations of inhibitors [32]. For the kinetic study on the inhibition of AChE, compound 7l was used at the concentrations of 0, 0.01, 0.05, and 0.1 µM. The Lineweaver–Burk reciprocal plot was constructed by plotting 1/V against 1/[S] at variable concentrations of the substrate acetylthiocholine (187.5, 750, 1500, 3000 µM). The inhibition constant Ki was calculated by the plot of slopes versus the corresponding concentrations of compound 7l. The same method was performed for the kinetic study on the inhibition of BChE using different concentrations of compound 7b (0, 0.01, 0.04, and 0.08 µM) and butyrylthiocholine (187.5, 750, 1500, 3000 µM).
Investigation of beta-secretase (BACE1) inhibitory effects
The BACE-1 inhibitory activity was studied via a fluorescence resonance emission transfer (FRET) method. The used kit consisted of BACE1 enzyme and APP peptide-based substrate (Rh-EVNLDAEFK- quencher) and the evaluation was performed according to our previous study [32].
Inhibition of AChE-induced and self-induced Aβ aggregation
The test was performed against Aβ1-42 aggregation and AChE-induced Aβ1-40 peptide aggregation using the Thioflavin T (ThT) assay according to our previous work [33].
Neuroprotection effect against Aβ-induced damage
The neuroprotectivity assay for protecting neuronal PC12 cells against Aβ25–35 induced damage was examined according to our previous report [34].
Hepatoxicity
To evaluate the effect of selected compounds on cell viability, the MTT assay was performed using HepG2 cell line [32].
Metal chelating studies
To study the metal chelating ability of selected compounds, a mixture of the methanolic solutions of the related compound (1 mL) and metal ion (1 mL) with the same final concentrations of 20 µM was incubated at room temperature for 30 min in a quartz cuvette. The absorption spectra were then recorded in the range of 200–600 nm [29] and compared with those spectra obtained from compounds alone.
Molecular docking evaluation
Interactions between selected derivatives and studied enzymes were investigated by in silico molecular docking to explore the preferred orientation of the ligands in the corresponding receptors’ binding site. The crystal structures of AChE (PDB code: 4ey7), BChE (PDB code: 4bds), and BACE1 (PDB code: 1w51) were downloaded from the Protein Data Bank.
Before screening the most potent compounds, the docking protocol was validated. The structure of cognate ligands (known potent compounds) were sketched using Hyperchem software and minimized with molecular mechanics MM+ and AM1 methods. All the water molecules and the co-crystallized ligands were removed from the enzyme structures. Polar hydrogens and gasteiger charges were assigned to the structures with AutoDockTools1.5.2 (ADT) and the PDBQT file format was prepared. ADT was used to select a docking grid and the parameters for the X, Y, Z grid dimensions were defined as 60 by 60 by 60 A°. AutoDock 4.2 was applied with a total of 100 runs that were carried out with a population size of 150 individuals, a maximum of 25 million energies evaluations, a maximum of 270,000 generations, a gene mutation rate of 0.01, and a crossover rate of 0.8 [35]. After validation of protein − ligand performance, the structure of selected compounds were sketched, minimized, and docked with the validated method [36]. Schematic 3D representations of the ligand-receptor interactions were generated using Discovery Studio Visualizer software programs.
In vivo memory evaluation using the Morris water maze (MWM) test
Ethical considerations
The Ethics Committee of Tehran University of Medical Sciences approved this research by the code IR.TUMS.TIPS.REC.1398.091 in 2019.
Drugs and animals
The required drugs including scopolamine hydrobromide and donepezil hydrochloride were purchased from Sigma and dissolved in saline as a carrier for the drugs. Compound 7l was dissolved in 20% polyethylene glycol (PEG-400) as a carrier.
Rats (Male Albino Wistar) with an average weight of 80–120 g were obtained from the Faculty of Pharmacy, Tehran University of Medical Sciences. They were randomly divided into several experimental groups. Their 12 h sleep and wake cycle was maintained and food and water were placed in their special place. Room temperature was maintained between 25 ± 2 °C. The method of handling animals in our laboratory is approved by the Ethics Committee for Animal Behavior.
Morris water maze
The Morris water maze is a water tank with a diameter of 136 cm and a depth of 35 cm. The maze is hypothetically divided into four equal parts, and a 10 cm diameter fiberglass escape platform is placed in one of the four sections, 1 cm below the water surface and cannot be seen from the outside. The water temperature is set at 25° C. The maze is placed in a room with various spatial signs, fixed during the tests and visible to the animal in the maze. The set is monitored by a tracker camera, located above the center of the water maze, and the test related information is stored using the Ethovision software. The choice of starting location is random. Simultaneously with releasing the animal into the water, the start button of the program is pressed and the program starts recording the behavior of the animal inside the maze. The maximum time allowed for the animal to find the platform is 90 s.
If the animal is unable to find the platform during the time (usually seen in the early days), it will be guided to the platform after the program is stopped and allowed to stand there for 15 s. During the 15 s, the animal remembers its position in the tank according to the position of the platform and the signs installed in the room. If the animal finds the platform by any means (accidentally or by using the signs) before the end of 90 s, the recording will be stopped as soon as the animal stands on the platform and it will be given 15 s to remember the signs. Rats are taught for 4 consecutive days, 4 times a day from 4 different random directions in the water maze. In this stage, the learning process of the animal is studied based on the time and distance traveled to find the platform.
