
This article discusses the aspects of four-circle diffractometers that are used to identify the composition of substances and materials and to determine many mechanical, physicochemical, and biological functionally significant characteristics of substances at the arbitration level. When monitoring the operation and certification of diffractometers, a system of reference materials of diffraction properties, including the reference materials of the crystal lattice parameter, is used. Taking into account the new quantum reality of modern technologies, the characteristics of these reference samples (RSs) have been refined. According to the precision measurement results of the unit cell size of RSs of silicon’s diffraction properties, a change in its structural characteristics with a change in the sphere diameter of the samples was revealed. This size effect, detected for the first time in the millimeter range, is similar to the effect when changing the size of silicon nanoparticles and other substances with different types of chemical bonding of atoms. Hence, the size effect must be taken into account when certifying RSs used in testing four-circle diffractometers. The results are important for understanding the nanoconditions of matter and will be useful in various industries, such as robot engineering, solar energy, focusing laser beams, and developing navigation systems.
Similar content being viewed by others

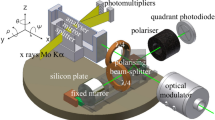
Avoid common mistakes on your manuscript.
Introduction
Materials for modern devices are developed according to a refined processing chain: “composition → structure → dispersion → properties”. Materials for gas turbine, robotic devices, and many electronics and solar energy devices have a single-crystal form. Unlike polycrystals, single crystals have a stricter order of the crystal lattice structure. That is, there are no high-angle boundaries and defects inherent in polycrystalline materials, which allows identifying reliably the control parameters for the formation of functionally significant characteristics. At all stages of the material development, the necessary and most complete information of the arbitration level is received from the results obtained using diffractometers. The single-crystal shape of a substance increases the intensity of reflected radiation by several orders of magnitude in diffraction measurement systems. As a result, an even number of small samples can be analyzed.
Among various diffractometers for characterizing single crystals, four-circle diffractometers are of special significance. Goniometers included in the structure of diffractometers of this type provide several degrees of freedom for sample rotation. Thus, data on all equivalent reflections for a family of atomic planes with different signs and permutations of Miller indices in each group can be separately obtained, which significantly increases the reliability and reproducibility of measurement results. In the diffractometers of this type, a high monochromatization and collimation of incident X-ray beams are achieved, which also increase the accuracy of measuring the angular distribution of the reflection intensity. Another important difference between the analysis of single-crystal and polycrystalline samples in these devices is the ability to collect data from the entire sample volume, not just from surface or near-surface layers.
Diffractometers are complex science-intensive and automated measuring systems in which measuring instruments (MIs), computer equipment, and auxiliary devices are functionally combined to obtain measurement information signals about physical quantities. Diffractometers include several devices, and the control of their operations is associated with different measurement units, reflecting different controlled characteristics (rotation step of goniometers in different directions, temperatures, and voltages). All characteristics are recorded during the experiment to take into account and introduce appropriate corrections if necessary.
The starting measurement model depends on the initial target. In high-precision experiments, to form a target, literature sources must be analyzed, which, as a rule, present conflicting information that requires clarification of the values of various physical quantities to characterize materials in the experiment performed. Moreover, in the starting model, the shape and perfection of the crystal structure of the sample should be taken into account. In four-circle diffractometers, the experimental conditions that reflect a complete, high-precision, and reliable model of the structure of matter can be set. The zero position on the goniometer scale is determined via calibration. Technically (according to the procedure), a trial short experiment is usually conducted to establish the correspondence of the starting model to the quality of the sample analyzed with the clarification of the model. During the experiment, when more than half of the given data array are obtained with a set of half of the possible reflection equivalents, the array is transferred to another computer for processing. Then, an additional task can be given to improve the statistical indicators. After processing large data arrays, characteristics are determined, which are also described by different measurement units, namely, composition, size of a unit cell, coordinates of atoms, defects (e.g., type and amount of a substance, its distribution over coordinates in the unit cell, and type and number of defects of various levels that violate the strict order in the initial structure model). Therefore, diffraction methods are classified as mixed types of measurements.
