
Abstract
Amyloid-beta (Aβ) interacts with the serine/threonine protein kinase AKT (also known as protein kinase B)/glycogen synthase kinase 3β (GSK3β) pathway and deactivates GSK3β signaling, which result in microtubule protein tau phosphorylation. Atorvastatin, a HMG-CoA reductase inhibitor, has been proven to improve learning and memory performance, reduce Aβ and phosphorylated tau levels in mouse model of Alzheimer’s disease (AD). However, it still remains unclear whether atorvastatin is responsible for regulation of AKT/GSK3β signaling and contributes to subsequent down-regulation of Aβ1-42 and phosphorylated tau in APP/PS1 transgenic (Tg APP/PS1) mice. Herein, we aimed to investigate the possible impacts of atorvastatin (10 mg/kg, p.o.) on the memory deficit by behavioral tests and changes of AKT/GSK3β signaling in hippocampus and prefrontal cortex by western blot test in Tg APP/PS1 mice. The results showed that treatment with atorvastatin significantly reversed the memory deficit in the Tg APP/PS1 mice in a novel object recognition and the Morris water maze tests. Moreover, atorvastatin significantly attenuated Aβ1-42 accumulation and phosphorylation of tau (Ser396) in the hippocampus and prefrontal cortex of Tg APP/PS1 mice. In addition, atorvastatin treatment also increased phosphorylation of AKT, inhibited GSK3β activity by increasing phosphorylation of GSK3β (Ser9) and decreasing the beta-site APP cleaving enzyme 1 (BACE1) expression. These results indicated that the memory ameliorating effect of atorvastatin may be, in part, by regulation the AKT/GSK3β signaling which may contribute to down-regulation of Aβ1-42 and tau hyperphosphorylation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, leading to progressive cognitive decline in the elderly population (Reitz and Mayeux 2014). It has a complex pathophysiology, which, although not completely understood, may be characterized by amyloid beta (Aβ)-containing plaques and neurofibrillary tangles composed of hyperphosphorylated microtubule protein tau, and synaptic and neuronal loss, along with progressive cognitive dysfunction (Selkoe 2001).
There is no effective pharmacotherapy for preventing the process of AD. Increasing evidence shows that the serine/threonine protein kinase AKT (also known as protein kinase B) and glycogen synthase kinase 3 β (GSK3β) are the key transducers of brain metabolic and mitogenic signals required for down-regulation of Aβ levels and dephosphorylation of tau, which suggest that AKT/GSK3β signaling is the potential mediators for AD (Ali and Kim 2015; Bao et al. 2013; Zeng et al. 2015). Notably, GSK3β plays a significant role as one of the major kinase that contributes to the hyperphosphorylated state of tau observed in pathology of AD (Deng et al. 2015). Previous study suggested that inhibition of GSK3β regulates β-site APP-cleaving enzyme 1 (BACE1) expression, leading to reducing Aβ neuropathology and alleviating memory deficits in mouse model of AD (Ly et al. 2013). Consistent with the above finding, the AKT and GSK3β levels are dysregulated in the brain of patients and animal models of AD (Ryder et al. 2004; Castri et al. 2007; Hernández et al. 2010; Dionisio et al. 2015), indicating the possibility that chemical compounds may be able to modulate the activity of the AKT/GSK3β signaling for preventing the progresses of AD.
There is accumulating evidence that cholesterol-lowering agents, particularly 3-hydroxy-3-methylglutarylcoenzyme A (HMG-CoA) reductase inhibitors (also known as statins), have many pleiotropic effects, i.e., reducing Aβ production, suppressing inflammatory responses, protecting neurons against excitotoxins, apoptosis and oxidative stress, and promoting synaptogenesis (Lu et al. 2004; Selley 2005; Shinohara et al. 2010; Zhang et al. 2013; Métais et al. 2014). Statins have been hypothesized to have beneficial effects on memory deficits in several clinical and animal studies (Javadi-Paydar et al. 2011; Bettermann et al. 2012; Tendolkar et al. 2012; Roy et al. 2015). However, all of the above therapies are still not used for preventing the progresses of AD.