On the fifth day after the training tests, a probe trial is performed. After removing the platform, the animal is randomly released inside the maze from one of the directions. The test is based on the assumption that the animal remembers the location of the platform, and must spend most of its time in the quarter where the platform was located. If this result is obtained, it will be clear that the animal did not accidentally find the platform, but it has determined the position of the platform based on the evidence and the signs in the room. This step is repeated once for each animal, for 90 s, and the duration of the time elapsed in the target quarter, where the platform was located, is a criterion of measuring the animal’s memory and learning abilities.
Experimental groups
In this study, five groups each consisting of 7 rats: control, scopolamine, synthesized compound at different concentrations plus scopolamine, and donepezil as positive control were investigated. All injections were done intraperitoneal and were given 30 min before the test. In the synthesized compound group, the compound was injected 1 h before the start of the test and the scopolamine was injected 30 min before the start of the test. For each group, the duration and distance traveled to find the platform and the speed of swimming in the first 4 days of training were recorded. On day 5 (probe day), the time elapsed in the target quarter where the platform was located, was used to measure the animal’s memory.
Control group
The rats in this group received normal saline (5 mL/kg) 30 min before the test during the first 4 learning days.
Scopolamine group
This group of rats received scopolamine hydrobromide (4 mg/kg) 30 min before the test in the first 4 days of learning.
Synthesized compound group
Compound 7l at different doses and scopolamine (4 mg/kg) was given to the animals 60 and 30 min before the test, respectively, in the first 4 learning days.
Donepezil group (positive control)
The rats in this group received donepezil (2.5 mg/kg) and scopolamine (4 mg/kg) 60 and 30 min before the test, respectively, in the first 4 days of learning.
Statistical analysis
The results were evaluated using the “one way anova” method, followed by a statistical comparison of “Bonferroni” and “Newman-Keuls”. Prism software version 6.0 was also used.
Results and discussion
Design of target compounds
To design novel multi-targeted tacrine analogues (Fig. 2), tacrine moiety was maintained as the main core and it was tried to incorporate a potent heterocyclic scaffold linked to tacrine moiety as heterocycles have shown versatile ChEI activity [37]. In this regard, isoxazole derivatives were selected based on our previous studies [29, 31, 38,39,40]. As can be seen in Fig. 2, compound F demonstrated good and selective AChEI activity (IC50 = 0.90 μM), however, it inhibited BChE with IC50 value of 31.20 µM [31]. In addition, compound G showed good and selective activity toward BChE (IC50 = 0.32 μM) and depicted lower activity toward AChE (IC50 = 16.82 μM). It also possessed negligible BACE1 inhibitory activity and neuroprotectivity [40]. Compound H showed selective AChEI activity (IC50 = 1.23 μM) and it was completely inactive against BChE. It also could inhibit BACE1 by 48.46% at 50 µM and moderate neuroprotectivity. Compound H was able to chelate Zn2+, Fe2+, and Cu2+ ions [30]. Apart from isoxazole moiety, the presence of the amide moiety (Fig. 2) has also been found as an important linker which is prone to construct desired interactions with the active site backbone and able to chelate metal ions.

Design of target compounds
Chemistry
Synthesis of tacrine analogues 7 has been schematically explained in Scheme 1. Initially, different derivatives of anthranilic acid 1 and cyclohexanone 2 reacted in POCl3 for 3 h under reflux conditions to afford chlorotetrahydroacridine derivative 3 [27]. Then, reaction of compound 3 and ethane-1,2-diamine 4 at reflux for 4 h led to the nucleophilic reaction of the amine and formation of compound 5 [28]. Finally, reaction of various derivatives of isoxazole-3-carboxylic acid 6 and compound 5 in dry acetonitrile, in the presence of EDCI and HOBT at room temperature for 24–48 h [29] gave the title compounds 7.

Synthesis of tacrine analogues 7a–l
Anti-cholinesterase activity
All synthesized compounds 7a–l were tested in vitro through the modified Ellman's method comparing with tacrine and donepezil as the reference drugs. Tacrine was selected as the positive control to compare the potency of synthesized hybrids based on structural similarity. For each compound, 50% inhibitory concentration (IC50) was determined and reported in Table 1. According to the presence or absence of the chlorine substitution in compounds 7, they were classified into two categories depending on the (i) absence and (ii) presence of Cl at 6- position of acridine moiety (7a–g and 7 h–l).
In the case of AChEI activity, the most potent compounds 7l and 7i with IC50 values of 0.050 and 0.052 µM belonged to the second category. These compounds have different substituents on the aryl ring connected to the isoxazole moiety, compound 7l contained meta-methoxy and compound 7i contained para-chloro aryl groups. Their counterparts in the first category, compounds 7f and 7b showed lower activity (IC50s = 0.129 and 0.075 µM, respectively) than compounds 7l and 7i. It should be noted that compound 7b having para-chloro aryl group showed potent anti-AChE activity indicating the efficacy of Cl even if it is not present on the acridine moiety. Compounds 7h and 7a containing para-fluoro aryl group also were found to be potent AChE inhibitors, however, 7h possessing Cl on the acridine moiety was a little more potent than 7a. The absence of substituents on the aryl ring (compounds 7j and 7d, IC50s = 0.100 and 0.299 µM, respectively) generally resulted in the reduction of activity. Also, replacement of F and Cl by large-size halogen (Br, 7c, IC50 = 0.175 µM) led to at least a twofold reduction in the anti-AChE activity. Finally, compounds 7e and 7k having para-methyl aryl group showed IC50s = 0.147 and 0.170 µM, respectively. Nevertheless, the presence of strong electron withdrawing group (NO2, 7g, IC50 = 10.26 µM) absorbed more attention since its activity was remarkably deteriorated. With these results in hand, it can be concluded that (i) the presence of Cl at 6- position of acridine usually improved AChEI activity (7l > 7f, 7i > 7b, 7h > 7a, 7j > 7d) which was in good accordance with our previous results (Fig. 1). However, an exception was observed in the case of 7e > 7k. (ii) The Cl substituent was not only effective when it was present on the acridine moiety but also when it was inserted into the aryl group connected to the isoxazole ring. (iii) The presence of strong electron donating group (OMe) and F on the aryl was found to be effective for inducing desired anti-AChE activity, while the presence of strong electron withdrawing group (NO2) was not beneficial at all.