The characterization results of single crystals using four-circle diffractometers depend on the accuracy and completeness of determining the values of various characteristics of the diffraction pattern and are associated with the structural aspects of the substance based on various patterns that describe the scientific and processing chain: “composition → structure → dispersion → properties.” The depth and quality of the characteristics (chain elements) are determined by the initial measurement models used. It is also essential to analyze the influence of external factors and subsequent models necessary to determine the structural characteristics at different levels (scales) of the structure.
The operation control of diffractometers is based on the use of reference samples (RSs) of diffraction properties [1–4]. In the USA (National Institute of Standards and Technology, NIST) and Russia (Research Institute for Metrological Service, RIMS), more than 12 RS types of diffraction properties with different types of characteristics have been developed. The NIST and RIMS have also created reference MI–RSs for the diffraction properties of single crystals. The RIMS developed RSs based on silicon dioxide spheres (α-quartz). During the development, the test measurement results with the four-circle diffractometer KM-4 were taken into account. Subsequent certification was performed based on the results of an interlaboratory experiment. Laboratories of the Russian Academy of Sciences and industry institutes took part in this experiment, where four-circle diffractometers of various types were used in the measurements.
Moreover, the RIMS developed and certified a new type of RSs for diffraction properties, namely, GSO 9575-2010 PRF-4(3)-silicon, which includes high-purity silicon single-crystal spheres. However, the results of determining the parameters of the crystal lattice of dispersed powder preparations prepared from the same single crystals noticeably differed from the data obtained for the original single-crystal wafers and from those of interlaboratory experiments. Nonetheless, the achieved measurement accuracy was maintained. A similar difference in the data was also noted for different batches of fine-crystalline silicon RSs certified by the NIST [4, 5]. This difference in the values of the lattice parameters for bulk single crystals and dispersed samples has not been explained yet.
In high-precision measurements that characterize the composition and size of the unit cell of substances using four-circle diffractometers, the analyzed samples are sphere-shaped. This form allows taking into account many corrections accurately in the intensity of recorded reflections in the diffraction pattern. In addition, sphere-shaped samples with a prepared spherical surface are highly compact. This characteristic increases the irradiated volume and the sensitivity when measuring weak reflections in the high-angle region, where the accuracy of determining the lattice parameters is high, and when performing precision measurements of even very small analyzed samples. Many modern crystalline products used in scientific and application fields also have a sphere or hemisphere shape, and their functional characteristics depend on the state of the surface.
The spherical surface and structural characteristics of crystalline products can be distorted. However, the author of this article did not find any information in the literature on the relationship between different values of the RS lattice parameters and the distortion of the crystal structure as a result of an increase in the surface curvature of single-crystal products. The concept of curvature has been known for a very long time. Its role and mathematical description were elaborated by K. F. Gauss who established the relationship between the degree of curvature of the surface of sphere-shaped specimens and their size.
This work aimed to measure the unit cell dimensions of single-crystal samples from a batch of RSs of diffraction properties GSO 9575-2010 PRF-4(3)-silicon with different sphere sizes and to analyze the influence of curvature on the surface condition of these crystalline materials.
Measurement of silicon unit cell sizes in the millimeter range using four-circle diffractometers
The maximum sample size was determined by the diameter of the diffractometer collimator. In the present study, an XCalibur device (Oxford Diffraction, Rigaku, Japan) with a 0.5-mm-wide collimator was used. The minimum sample size depends on the sensitivity of the highly sensitive detector used and on the number and type of scattering centers (atoms) in the materials analyzed. The sample preparation methods, performance of high-precision experiments, and processing of large data arrays, which were formed as a result of complete experiments using four-circle diffractometers, are described in detail in Refs. [6, 7] and are part of the general approach of a diffractometric work with crystalline substances [8].