Atorvastatin is a member of the statin family as a strong HMG-CoA reductase inhibitor, which is currently used in clinic worldwide. The safety of high doses of atorvastatin has been demonstrated by previous study (Waters 2005). In addition, previous studies also have been indicated that treatment with atorvastatin significantly reduces senile plaque and phosphorylated tau in vitro (Sui et al. 2015) or in vivo (Lu et al. 2010; Kurata et al. 2011). However, whether atorvastatin improves cognitive function and reduces Aβ1-42 levels and phosphorylated tau in Tg APP/PS1 mice remain unknown. The present study investigated whether atorvastatin might attenuate memory deficit through the inhibition of Aβ1-42 production and hyperphosphorylated tau in the hippocampus and prefrontal cortex of Tg APP/PS1 mice. Additionally, whether AKT/GSK3β signaling changes were paralleled with alterations in Aβ1-42 production and hyperphosphorylated tau were not explored in the previous studies (Lu et al. 2010; Kurata et al. 2011; Sui et al. 2015). In present study, we mainly intended to investigate the possible roles of atorvastatin on regulation of AKT/GSK3β signaling in Tg APP/PS1 mice.
Materials and methods
Animals
The amyloid precursor protein (APP)/presenilin 1(PS1) heterozygous mice were purchased from the Model Animal Research Center of Nanjing University. A total of 40 male mice (30 Tg APP/PS1 mice, 10 wild type (WT) mice) aged 9 months were randomly assigned into four groups: WT + Vehicle group; Tg APP/PS1 + Vehicle group; Tg APP/PS1 + Atorvastatin group; and Tg APP/PS1 + Donepezil group. The animals were housed in a temperature-controlled animal facility with a 12 h light–dark cycle (lights on at 6:00 a.m.). Water and food were freely available in their home cages. All procedures followed the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23, revised 1996) and were approved by the Animal Care and Use Committee of Ningbo University (Ningbo, China). All efforts were made to minimize animal suffering.
Drugs and Treatment
Atorvastatin (Lipitor Atorvastatin calcium, Pfizer, NY, USA) and donepezil (Sigma Aldrich, St. Louis, MO, USA) dosage were dissolved in saline (0.9 %) containing 0.5 % w/v carboxymethyl cellulose (CMC). Both of atorvastatin and donepezil were given orally daily with 10 mg/kg respectively, and control groups received an equal volume of vehicle. Volume of administration was 10 ml/kg. The schedule of drug treatment and test orders was shown in Fig. 1.

Schedule of drug treatment and test orders. Atorvastatin (10 mg/kg), Donepezil (10 mg/kg) or their vehicle (saline containing 0.5 % w/v CMC) was injected (p.o.) once daily, 7 days before behavioral experiments and continuing until day 16 when the animals were sacrificed (SAC) for biochemical assays. On day 8, the behavioral experiments were performed, including the open field test (OFT), novel object recognition test (NORT), and the Morris water-maze (MWM) tests
Open field test (OFT) and Novel object recognition test (NORT)
This task is based on the spontaneous tendency of rodents to explore novel objects (Ennaceur and Delacour 1988). The test was performed in an apparatus made of a white Plexiglas box (50 × 50 × 39 cm) with the floor divided into four identical squares in a dim room. Mice were habituated to an empty apparatus for 5 min to test locomotor activity and anxiety-like behavior 24 h prior to exposure to objects, in order to habituate them to the apparatus and test room. The number of rearing events (forepaws elevated from the floor) and line crossings (with all four paws placed into a new square), movement distance length and movement speed were considered to be an index of exploratory behaviour. In addition, the number of grooming sessions, amount of time spent in the centre and periphery were recorded as measures of anxiety in mice. Twenty-four hours after habituation, mice were acclimated in the testing room during 1 h before the beginning of the sessions. Firstly, mice completed an acquisition trial (24 h after habituation) that consisted of leaving the animals in the apparatus containing two identical objects (A and A’). After a 3 h retention interval, the mice were placed back into the arena and exposed to the familiar object (A) and to a novel object (B) for short-term recognition memory test. 24 h later, long-term recognition memory was evaluated and a different pair of dissimilar objects (a familiar and a novel one; A and C, respectively) were presented. In all sessions, each mouse was always placed in the apparatus facing the wall and allowed to explore the objects for 5 min, after which the mouse was returned to its home cage. Behavior was recorded by a video camera mounted vertically above the test arena and analyzed using appropriated video-tracking software (Duoyi, Shanghai, China). Between trials, the apparatus was cleaned with 5 % ethanol solution to hide animal clues. The light inside the apparatus was maintained at a minimum to avoid any anxiety behavior. A recognition index, a ratio of the amount of time spent exploring any one of the two objects (training session) or the novel object (retention session) over the total time spent exploring both objects, was used to measure cognitive function.