In the case of anti-BChE activity, compound 7b from the first category having para-chloroaryl group showed the most potent activity (IC50 = 0.039 µM). However, its counterpart in the second category 7i depicted approximately a tenfold decrease in the inhibitory activity (IC50 = 0.372 µM). Replacement of Cl by F in compounds 7a and 7h led to the reduction of anti-BChE activity (IC50s = 0.047 and 0.202 µM, respectively), still the compound from the first category was much more active than its counterpart in the second category. Moderate electron donating group (Me) was found to be important, ranking after Cl and F since compound 7e showed good activity toward BChE with IC50 value of 0.051 µM. Introduction of a strong electron donating substituent (OMe) decreased activity (compound 7f, IC50 = 0.075 µM) and it should not be forgotten that compounds 7e and 7f were significantly more potent than those counterparts in the second category (IC50s = 0.104 and 0.413 µM, respectively). Compound 7c containing para-bromoaryl group showed moderate activity (IC50 = 0.087 µM) as potent as compound 7d with no substituents on the aryl group (IC50 = 0.088 µM). Interestingly, the counterpart of 7d, compound 7j showed the same BChEI activity (IC50 = 0.088 µM). Compound 7g possessing para-nitroaryl group was the weakest inhibitor of BChE in the series 7a–l (IC50 = 1.064 µM). In summary, i) anti-BChE activity of synthesized compounds lacking Cl on the acridine moiety led to better activity than those possessing it, as the order of activity 7b > 7i, 7a > 7h, 7e > 7k, 7f > 7l was observed. ii) The effect of substituents on the aryl group connected to the isoxazole moiety was completely in relation with the absence or presence of Cl on the acridine moiety. In the first category (7a–g), the presence of Cl, F, and Me was found to be important but in the second category of compounds (7h–l), the absence of substituents led to higher activity. However, the presence of methyl group was effective.
Comparing AChEI and BChEI activity of synthesized compounds 7 indicated that the first category of compounds 7a–g were better inhibitor of BChE than AChE while in the second category 7h–l, the results were generally reversed. For the first category, AChEI and BChEI activity was improved by the presence of Cl and F on the aryl ring connected to the isoxazole moiety. However, in the case of the second category, no definite rule was followed. Also, the presence of strong electron withdrawing group (NO2) deteriorated both AChE and BChE inhibitory activity.
The presence of Cl on the acridine moiety often improved AChEI activity comparing with their analogues lacking this substituent, while it decreased BChEI potency. The AChEI activity of chlorinated tacrine derivatives has been previously discussed in the literature [28, 41,42,43], which can be explained by the formation of halogen bonding with the receptor [44].
Comparing our results with those reported for tacrine-hybrids (Fig. 1) showed that synthesized tacrine-isoxazoles 7 were found to be generally more potent than compounds B–E. It seems that the hybridization of isoxazole with tacrine was more efficient than the presence of thidiazole (compound B) [23], pyrazolo[3,4-b]pyridine (compound C) [24], and 1,2,3-triazole (compound D and E) [26]. However, oxindole hybrids [22] were found to be more potent than isoxazole.
Kinetic studies
Kinetic studies were performed to investigate the mechanism of inhibition by the most potent inhibitors of AChE and BChE, compounds 7l and 7b, respectively. Graphical analysis of the reciprocal Lineweaver–Burk plot of both inhibitors indicated a competitive inhibition (Fig. 3 and 4) describing that both compounds had exclusive affinity for the enzymes and competed with the substrate for binding to the active site. In addition, the Ki values were calculated using the secondary plot as Ki = 0.035, 0.009 µM for the inhibition of AChE and BChE, respectively.

Kinetic study of compound 7l [I] against AChE. Lineweaver–Burk plot and double reciprocal Lineweaver–Burk plot are shown

Kinetic study of compound 7b against BChE. Lineweaver–Burk plot and double reciprocal Lineweaver–Burk plot are shown
Beta-secretase 1 (BACE1) inhibitory activity of compound 7l
Small molecule nonpeptide inhibitor of BACE1 is a promising target for the treatment of AD. This enzyme functions in the first step of the pathway of production and deposition of Aβ [45]. In this respect, BACE1 inhibitory activity of compound 7l was evaluated and compared with OM99-2 (IC50 = 0.014 ± 0.003 µM). Our results indicated that 7l had good inhibitory activity with IC50 = 1.65 ± 0.005 µM.
Inhibition of AChE-induced and self-induced Aβ aggregation
Aβ peptide has been detected as the major component of the amyloid plaques found in the brains of people with AD. Thus, the effect of potent inhibitors 7b and 7l were investigated for their inhibition ability against Aβ1-42 aggregation and AChE-induced Aβ1-40 peptide aggregation comparing with donepezil and tacrine as the reference compounds.