Cubic billets were made from plates of high-purity silicon single-crystal sphere from a batch for GSO 9575-2010 PRF-4(3)-silicon. Then, spheres of various diameters were obtained from these billets in cylindrical chambers, on the walls of which abrasive cloths with a diamond coating were glued. Under the influence of the air flow, the billets, moving inside the chamber, changed their shape from cubic to spherical. Two spherical samples were made with a large spread of sphere diameters (0.43 and 0.06 mm) and, consequently, with a significantly different curvature of the surface and crystal space.
The samples were placed on a small goniometric diffractometer head mounted on a large goniometer. As a result of the adjustment, the sample fell as accurately as possible into the center (crossing of all axes) of the goniometer (with a deviation of no more than 5–10 μm). The large goniometer design of the diffractometer provided a range of possible sequential rotations of the detector by 0°–120°. The sample was sequentially rotated with a step of 0.5° to fill very densely the space inside the reflection sphere and, consequently, the entire space of the unit cell. We used a penetrating short-wavelength monochromatic MoKα1 radiation to analyze the entire volume of the sample.
The experiment duration for a 0.43-mm-wide sample is very long (i.e., 66 h) compared to those of conventional experiments. Due to the high intensity of the reflected radiation for silicon, the X-ray tube voltage was reduced from 50 to 33 kV to preclude the appearance of reflections from the second harmonic of the incident radiation. The temperature was maintained at a level of (298.0±0.2) K throughout all measurements.
In the experiment, 6196 frames were recorded, which were divided into 32 sectors according to the angular characteristics of the diffraction pattern. Depending on the intensity of the emerging Bragg reflections, which are different in each angular range, the exposure time at each step of rotation of the goniometer with the sample varied, amounting to 30 s at high angles. This finding shows the high reliability in the analysis of angular positions of reflections in this high-angle range, which is most favorable for the high-precision determination of the crystal lattice parameter (unit cell size). In total, in this experiment, when studying the diffraction pattern, 8791 reflections were detected, corresponding to the unit cell of silicon with cubic symmetry (space group Fd3m). The number of independent sets of reflections ranges from 6 to 48 groups, depending on the Miller indices for these reflections. The comparison of the results between these groups ensures a high reproducibility of the results of each experiment.
The diameter of test specimen 2 was 0.06 mm. The same goniometric head and X-ray tube were used with the same radiation parameters as in the analysis of sample 1. However, due to a noticeable decrease in the analyzed volume of the sample, the intensity of the reflected radiation was reduced. In this regard, the exposure time at each step was increased, and the experiment duration was 160 h continuously, which provided the necessary statistics when collecting pulses to determine the coordinates of each reflection maximum.
To take into account possible time drifts, the same two control reflexes were recorded every 50 frames. The increased stability of the high voltage of the X-ray tube (voltage change was no more than one tenth of a volt at 32 kV) was achieved using additional stabilizing power sources. The reflections for this sample, which are weakest in the intensity, comparable with the background, were excluded by the program for processing frames with a diffraction pattern. The lattice parameters of each single-crystal sample at the last stage of processing the data array were sequentially clarified. First, the parameter value was determined over the entire data array until the results completely converged after several clarification cycles. Then, several additional clarification cycles were performed over the data array in the high-angle range.
The measurement results of two samples, namely, the change in the parameters of the crystal lattice of samples with different curvatures in the crystal space (according to Gauss, the curvature degree k is described by the inverse square of the radius R of the sphere), generalized for all independent groups with different Miller indices, are presented in Table 1, where dс is the sphere diameter and a is the lattice parameter. In the table, the difference in values is presented in bold.
Discussion
An additional increase in the accuracy of measurements with four-circle diffractometers enabled to register for the first time in the present study a change in the size of a silicon unit cell in the millimeter range, which has an exponential nature.