Morris water maze (MWM)
Spatial learning and memory were assessed using the MWM, as previously described (McKee et al. 2008) with minor modification. Briefly, a circular plastic pool was filled with water (22 ± 3 °C) to obscure the location of a submerged platform. Four visual cues were placed around the tank to orient the mice, with the platform remaining in a fixed location. The platform location was kept constant for each mouse during training and probe trials test, and it was 1.5 cm beneath the surface of the water. Four trials starting from 4 different starting positions were performed each day (total 5 consecutive days) with a trial interval of 30 min. When mice failed to find the hidden platform within 90 s, they were guided to the platform and were left there for 20 s, before being returned to their cages. The average data of latency for the platform in four trials of each mouse was counted for all tested mice per group per day. To evaluate retention memory, probe trials were conducted 24 h after last acquisition training. During these probe trials, the platform was removed, and the swimming time in the target quadrant and the number of platform crossings were recorded during 90 s. The recorded data were used to analyze mice performance.
Western blot analysis
Total proteins were extracted from hippocampal and prefrontal cortex tissues of each group (n = 4) mice by using ice cold radio immuno precipitation assay (RIPA) lysis buffer (Pierce, Rockford, IL, U.S.A.) containing (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 % NP-40, 0.5 % sodium deoxycholate, 0.1 % SDS; Upstate, Temecula, CA, USA) and then the lysate was centrifuged (15,000×g, 30 min, 4 °C). Protein concentration of each sample lysate was determined using the BCA kit (Thermo Scientific, Rockford, IL, USA). The lysates (25 μg total protein) were separated on 10 % SDS-polyacrylamide gel electrophoresis (PAGE) gels and transferred to PVDF membranes (0.22 μm; Millipore, CA, USA). Membranes were then incubated with rabbit anti-phospho-AKT-Ser473 (1:1000; Cell Signaling, Danvers, MA, USA), rabbit anti-total-AKT (1:1000; Cell Signaling, USA), rabbit antiphospho-GSK3β-Ser9 (1:500; Abcam, Cambridge, MA, USA), rabbit anti-GSK3β (1:300; Abcam, Cambridge, MA, USA), rabbit anti-phospho-Tau-Ser396 (1:800; Invitrogen, Grand Island, NY, USA), rabbit anti-Tau (1:1000; Millipore, CA, USA), rabbit anti-BACE1 (1:1000; Millipore, CA, USA), rabbit anti-Aβ1-42 (1:1000; Millipore, CA, USA) or rabbit anti-GAPDH (1:2000; Millipore, CA, USA) at 4 °C overnight. The membranes were then incubated with Alexa Fluor 700-conjugated goat anti-rabbit antibody (1:10000; Invitrogen, Eugene, OR) for 60 min. Target bands were detected and quantified using a fluorescence scanner (Odyssey Infrared Imaging System, LI-COR Biotechnology, Lincoln, NE).
Statistical analysis
All measurements were performed by an independent investigator blinded to the experimental conditions. Data are expressed as the mean ± standard error of means (SEM). Data were analyzed by an one-way ANOVA or a two-way ANOVA followed by Newman-Keuls post hoc test using the GraphPad Prism software (Version 5.0, Prism software for PC, GraphPad, USA). The criterion for significance was p < 0.05.