Compound 7b showed 61.8 ± 3.55% and 58.6 ± 2.9% potency for the inhibition of Aβ1-42 self-aggregation and AChE-induced Aβ aggregation, respectively. Those values for compound 7l were 79.4 ± 1.9 and 71.4 ± 1.8%, respectively. However, donepezil showed 15.9 ± 1.2 and 28.4 ± 3.6% inhibition and tacrine’s values were found to be 7.9 ± 0.5 and 7.0 ± 1.4%, respectively. Our results indicated high activity of compounds 7b and 7l for the inhibition of AChE-induced and self-induced Aβ aggregation.
Neuroprotection effect against Aβ-induced damage
The neuroprotective role of the strongest AChE inhibitor 7l against Aβ-induced PC12 cell damage was investigated comparing with caffeic acid as a positive control which protected the PC12 cells with 33.85% at 100 μM. Unfortunately, no significant protection was observed by compound 7l at different concentrations.
In vitro hepatotoxicity test
The clinical use of tacrine has been restricted due to its poor oral bioavailability and severe hepatotoxicity [20]. Therefore, development of tacrine analogues with low hepatotoxicity is in the center of attention.
Compounds 7b and 7l were selected to be evaluated in vitro toward HepG2 cells (Fig. 5). It was found that the cytotoxicity of 7l was comparable with that of tacrine. It demonstrated lower toxicity since the cell viability values were calculated as 100, 100, 87.5, and 54.3% at concentration of 2.5, 5, 10, and 20 μg/mL, respectively compared to tacrine which showed cell viability (%) of 100, 100, 89.4, and 65.8% at the same concentrations. However, compound 7b was found to be more toxic than 7l and tacrine based on cell viability (%) values of 54, 50, 42, and 44% at the similar concentrations.

Cytotoxicity of compounds 7b and 7l against HepG2
Metal chelating studies
Compounds 7l and 7b were tested for their metal chelating abilities toward Fe2+, Cu2+, and Zn2+ ions (Fig. 6a and b). In the case of compound 7l (Fig. 6a), UV spectrum of its methanolic solution (20 µM) showed two absorption peaks at 254.4 and 333.3 nm. After interaction of compound 7l with Zn2+ (7l-Zn2+) for 30 min, those peaks shifted to 250.1 and 331.2 nm, respectively. The similar interaction with Fe2+ ions (7l-Fe2+) resulted in the shift of those peaks to 252.3 and 329.1 nm. In the case of interaction of 7l and Cu2+ ions, only one shift to 329.1 nm was observed and the peak at 254.4 did not change.

The absorbance change of compounds 7l (A) and 7b (B) alone and in the presence of Zn2+, Fe2+, and Cu2+ ions at wavelength of 200–600 nm
Compound 7b (Fig. 6b) depicted three characteristic peaks at 248.0, 273.6, and 335.5 nm. After interaction of 7b and metal ions, the peak at 248.0 nm shifted to 245.9 nm for 7b-Zn2+, however 7b-Cu2+ and 7b-Fe2+ showed no change. Also, the peak at 273.6 nm was similarly observed for 7b-Zn2+ and 7b-Cu2+ but it shifted to 275.7 nm for 7b-Fe2+. It should be noted that all 7b complexes showed peaks at 335.5 nm with different intensity.
Docking studies
To gain further insight into the interactions and related inhibitory effects, in silico simulations of molecular docking were carried out on the crystal structures of AChE (PDB code: 4ey7), BChE (PDB code: 4bds), and BACE1 (PDB code: 1w51).
Validation of protein − ligand docking was performed and recorded the RMSD values of 0.73 Å, 0.36 Å, and 1.39 Å between the docked structure and crystallographic inhibitors of AChE, BACE1, and BChE, respectively (RMSD < 3 Å is the commonly acceptable limit). The results indicated that the parameters for docking simulations were good in the prediction and validation of protein–ligand docking performance.
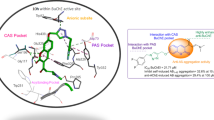
Docking study results of the most potent AChE inhibitor (compound 7l) in the AChE active site (Fig. 7) showed that 7l displayed three conventional hydrogen bonding interactions between the oxygen of the amide group and Tyr124, the amine group of tetrahydroacridin moiety and Tyr124, and the oxygen of 3-methoxyaryl group and Tyr72, respectively. It was found that tetrahydroacridin core established effective π-π and π-aryl interactions with side chains of Trp86 and Tyr124 (important residues of the choline-binding site). The same π-π interactions were evident between Trp286, Tyr341 (residue of the PAS pocket), and 3-methoxyaryl and isoxazole moieties. Also, the AChE residues, His447 (critical residue of catalytic triads) and Gly448 showed π-donor and halogen interactions with the chlorotetrahydroacridin-9-amine ring [45]. Taking into account all of these interaction modes confirmed the higher stability of 7l-AChE (-9.52 kcal mol−1) and its potent activity.