In the complete experiments performed in this work, all possible reflections were sequentially recorded, in contrast to the short dataset, which is used often, when only one or two sets of possible equivalents were analyzed. This is also a significant difference between the measurement technique used and accelerated approaches to measuring the characteristics of single crystals using diffractometers of this type. Usually, only a part of the reflections possible for single crystals, corresponding to an independent part of the possible equivalents in the reflection sphere, is analyzed. A short set with traditional measurements provides only a general idea of the type and size of the unit cell. A multiple increase in the amount of data in both experiments performed in the present study compared with measurements using powder diffractometers or plates, respectively, significantly increased the accuracy of the diffraction pattern characteristics. At the same time, random errors associated with the instrumental aspects of X-ray radiography in a given geometry of the optical scheme of a four-circle diffractometer and with the detection of a diffraction pattern and subsequent processing of the resulting large data arrays were taken into account.
The noticeable difference revealed in the crystal lattice parameters of the two studied samples (size effect) exceeds the statistical error by two orders of magnitude. In addition, both samples were measured using the same device using the same X-ray tube. This also led to the high accuracy and reliability of the results for studying the relative changes in the lattice parameter with a decrease in the size of the globule and a change in the curvature of its sphere.
The influence of the size of samples analyzed on their physical properties has been the subject of research for a long time [9–11]. The transition to the ultradispersed state of a substance or finely dispersed inclusions in the matrix leads to an increase in strength and a change in many other functional characteristics of materials [11–14]. For materials with particle sizes less than 100 nm, isolated from a variety of ultrafine substances, an increase in the proportion of the surface area relative to the volume is used in hypotheses to explain the unique properties of matter in the nanostate. At the same time, direct X-ray methods for studying the geometric and thermodynamic characteristics of nanomaterials have limitations in terms of the accuracy of the results of the experimental foundation or confirmation of hypotheses. This can be attributed to different influencing factors, including physical (e.g., abnormal broadening of Bragg reflections) and instrumental (when measurements are performed directly in the lower range of nanoparticle sizes) limitations.
In particular, for small particles in the form of powders, when measured in the nanometer size range [15], a similar and even stronger change in the crystal lattice parameters of silicon and other elements and chemical compounds based on them is noted. The detection of a similar size effect in the millimeter range can be used to understand the similar nature (mechanism) of the existing difference in data for the same substance in the form of single crystals and powders. Polycrystals are usually radiographed in a crushed form with particles ranging in size from several units to tens of micrometers to obtain a more average pattern and a correct description of the Bragg reflection profiles. After a combination of the processes of lattice dilatation and broadening of Bragg reflections, the determined angular positions were shifted. This condition changes the measured values of the lattice parameters of such polycrystalline objects. This change is also most noticeable for the frequently used beginning angular range, especially in the phase analysis of poly- and single crystals [16–19].
Numerous hypotheses of the existence of size effects and the theory of the state of matter in the nanometer range have not yet formulated a substantial experimental confirmation via direct X-ray methods due to the large number of influencing factors. As a result, the accuracy of measurements of the diffraction pattern characteristics in the nanometer range of sample sizes, especially in the small-angle region, is two to three orders of magnitude lower than that in the millimeter range used in this study.
The achieved significant increase in the accuracy of the measurement results in the millimeter range using four-circle diffractometers in comparison with similar data for powders in the nanometer size range suggests that the results for single crystals with different sphere sizes will provide a complete identification of the main mechanisms for the appearance of size effects in the nanometer range of particle sizes.
The curvature of the sphere surface of a smaller sample (0.06 mm in diameter) and the resulting distortions in the crystal lattice space on the surface of the samples are much higher than those for a sample with 0.43 mm in size, and an increase in the lattice parameter is also evident (Table 1). It can be assumed that “broken” chemical bonds on the sample surface increase the amplitude of vibrations of atoms with such bonds with an increase in their curvature and can lead to the reconstruction of these bonds. This statement agrees well with the fact that soft modes of vibrations of unit cell atoms are accompanied by an increase in the catalytic activity and self-organization processes on the crystal surface [20]. Among other hypotheses of the size effect registered, a change in the charge state of chemical bonds should also be noted.