Results
Effects of atorvastatin treatment on open field test (OFT) behaviour in WT and Tg APP/PS1 mice
To exclude the possibility that atorvastatin induced locomotor activity alterations in OFT, we measured the effects of atorvastatin on locomotor activity on day 8 after eight consecutive days drug treatment. The mice treated with atorvastatin (10 mg/kg, p.o.) or donepezil (10 mg/kg, p.o.) did not differ from mice treated with vehicle in path length [F (3, 39) = 0.08655, p = 0.9670; Fig. 2a], average movement speed [F (3, 39) = 0.1170, p = 0.9495; Fig. 2b], number of line crossing [F (3, 39) = 0.1453, p = 0.9320; Fig. 2c], rearing [F (3, 39) = 0.1452, p = 0.9321; Fig. 2d], number of grooming episodes [F (3, 39) = 0.1489, p = 0.9297; Fig. 2e], amount of time spent in the centre [F (3, 39) = 0.2153, p = 0.8851; Fig. 2f] and periphery of the arena [F (3,39) = 0.1509, p = 0.9284; Fig. 2f]. Our data indicated that anxiety levels and locomotor activity are not affected by repeated administration of atorvastatin or donepezil in mice.

Effects of atorvastatin on locomotor activity and anxiety-like behaviors of mice. Path length (a), Speed (b), number of line crosses (c), number of rearing events (d), number of grooming episodes (e) and amount of time spent in the center of the arena vs. the periphery (f), were recorded in an open field test (OFT), over a 5 min observation time, 60 min after injection (p.o.) of vehicle, Atorvastatin (10 mg/kg) or Donepezil (10 mg/kg) in mice. Values are shown as means ± S.E.M. (n = 10 per group)
Effects of repeated treatment with atorvastatin on memory deficit in Tg APP/PS1 mice
Novel object recognition test (NORT)
The NORT is very useful to determine short-term memory and long-term memory of rodents. In order to examine whether sub-chronic administration of atorvastatin or donepezil is able to prevent memory impairment in the NORT of Tg APP/PS1 mice. In the current work, we evaluated the effects of repeated treatment with atorvastatin or donepezil on recognition memory by submitting the animals to a NORT. As shown in Fig. 3a, the exploration time of the two objects was recorded in the training sessions 1 h after the atorvastatin or donepezil treatment on day 9. In the training session, all the treatment groups were shown to have similar exploratory preference for the two identical objects (object A and object A’) [Student’s t-test, p > 0.05, Fig. 3a]. The retention session was conducted 3 h (short-term memory test) and 24 h (long-term memory test) after the training session respectively. As is shown in Fig. 3b, when the animals were placed in the arena 3 h after first exploration period (training session), the Tg APP/PS1 mice treated with vehicle were not able to discriminate between the familiar (object A) and novel object (object B), as indicated by similar exploration times for both objects (Student’s t-test, p > 0.05). However, the exploratory preference for the novel object (object B) in the Tg APP/PS1 mice treated with vehicle was significantly decreased [Student’s t-test, p < 0.05; Fig. 3b] compared with WT mice treated with vehicle. In addition, Tg APP/PS1 mice treated with atorvastatin or donepezil were able to improve short-term recognition memory compared with Tg APP/PS1 mice treated with vehicle (p < 0.05 and p < 0.05, respectively; Fig. 3b). Further, similar results were found when long-term recognition memory was evaluated, indicating that mice treated with atorvastatin [Student’s t-test, p < 0.05; Fig. 3c] or donepezil [Student’s t-test, p < 0.05; Fig. 3c] also significantly reversed the long-term memory deficit of Tg APP/PS1 mice.