Docking conformations of compound 7l (green line) in the AChE. Hydrogen bonds are depicted in green dashed lines, π-π stacked interactions are depicted in dark pink dashed lines, π-aryl interactions are depicted in pink dashed lines, π-donor interactions are depicted in pale green dashed lines, and halogen interactions are depicted in blue dashed lines
Next, to confirm the potential of 7b as a potent BChE inhibitor, molecular docking was also developed. Regarding the results depicted in Fig. 8, it can be understood that 7b was mainly stabilized by hydrogen bonding interactions between the nitrogen of tetrahydroacridin ring and Ser198 (catalytic triads of the BChE) as well as nitrogen of amide linker and Pro285. Additionally, 4-chloroaryl substituent contributed to binding by π-π-T-shape interactions with Pro285 residue in the PAS pocket. Moreover, hydrophobic interactions were established between the tetrahydroacridin ring and Trp231 (π-alkyl), Leu286 (π-alkyl), and Phe329 (π-π) which described the stability of the 7b-BChE complex (−9.74 kcal mol−1). The molecular docking was consistent with the results of enzyme assays. The above results suggested sufficient explanation for 7b as a potent BChE inhibitor.

Docking conformations of compound 7b (gray line) in BChE. Hydrogen bonds are depicted in green dashed lines, π-π stacked interactions are depicted in dark pink dashed lines, and π-aryl interactions are depicted in pink dashed lines
Figure 9 shows the docking interactions of compound 7l within the BACE1 active site. Chloro-tetrahydroacridin-9-amine moiety made H-bonding interaction with Gly230 (2.05 Å) as well as π-π stacked interaction with Tyr71. Chlorotetrahydroacridine ring also showed a π–alkyl interaction with Ile110. On the other side of the molecule, 3-methoxyaryl pendant formed three Vander Waals interactions with Gly34, Asp228, and Thr329. π-Sigma and π-alkyl interactions were also observed between the methoxyaryl substituent and Val332 and Ile226 residues, respectively. Two other H-bonding interactions were also seen between the nitrogen of amide linker and Thr231 (1.86 Å) as well as oxygen of carbonyl group with Gln73 (3.00 Å). Also, isoxazole core was fixed through two π-alkyl and one Vander Waals interactions with Arg235, Val332 and Thr72, respectively. These results confirmed the high potential of 7l as a potent BACE1 inhibitor.

Docking conformations of compound 7l (green line) in the BACE1 enzyme. Hydrogen bonds were depicted in green dashed lines, van der Waals interactions were depicted in pale green dashed lines, π-alkyl and alkyl interactions were depicted in pale purple dashed lines, and π-sigma interactions were depicted in purple dashed lines
Morris water maze test
Morris water maze (MWM) has been developed to evaluate the efficacy of drug candidates for the improvement of memory deficits. Practically, scopolamine which is an anticholinergic compound, is used to induce amnesia and memory loss in animal models [47]. In this work, the effect of compound 7l (the most potent AChE inhibitor) was evaluated for its ability to reverse scopolamine-induced memory deficit in rats.
The effect of prescribing 7l on the escape latency time, traveled distance and the swimming speed during learning days
Figure 10 (A-C) shows the escape latency time, traveled distance, and swimming speed during learning days, respectively for all groups in the first 4 day of trials. As can be seen in Fig. 10A and 10B, the scopolamine group depicted a significant increase of escape latency time (P < 0.01) and traveled distance (P < 0.01) comparing with the control group. Group administered by compound 7l at the dose of 1.25 mg/kg plus scopolamine showed a smaller significant difference (P < 0.05) in terms of the escape latency time and traveled distance than scopolamine group comparing with control group.

(A) The effect of prescribing 7l on the escape latency time during learning days. (B) The effect of prescribing 7l on the traveled distance during learning days. (C) The effect of prescribing 7l on the swimming speed during learning days. Each column displays the mean ± SEM for seven rats in all 4 training days for each group. Sco, scopolamine; *Significantly different from control group. *P < 0.05; **P < 0.01. #Significantly different from scopolamine group. # P < 0.05; ## P < 0.01

The effect of prescribing 7l on the probe test in the different groups. Values are expressed as mean ± SEM for seven animals in each group. Sco, scopolamine; *Significantly different from control group. **P < 0.01, # Significantly different from scopolamine group. ##P < 0.01
This result indicated that compound 7l at the dose of 1.25 mg/kg could slightly improve learning memory impairment made by scopolamine. However, the group administered by compound 7l at the dose of 2.50 mg/kg plus scopolamine showed significant improvement of both escape latency time and traveled distance (P < 0.05 and P < 0.01, respectively) comparing with scopolamine group. It should be noted that all groups showed no significant differences in swimming speed (Fig. 10c) due to MWM learning impairment are independent of locomotor effects [48, 49].
The effect of prescribing 7l on the probe test
After training rats for 4 days, the probe test was performed on the fifth day. The trained rats were released into the water tank for 90 s and the hidden platform was removed and the time spent in the target quarter was recorded.
As shown in Fig. 11, this parameter was significantly reduced in the groups receiving scopolamine and compound 7l at the dose of 1.25 mg/kg plus scopolamine comparing with control group (P < 0.01). However, it was increased in the groups administrated by compound 7l at the dose of 2.50 mg/kg and donepezil plus scopolamine comparing with the scopolamine group (P < 0.01). These data indicated that memory impairment was occurred in two first groups, whereas in the two-latter groups, compound 7l and donepezil could reverse memory deficit made by scopolamine.