The size effect for macrosized samples used in the practice of diffractometric measurements is of interest from the fundamental and applied viewpoints on the structure and properties of crystalline spheres.
High curvature and distortions arising in the space of the crystal lattice have also been established under severe plastic deformation, especially for products made of substances with a metallic type of chemical bond [21, 22].
In nature, the curvature of the shape of crystals and accompanying phenomena are noted for substances of many types during the growth of crystals under various environmental conditions [23]. For example, some forms of the unit cell and crystals of bioactive selenium resemble the helical structure of DNA. For this element, the size effect was also found [24], which is observed for silicon and other elements. More detailed studies of the size effect registered and accompanying phenomena in substances with different types of chemical bonds will reveal the generality of its influence on the characteristics of the material nanostate, where the contributions of surface phenomena are amplified many times.
The use of certified samples and measurement techniques increases the accuracy and reliability of results. In applied metrology, the study on the size effect is important for characterizing the RSs of diffraction properties of single crystals and identifying bioactive materials [18, 24]. Silicon is used not only as a material for semiconductor electronic products where quality is controlled by X-ray single-crystal diffractometry, including the analysis of the surface curvature of products. It is also an important structural material because it has excellent mechanical characteristics [25]. Hemispheres and spheres made of single-crystal silicon have long been used in the manufacture of navigation and measuring devices, including low-cost high-performance sensors and transducers that are easily connected to microprocessors [26, 27]. The use of silicon nanowires determines the possibility of creating a new generation of micromechanical sensors and actuators. Silicon hemispheres and curved single crystals are components of solar energy devices [28] and also serve as focusing monochromators and collimators included in various X-ray optical and laser systems. Nanosized silicon spheres and filaments are used in various topical biomedical and other devices.
Conclusions
The high accuracy of the experimental data obtained in this study, i.e., the measured parameters of the crystal lattice (dimensions of the unit cell of the substance) of silicon samples with different curvatures of the surface of the spheres, enabled to detect the size effect. For applications where a precise measurement accuracy is required, a size factor must be introduced. This case is especially significant in the certification of RSs of diffraction properties when measured using four-circle diffractometers. For materials science, a comprehensive study of the size effect will further reveal in more detail the formation mechanisms of the functional characteristics of spherical products used in various modern industries and clarify the nature of the aspects of the nanosized condition.
References
W. Wong-Ng et al., J. Res. Natl. Inst. Stand. Technol., 106, No. 6, 1071 (2001). https://doi.org/10.6028/jres.106.058
B. N. Kodess, “Metrological assurance of high-accuracy measurements of key materials characteristics for modern technology and their certified reference materials of composition and properties,” Hist. Sci. Eng., No. 9, 29–36 (2010).
B. Kodess, et al., Meeting Report, Neutron News, 20, No. 3, 4 (2009). https://doi.org/10.1080/10448630903114026.
C. R. Hubbard, J. Appl. Crystallogr., 16, No. 3, 285–288 (1983). https://doi.org/10.1107/S0021889883010456
D. Yoder-Short, J. Appl. Crystallogr., 26, No. 2, 272–276 (1993). https://doi.org/10.1107/S0021889892011610
B. N. Kodess, O. P. Lazukina, E. N. Volkova et al., Inorg. Mater., 56, 512–517 (2020). https://doi.org/10.1134/S0020168520050076.
B. N. Kodess, F. A. Sidorenko, Phys. Met. Metallogr., 122, No. 4, 345–350 (2021). https://doi.org/10.1134/S0031918X21040037.