Atorvastatin reversed the short-term and long-term memory deficit of Tg APP/PS1 mice in novel object recognition test (NORT). a The novel object recognition test (NORT) training session was measured after 9 days of Atorvastatin (10 mg/kg) or Donepezil (10 mg/kg). b Short-term memory test session performed 3 h after training on day 9; c Long-term memory test session performed 24 h after training session on day 10. Columns are indicated as mean ± S.E.M, n = 10 animals in each experimental group. **p < 0.01, significant differences between familiar and new object; △ p < 0.05, compared to Tg APP/PS1 mice treated with vehicle
Morris water maze (MWM)
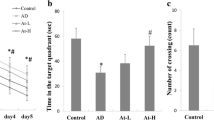
The MWM is a behavioral task to test spatial learning and memory. It has been widely used in the study of AD models. The spatial effect of atorvastatin or donepezil on learning and memory was investigated using the MWM test. As shown in Fig. 4a, the mean escape latency of Tg APP/PS1 mice treated with vehicle was significantly increased compared with WT mice treated with vehicle on day 3 [F (4144) =194.5, p < 0.01], day 4 [F (4144) =194.5, p < 0.01] and day 5 [F (4144) =194.5, p < 0.01], while Tg APP/PS1 mice treated with atorvastatin or donepezil showed significant improvement compared with Tg APP/PS1 mice treated with vehicle on training day 4 [F (4144) =194.5, p < 0.01 for atorvastatin; p < 0.01 for donepezil] and day 5 [F (4144) =194.5, p < 0.01 for atorvastatin; p < 0.01 for donepezil]. On the day 6, the platform was removed and the probe trial test was conducted. As shown in Fig. 4b and c, compared with WT mice treated with vehicle, Tg APP/PS1 treated with vehicle significantly decreased the swimming time in the target quadrant [F (3,39) = 16.15, P < 0.01; Fig. 4b] and the number of platform crossings [F (3,39) = 9.117, P < 0.01; Fig. 4c]. However, atorvastatin or donepezil-treatment significantly increased the time spent in the target quadrant [F (3,39) = 16.15, p < 0.01; Fig. 4b] and number of platform crossing [F (3,39) = 9.117, p < 0.01; Fig. 4b] compared with Tg APP/PS1 mice treated with vehicle.

Atorvastatin attenuated the memory impairment of Tg APP/PS1 mice in Morris water maze (MWM) task. a Escape latency for escape to a submerged platform in the training trials. b Time in the target quadrant in the probe trail. c Platform crossing times in the probe trail. Columns are indicated as mean ± S.E.M, n = 10 animals in each experimental group. **p < 0.01, compared to vehicle-treated WT mice; # p < 0.05, ##p <0.01, compared to Tg APP/PS1 mice treated with atorvastatin or donepezil
Effects of atorvastatin on Aβ1-42, BACE1, pTau (Ser396) and Tau expression in the hippocampus and cortex of Tg APP/PS1 mice
We further detected the expression of Aβ1-42, BACE1, pTau (Ser396) and Tau in the hippocampus and prefrontal cortex of mice by immunoblotting. Our results revealed that the expression of Aβ1-42 [hippocampus, F (3,15) = 53.12, p < 0.001, Fig. 5b; cortex, F (3,15) = 11.07, p = 0.0009, Fig. 5g], BACE1 [hippocampus, F (3,15) = 13.09, p = 0.0003, Fig. 5c; cortex, F (3,15) = 6.726, p = 0.0065, Fig. 5h] and pTau (Ser396) [hippocampus, F (3,15) = 42.52, p < 0.0001, Fig. 5d; cortex, F (3,15) =15.02, p = 0.0002, Fig. 5i] were significantly increased in Tg APP/PS1 mice treated with vehicle. However, compared with Tg APP/PS1 mice treated with vehicle group, atorvastatin or donepezil treatment significantly decreased the expression of Aβ1-42 (atorvastatin, hippocampus, p < 0.01; cortex, p < 0.01; donepezil, p < 0.01; cortex, p < 0.01), BACE1 (atorvastatin, hippocampus, p < 0.01; cortex, p < 0.01; donepezil, p < 0.01; cortex, p < 0.01), pTau (Ser396) (atorvastatin, hippocampus, p < 0.01; cortex, p < 0.01; donepezil, p < 0.01; cortex, p < 0.01) in hippocampus and cortex respectively. However, none of the treatments affected the expression of tau in the hippocampus [F (3,15) = 0.6041, p = 0.6248] and cortex [F (3,15) = 0.3681, p =0.7774] of mice.