Conclusion
In conclusion, searching efficient multi-targeted ligands against AD led us to the design and synthesis of novel arylisoxazole-tacrine hybrids. According to the Ellman’s method, all compounds demonstrated excellent in vitro ChE inhibitory activity and among tested compounds, 7l and 7b depicted the most potent AChEI (IC50 = 0.050 μM) and BChEI (IC50 = 0.039 μM) activity, respectively. It was perceived that anti-AChE activity of all compounds was more potent than tacrine and their activity was comparable with that of donepezil. Also, synthesized compounds showed dual activity toward ChEs. Their anti-BChE activity was much more potent than donepezil and found to be weaker than tacrine.
The important aspect of this work comes back to the potency of target compounds for the inhibition of βA aggregation and BACE1 as the main mechanisms involved in the onset of AD. Self-induced and AChE-induced Aβ aggregation inhibitory activity of compounds 7l and 7b were much more potent than those of donepezil and tacrine. It should be noted that compound 7l depicted better results than compound 7b. Evaluation of the inhibitory activity of compound 7l toward BACE1 indicated its high efficacy (IC50 = 1.65 µM).
Compounds 7l and 7b were found to be efficient chelators of zinc, copper, and iron ions. However, in vitro hepatotoxicity assay on HepG2 cells revealed that compounds 7l and 7b were not safer than tacrine. Furthermore, compound 7l lacked satisfactory neuroprotectivity against Aβ-induced damage in PC12 cells. The effect of compound 71 on the improvement of cognitive and spatial memory deficits in AD was investigated through MWM test which remarkably reversed scopolamine-induced memory deficit in rats.
All results suggested that synthesized tacrine hybrids particularly compounds 7l and 7b can be considered as the versatile drug candidates against AD. Although various benefits were achieved in this work, more complementary animal and human tests are in demand to show desired efficacy of 7l as a lead compound.
References
Patterson C (2018) World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers. Alzheimer’s Disease International (ADI), London
Deture MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 5:1–18. https://doi.org/10.1186/s13024-019-0333-5
https://alz-journals.onlinelibrary.wiley.com/doi/full/https://doi.org/10.1002/alz.12068
Piton M, Hirtz C, Desmetz C, Milhau J, Dominique-Lajoix A, Bennys K, Lehmann S, Gabelle A (2018) Alzheimer’s disease: advances in drug development. J Alzheimers Dis 65(1):3–13. https://doi.org/10.3233/JAD-180145
Oxford AE, Stewart ES, Rohn TT (2020) Clinical trials in Alzheimer's disease: A hurdle in the path of remedy. Int J Alzheimers Dis e5380346. https://doi.org/10.1155/2020/5380346.
Dos-Santos-Picanco LC, Ozela PF, de-Fatima-de-Brito M, Pinheiro AA, Padilha EC, Braga FS, de-Paula-da-Silva CHT, Dos-Santos CBR, Rosa JMC, da-Silva-Hage-Melim LI (2018) Alzheimer's disease: A review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr Med Chem 25(26):3141-3159. https://doi.org/10.2174/0929867323666161213101126
Tobor TO (2019) On the etiopathogenesis and pathophysiology of Alzheimer’s disease: a comprehensive theoretical review. J Alzheimer’s Dis 68(2):417–437. https://doi.org/10.3233/JAD-181052
Teipel SJ, Fritz HC, Grothe MJ (2020) Alzheimer's disease neuroimaging initiative. Neuropathologic features associated with basal forebrain atrophy in Alzheimer disease. Neurology 95(10):1301–1311. https://doi.org/10.1212/WNL.0000000000010192.
Colautti J, Nagales K (2020) Tau and beta-amyloid in Alzheimer’s disease: Theories, treatments strategies, and future directions. Meducator 1(37):12–15. https://doi.org/10.15173/m.v1i37.2502
Castellani RJ, Plascencia-Villa G, Perry G (2019) The amyloid cascade and Alzheimer’s disease therapeutics: theory versus observation. Lab Invest 99:958–970. https://doi.org/10.1038/s41374-019-0231-z
Moussa-Pacha NM, Abdin SM, Omar HA, Alniss H, Al-Tel TH (2020) BACE1 inhibitors: current status and future directions in treating Alzheimer’s disease. Med Res Rev 40:339–384. https://doi.org/10.1002/med.21622
Bruni AC, Bernardi L, Gabelli C (2020) From beta amyloid to altered proteostasis in Alzheimer’s disease. Ageing Res Rev. https://doi.org/10.1016/j.arr.2020.101126
Cacabelos R (2020) Pharmacogenetic considerations when prescribing cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin Drug Metab Toxicol 16(8):673–701. https://doi.org/10.1080/17425255.2020.1779700
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215(4537):1237–1239. https://doi.org/10.1126/science.7058341
Agatonovic-Kustrin S, Kettle C, Morton DW (2018) A molecular approach in drug development for Alzheimer’s disease. Biomed Pharmacother 106:553–565. https://doi.org/10.1016/j.biopha.2018.06.147
Sharma K (2019) Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol Med Rep 20(2):1479–1487. https://doi.org/10.3892/mmr.2019.10374