Y. S. Belozerov, et al., Inorg. Mater., 57, 1135–1139 (2021). https://doi.org/10.1134/S0020168521110029
I. D. Morokhov, V. I. Petinov, L. I. Trusov, and V. F. Petrunin, Sov. Phys. Usp., 24, No. 4, 295–317 (1981). https://doi.org/10.1070/PM1981v024n04ABEH004800.
V. I. Zubov, “Some nano-size effects and properties of ultradispersive systems,” Zhur. Vsesoyuz. Himich. Obsh. im. D. I. Mendeleeva, 36, 133–136 (1991).
M. A. Filippov, B. N. Kodess, “Influence of high-pressure shock waves on the fine structure of manganese steel,” Phys. Met. Metallogr., 31, No. 1, 171–175 (1971).
B. Kodess, P. Kodess, “Investigation of nanostructure materials for developing new x-ray reference materials,” Proceedings of the Denver X-ray Conference, ICDD, 59, 243–254 (2015).
P. Aleksyeyev et al., Acta Phys. Pol. A, 4, No. 109, 555–559 (2006). 10.12693/APhysPolA.109.55.
A. Nabialek, et al., J. Appl. Phys., 105(6), 063918 (2009). https://doi.org/10.1063/13093696.
M. Y. Gamarnik, Phys. Status Solidi B, 161, 2, 457–462 (1990). https://doi.org/10.1002/pssb.2221610202.
B. N. Kodess, A. Y. Kuzin, Ind. Lab.: Diagn. Mater., 83 (12), 61–70 (2017). 10.26896/1028-6861-2017-83-12-61-70.
B. N. Kodess, V. A. Sarin, Meas. Tech., 57, No. 11, 1299–1303 (2015). https://doi.org/10.1007/s11018-015-0624-3.
B. N. Kodess, et al., “Metrological assurance of the quality control of pharmaceutical production using XRD methods,” Proceeding the 8th Pharmaceutical X-ray Diffraction Symposium, 4–7 May, Glasgow, Scotland, UK, 9–10 (2009).
M. T. Medetbekov et al., “Phase transitions, elastic constants and electron distribution in KDP-family,” Acta Сrystallogr. A: Found Crystallogr., 55, 61 (1999).
B. Kodess, P. Kodess, Mater. Res. Proc., 21, 259–263 (2022). 10.21741/978164 4 901755-46.
A. N. Tyumentsev et al., Phys. Mesomech., 16, No. 4, 319–334 (2013). https://doi.org/10.1134/S1029959913040061.
V. E. Panin et al., Phys. Mesomech., 18, No. 2, 89–99 (2015). https://doi.org/10.1134/S1029959915020010.
V. Vitelli, J. B. Lucks, and D. R. Nelson, Proceedings of the National Academy of Sciences, 103, No. 33, 12323–12328 (2006). https://doi.org/10.1073/pnas.0602755103.
M. V. Sukhanov et al., Doklady Chem., 466, No. 1, 11–14 (2016). https://doi.org/10.1134/S0012500816010079.
K. E. Petersen, Proceedings of the IEEE, 70, No. 5, 420–457 (1982). https://doi.org/10.1109/PROC.1982.12331.
V. A. Pogorelov, S. V. Sokolov, Cosm. Res., 53, No. 6, 458–468 (2015). https://doi.org/10.1134/S0010952515060040.
V. S. Shebashaevich et al., Setev. Sputnik. Radionavig. Sistemy, Radio i svyaz', Moscow (1993).
L. Tsakalakos, J. Balch, J. Fronheiser, B. A. Korevaar, O. Sulima, and J. Rand, Appl. Phys. Lett., 91, No. 23, 233117 (2007). https://doi.org/10.1063/12821113.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Izmeritel’naya Tekhnika, No. 5, pp. 35-41, May, 2022.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kodess, B.N. Influence of the Surface Curvature of Silicon Reference Materials on their Structural Characteristics. Meas Tech 65, 346–351 (2022). https://doi.org/10.1007/s11018-022-02086-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11018-022-02086-5