Atorvastatin decreased the expression of Aβ1-42, BACE1, pTau (Ser396) and Tau in the hippocampus and prefrontal cortex of mice. (a and f) represent immunoblots of Aβ1-42, BACE1 and pTau (Ser396) expression detected by Western blotting with tissues from the hippocampus (a) and prefrontal cortex (f); the rest panels are quantification of the immuno blotting bands of Aβ1-42 (b and g), BACE1 (c and h), pTau (Ser396) (D and I), and Tau (E and J). The data are expressed as mean ± S.E.M (n = 4 per group). **p < 0.01, compared to WT mice treated with vehicle; ##p < 0.01, compared to Tg APP/PS1 mice treated with atorvastatin or donepezil
Effects of atorvastatin on pAKT (Ser473), AKT, pGSK3β (Ser 9), GSK3β, expression in the hippocampus and cortex of Tg APP/PS1 mice
As shown in Fig. 6, the results showed that phosphorylated AKT (Ser473) levels were significantly decreased in the hippocampus [F (3,15) =7.155, p = 0.0052, Fig. 6b] and cortex [F (3,15) = 6.277, p = 0.0083, Fig. 6g] of Tg APP/PS1 mice treated with vehicle compared with WT mice treated with vehicle. However, treatment with atorvastatin or donepezil significantly inhibited the reduction of phosphorylated AKT (Ser473) activity in the hippocampus [atorvastatin, p < 0.01; donepezil, p < 0.01] and cortex [atorvastatin, p < 0.01; donepezil, p < 0.05] compared with Tg APP/PS1 mice treated with vehicle. We did not find a significant difference in the total AKT levels among the groups in hippocampus and cortex respectively [hippocampus, F (3,15) = 0.1281, p = 0.9416, Fig. 6c; cortex, F (3,15) = 0.04748, p = 0.9856, Fig. 6h]. In addition, our results also showed that phosphorylation of GSK3β at Ser9 was significantly decreased in the hippocampus [F (3,15) =5.173, p =0.0159, Fig. 6d] and cortex [F (3,15) =8.648, p = 0.0025, Fig. 6i] in Tg APP/PS1 mice treated with vehicle compared with the WT mice treated with vehicle. The AKT activation induced by atorvastatin or donepezil treatment was accompanied by the de-activation of GSK3β, revealed by the increased phosphorylation of GSK3β (Ser9) in hippocampus [atorvastatin, p < 0.05; donepezil, p < 0.05] and cortex [atorvastatin, p < 0.01; donepezil, p < 0.01] of Tg APP/PS1 mice respectively. However, none of the treatments affected the expression of GSK3β in the hippocampus [F (3,15) = 0.0699, p = 0.9749, Fig. 6e] and cortex [F (3,15) = 0.07149, p = 0.9741, Fig. 6j] of mice.

Effect of atorvastatin on expression of pAKT, AKT, pGSK3β (Ser9) and GSK3β in the hippocampus and prefrontal cortex of mice. (a) and (f) represent immunoblots of pAKT, AKT, GSK3β and pGSK3β (Ser9) expression detected by Western blotting with tissues from the hippocampus (a) and prefrontal cortex (f); the rest panels are quantification of the immunoblotting bands of pAKT (b and g), AKT (C and H), pGSK3β (Ser9) (D and I) and GSK3β (E and J). The data are expressed as mean ± SEM (n = 4 per group). *p < 0.05, **p < 0.01, compared to WT mice treated with vehicle; #p < 0.05, ##p < 0.01, compared to Tg APP/PS1 mice treated with atorvastatin or donepezil
Discussion
Accumulating evidence suggests that high levels of serum cholesterol may promote the pathological processes leading to AD (Notkola et al. 1998; Tschäpe and Hartmann 2006; Cedazo-Mínguez et al. 2011. Statins are well-known cholesterol-lowering drugs for treatment of AD (Sparks et al. 2006; McGuinness and Passmore 2010; Javadi-Paydar et al. 2011; Bettermann et al. 2012; Tendolkar et al. 2012; Roy et al. 2015). Statins have lots of pharmacological properties, such as anti-oxidative, anti-inflammatory, decrease Aβ formation and as ligands of peroxisome proliferator-activated receptor α (PPARα) (Sparks et al. 2006; Reiss and Wirkowski 2007; Kurinami et al. 2008; Butterfield 2011; Lappegård et al. 2013; Roy et al. 2015). However, the exact mechanism underlying the statins on AD-related cognitive disorders is still unclear. We chose to use atorvastatin in the present study, which is a strong 3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) reductase inhibitor currently being used in AD patients worldwide. The results showed that atorvastatin is effective in reversing learning and memory deficit in AD mouse model via reducing brain Aβ production and Tau hyperphosphorylation (Lu et al. 2010).