Kabir MT, Uddin MS, Begum MM, Thangapandiyan S, Rahman MS, Aleya L, Mathew B, Ahmed M, Barreto GE, Ashraf GM (2019) Cholinesterase inhibitors for Alzheimer’s disease: multitargeting strategy based on anti-Alzheimer’s drugs repositioning. Curr Pharm Des 25(33):3519–3535. https://doi.org/10.2174/1381612825666191008103141
Martinez A, Castro A (2006) Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer’s disease. Expert Opin Invest Drugs 15(1):1–12. https://doi.org/10.1517/13543784.15.1.1
Ismaili L, Refouvelet B, Benchekroun M, Brogi S, Brindisi M, Gemma S, Campiani G, Filipic S, Agbaba D, Esteban G, Unzeta M, Nikolic K, Butini S, Marco-Contelles J (2017) Multitarget compounds bearing tacrine-and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog Neurobiol 151:4–34. https://doi.org/10.1016/j.pneurobio.2015.12.003
McEneny-King A, Osman W, Edginton AN, Rao PPN (2017) Cytochrome P450 binding studies of novel tacrine derivatives: predicting the risk of hepatotoxicity. Bioorg Med Chem Lett 27(11):2443–2449. https://doi.org/10.1016/j.bmcl.2017.04.006
Sameem B, Saeedi M, Mahdavi M, Shafiee A (2017) A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur J Med Chem 128:332–345. https://doi.org/10.1016/j.ejmech.2016.10.060
Riazimontazer E, Sadeghpour H, Nadri H, Sakhteman A, Tüylü Küçükkılınç T, Miri R, Edraki N (2019) Design, synthesis and biological activity of novel tacrine-isatin Schiff base hybrid derivatives. Bioorg Chem 89:103006. https://doi.org/10.1016/j.bioorg.2019.103006
Makhaeva GF, Kovaleva NV, Boltneva NP, Lushchekina SV, Rudakova EV, Stupina TS, Terentiev AA, Serkov IV, Proshin AN, Radchenko EV, Palyulin VA, Bachurin SO, Richardson RJ (2020) Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer’s disease treatment: synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg Chem 94:103387. https://doi.org/10.1016/j.bioorg.2019.103387
Korabecny J, Musilek K, Zemek F, Horova A, Holas O, Nepovimova E, Opletalova V, Hroudova J, Fisar Z, Jung YS, Kuca K (2011) Synthesis and in vitro evaluation of 7-methoxy-N-(pent-4-enyl)-1,2,3,4-tetrahydroacridin-9-amine-new tacrine derivate with cholinergic properties. Bioorg Med Chem Lett 21:6563–6566. https://doi.org/10.1016/j.bmcl.2011.08.042
Pan T, Xie S, Zhou Y, Hu J, Luo H, Li X, Huang L (2019) Dual functional cholinesterase and PDE4D inhibitors for the treatment of Alzheimer’s disease: design, synthesis and evaluation of tacrine-pyrazolo[3,4-b]pyridine hybrids. Bioorg Med Chem Lett 29(16):2150–2152. https://doi.org/10.1016/j.bmcl.2019.06.056
Najafi Z, Mahdavi M, Mahdavi M, Saeedi M, Karimpour-Razkenari E, Asatouri R, Vafadarnejad F, Homayouni-Moghadam F, Khanavi M, Sharifzadeh M, Akbarzadeh T (2017) Novel tacrine-1,2,3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur J Med Chem 125:1200–1212. https://doi.org/10.1016/j.ejmech.2016.11.008
Hu MK, Lu CF (2000) A facile synthesis of bis-tacrine isosteres. Tetrahedron Lett 41(11):1815–1818. https://doi.org/10.1016/S0040-4039(00)00036-8
Najafi Z, Mahdavi M, Saeedi M, Karimpour-Razkenari E, Edraki N, Sharifzadeh M, Khanavi M, Akbarzadeh T (2019) Novel tacrine-coumarin hybrids linked to 1,2,3-triazole as anti-Alzheimer’s compounds: In vitro and in vivo biological evaluation and docking study. Bioorg Chem 83:303–316. https://doi.org/10.1016/j.bioorg.2018.10.056
Saeedi M, Rastegari A, Hariri R, Mirfazli SS, Mahdavi M, Edraki N, Firuzi O, Akbarzadeh T (2020) Design and synthesis of novel arylisoxazole-chromenone carboxamides: investigation of biological activities associated with Alzheimer’s disease. Chem Biodivers 17(5):e1900746. https://doi.org/10.1002/cbdv.201900746
Vafadarnejad F, Mahdavi M, Karimpour-Razkenari E, Edraki N, Sameem B, Khanavi M, Saeedi M, Akbarzadeh T (2018) Design and synthesis of novel coumarin-pyridinium hybrids: In vitro cholinesterase inhibitory activity. Bioorg Chem 77:311–319. https://doi.org/10.1016/j.bioorg.2018.01.013
Najafi Z, Mahdavi M, Saeedi M, Sabourian R, Khanavi M, Safavi M, Tehrani MB, Shafiee A, Foroumadi A, Akbarzadeh T (2017) 1,2,3-Triazole-Isoxazole based acetylcholinesterase inhibitors: synthesis, biological evaluation and docking Study. Lett Drug Des Discov 14:58–65. https://doi.org/10.2174/1570180813666160628085515
Saeedi M, Safavi M, Allahabadi E, Rastegari A, Hariri R, Jafari S, Bukhari SNA, Mirfazli SS, Firuzi O, Edraki N, Mahdavi M, Akbarzadeh T (2020) Thieno[2,3-b]pyridine amines: synthesis and evaluation of tacrine analogs against biological activities related to Alzheimer’s disease. Arch Pharm 353:e2000101. https://doi.org/10.1002/ardp.202000101
Karimi-Askarani H, Iraji A, Rastegari A, Abbas-Bukhari SN, Firuzi O, Akbarzadeh T, Saeedi M (2020) Design and synthesis of multi-target directed 1,2,3-triazole-dimethylaminoacryloyl-chromenone derivatives with potential use in Alzheimer's disease. BMC Chem 14(1):pe64. https://doi.org/10.1186/s13065-020-00715-0.