The amyloid hypothesis is supported by a large amount of experimental evidence (Barage and Sonawane 2015; Ferreira et al. 2015; Musiek and Holtzman 2015). As a model of AD, Tg APP/PS1 mice share similarity with clinical AD patients in pathogenesis, pathology and symptoms (Heneka et al. 2013; Biallosterski et al. 2015). Tg APP/PS1 mice begin to develop Aβ deposition at 6 months of age, and the impairment worsens in an age-dependent manner (Reiserer et al. 2007). Our study evaluated the therapeutic effects of atorvastatin on Aβ-associated pathogenesis in 9-month-old APP transgenic mice. Consistent with the previous studies (Kurata et al. 2011), our results showed that atorvastatin could attenuate the learning and memory deficit in Tg APP/PS1 mice, and partly inhibit the Aβ1-42 production in the hippocampus and cortex of the Tg APP/PS1 mice. These results strongly support our hypothesis that atorvastatin could exert non-cholesterol-lowering activity in AD progression. To investigate whether the decrease in Aβ1-42 expression by atorvastatin shown in our results was regulated by direct inhibition of Aβ1-42 production and/or secretion. We then examined the effects of atorvastatin on the enzymes that contribute to the production of Aβ. We found that atorvastatin treatment markedly decreased the expression of BACE1, the key enzyme that is responsible for APP processing and Aβ production (Liang et al. 2010; Vassar 2014), in hippocampus and cortex of Tg APP/PS1 mice. This effect may be closely related to the beneficial effects of atorvastatin on cognitive dysfunction.
Interestingly, several studies have shown that the incidence tau phosphorylation was triggered by the Aβ1-42 peptide, which is the activated form of APP (Ott et al. 2011; Stancu et al. 2014). Tau is a microtubule-associated protein mainly expressed in neurons, and its primary role is to stabilize the neuronal cytoskeleton. Intraneuronal aggregation of abnormally phosphorylated tau in neurofibrillary tangles (NFTs) constitutes a major neuropathological hallmark of AD (Duan et al. 2012; Kumar et al. 2015). Ser396 was identified as one hyperphosphorylated tau protein site in AD patients (Duka et al. 2013). We investigated Ser396, the individual site of hyperphosphorylated tau protein in Tg APP/PS1 mice. Our results indicated that atorvastatin effectively protected against tau phosphorylation at Ser396 site, suggesting that reduction of tau phosphorylation by decreasing cholesterol levels may be another one possible mechanisms of atorvastatin for improvement in cognitive function.
Previous studies showed that the serine/threonine protein kinase AKT and GSK3β are the key transducers of brain metabolic and mitogenic signals required for down-regulation of Aβ and tau dephosphorylation (Huang et al. 2014; Ali and Kim 2015). Importantly, GSK3β is activated through the phosphorylation at Tyr216 and inhibited by phosphorylating Ser9. Active GSK3β contributes to phosphorylate tau and APP, which probably in turn contributes to NFTs formation and amyloidogenic processing of APP (Aplin et al. 1996; Durairajan et al. 2012; Deng et al. 2015). In addition, increasing evidence suggests that the AKT/GSK3β signaling pathway is directly impacted by Aβ exposure in vitro and in vivo studies (Liu et al. 2015; Tiwari et al. 2015; Ghasemi et al. 2015; Kim et al. 2015). We reported that pAKT (Ser473) and pGSK3β (Ser9) were significantly decreased in hippocampus and cortex of Tg APP/PS1 mice, this dysregulation of AKT/GSK3β signaling correlated with impairment of cognitive function. Our current study consistent with the previous work that shows the phosphorylation and consequently activation of AKT when AD mice were treated with atorvastatin (10 mg/kg) for 7 days (Piermartiri et al. 2009). Therefore, up-regulation of pAKT (Ser473) and pGSK3β (Ser9) produced by atorvastatin paralleled with memory improvement, supporting the notion that dysregulation of the AKT/GSK3β pathway is critical for the Aβ1-42 production and phosphorylation of Tau dysfunction that characterizes AD.