Iraji A, Firuzi O, Khoshneviszadeh M, Tavakkoli M, Mahdavi M, Nadri H, Edraki N, Miri R (2017) Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur J Med Chem 141:690–702. https://doi.org/10.1016/j.ejmech.2017.09.057
Iraji A, Firuzi O, Khoshneviszadeh M, Nadri H, Edraki N, Miri R (2018) Synthesis and structure-activity relationship study of multi-target triazine derivatives as innovative candidates for treatment of Alzheimer’s disease. Bioorg Chem 77:223–235. https://doi.org/10.1016/j.bioorg.2018.01.017
Edraki N, Firuzi O, Fatahi Y, Mahdavi M, Asadi M, Emami S, Divsalar K, Miri R, Iraji A, Khoshneviszadeh M, Firoozpour L, Shafiee A, Foroumadi A (2015) N-(2-(Piperazin-1-yl)phenyl)arylamide derivatives as β-secretase (BACE1) inhibitors: simple synthesis by Ugi Four-component reaction and biological evaluation. Arch Pharm 348(5):330–337. https://doi.org/10.1002/ardp.201400322
Martorana A, Giacalone V, Bonsignore R, Pace A, Gentile C, Pibiri I, Buscemi S, Lauria A, Piccionello AP (2016) Heterocyclic scaffolds for the treatment of Alzheimer’s disease. Curr Pharm Des 22:3971–3995
Saeedi M, Mohtadi-Haghighi D, Mirfazli SS, Mahdavi M, Hariri R, Lotfian H, Edraki N, Iraji A, Firuzi O, Akbarzadeh T (2019) Design and synthesis of selective acetylcholinesterase inhibitors: Arylisoxazole-Phenylpiperazine derivatives. Chem Biodivers 16:e1800433. https://doi.org/10.1002/cbdv.201800433
Vafadarnejad F, Saeedi M, Mahdavi M, Rafinejad A, Karimpour-Razkenari E, Sameem B, Khanavi M, Akbarzadeh T (2017) Novel indole-isoxazole hybrids: synthesis and in vitro anti-cholinesterase activity. Lett Drug Des Discov 14:712–717. https://doi.org/10.2174/1570180813666161018124726
Vafadarnejad F, Karimpour-Razkenari E, Sameem B, Saeedi M, Firuzi O, Edraki N, Mahdavi M, Akbarzadeh T (2019) Novel N-benzylpyridinium moiety linked to arylisoxazole derivatives as selective butyrylcholinesterase inhibitors: Synthesis, biological evaluation, and docking study. Bioorg Chem 92:103192. https://doi.org/10.1016/j.bioorg.2019.103192
Ragab HM, Teleb M, Haidar HR, Gouda N (2019) Chlorinated tacrine analogs: Design, synthesis and biological evaluation of their anti-cholinesterase activity as potential treatment for Alzheimer’s disease. Bioorg Chem 86:557–568. https://doi.org/10.1016/j.bioorg.2019.02.033
Ragab HM, Ashour HMA, Galal A, Ghoneim AI, Haidar HR (2016) Synthesis and biological evaluation of some tacrine analogs: study of the effect of the chloro substituent on the acetylcholinesterase inhibitory activity. Monatsh Chem 147:539–552. https://doi.org/10.1007/s00706-015-1641-2
Keri RS, Quintanova C, Chaves S, Silva DF, Cardoso SM, Santos MA (2016) New tacrine hybrids with natural-based cysteine derivatives as multitargeted drugs for potential treatment of Alzheimer’s disease. Chem Biol Drug Des 87(1):101–111. https://doi.org/10.1111/cbdd.12633
Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM (2013) Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem 56(4):1363–1388. https://doi.org/10.1021/jm3012068
Iraji A, Khoshneviszadeh M, Firuzi O, Khoshneviszadeh M, Edraki N (2020) Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg Chem 97:e103649. https://doi.org/10.1016/j.bioorg.2020.103649
Yazdani M, Edraki N, Badri R, Khoshneviszadeh M, Iraji A, Firuzi O (2020) 5,6-Diphenyl triazine-thio methyl triazole hybrid as a new Alzheimer’s disease modifying agents. Mol Divers 24:641–654. https://doi.org/10.1007/s11030-019-09970-3
Liao J, Nai Y, Feng L, Chen Y, Li M, Xu H (2020) Walnut oil prevents scopolamine-induced memory dysfunction in a mouse model. Molecules 25:pe1630. https://doi.org/10.3390/molecules25071630.
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. https://doi.org/10.1038/nprot.2006.116
Brandeis R, Brandys Y, Yehuda S (1989) The use of the Morris Water Maze in the study of memory and learning. Int J Neurosci 48(1–2):29–69. https://doi.org/10.3109/00207458909002151
Acknowledgements
This work was supported by grants from the Research Council of Tehran University of Medical Sciences with project No. 98-01-33-41955.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of financial or personal interests that could have appeared to influence the content of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper is dedicated to our unique teacher in chemistry and medicinal chemistry (1937-2016)
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rastegari, A., Safavi, M., Vafadarnejad, F. et al. Synthesis and evaluation of novel arylisoxazoles linked to tacrine moiety: in vitro and in vivo biological activities against Alzheimer’s disease. Mol Divers 26, 409–428 (2022). https://doi.org/10.1007/s11030-021-10248-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10248-w