Taken together, the present study demonstrated that atorvastatin ameliorated memory impairment in behavioral tasks including in the NOR and MWM tasks in mice. Moreover, the memory enhancing effects of atorvastatin were mediated, in part, by attenuating Aβ1-42 production and tau pathology. Atorvastatin increased the levels of phosphorylated AKT and GSK3β (Ser9) in the hippocampus and cortex of mice, may indicate that the memory enhancing effects of atorvastatin are likely related to the activation of the AKT/GSK3β (Ser9) signaling pathway. Atorvastatin may represent a promising therapeutic strategy to attenuate AD progression.
References
Ali T, Kim MO (2015) Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3β pathway in the mouse hippocampus. J Pineal Res 59(1):47–59
Aplin AE, Gibb GM, Jacobsen JS, Gallo JM, Anderton BH (1996) In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J Neurochem 67(2):699–707
Bao XQ, Li N, Wang T, Kong XC, Tai WJ, Sun H, Zhang D (2013) FLZ alleviates the memory deficits in transgenic mouse model of Alzheimer’s disease via decreasing beta-amyloid production and tau hyperphosphorylation. PLoS One 8(11), e78033
Barage SH, Sonawane KD (2015) Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 52:1–18
Bettermann K, Arnold AM, Williamson J, Rapp S, Sink K, Toole JF, Carlson MC, Yasar S, Dekosky S, Burke GL (2012) Statins, risk of dementia, and cognitive function: secondary analysis of the ginkgo evaluation of memory study. J Stroke Cerebrovasc Dis 21(6):436–44
Biallosterski BT, Prickaerts J, Rahnama’i MS, de Wachter S, van Koeveringe GA, Meriaux C (2015) Changes in voiding behavior in a mouse model of Alzheimer’s disease. Front Aging Neurosci 7:160
Butterfield DA (2011) Atorvastatin and Aβ(1–40): not as simple as cholesterol reduction in brain and relevance to Alzheimer disease. Exp Neurol 228(1):15–18
Castri P, Iacovelli L, De Blasi A, Giubilei F, Moretti A, Tari Capone F, Nicoletti F, Orzi F (2007) Reduced insulin-induced phosphatidylinositol-3-kinase activation in peripheral blood mononuclear leucocytes from patients with Alzheimer’s disease. Eur J Neurosci 26(9):2469–2472
Cedazo-Mínguez A, Ismail MA, Mateos L (2011) Plasma cholesterol and risk for late-onset Alzheimer’s disease. Expert Rev Neurother 11(4):495–498
Deng J, Habib A, Obregon DF, Barger SW, Giunta B, Wang YJ, Hou H, Sawmiller D, Tan J (2015) Soluble amyloid precursor protein alpha inhibits tau phosphorylation through modulation of GSK3β signaling pathway. J Neurochem 135(3):630–637
Dionisio PA, Amaral JD, Ribeiro MF, Lo AC, D’Hooge R, Rodrigues CM (2015) Amyloid-β pathology is attenuated by tauroursodeoxycholic acid treatment in APP/PS1 mice after disease onset. Neurobiol Aging 36(1):228–240
Duan Y, Dong S, Gu F, Hu Y, Zhao Z (2012) Advances in the pathogenesis of Alzheimer’s disease: focusing on tau-mediated neurodegeneration. Transl Neurodegener 1(1):24
Duka V, Lee JH, Credle J, Wills J, Oaks A, Smolinsky C, Shah K, Mash DC, Masliah E, Sidhu A (2013) Identification of the sites of tau hyperphosphorylation and activation of tau kinases in synucleinopathies and Alzheimer’s diseases. PLoS One 8(9), e75025
Durairajan SS, Liu LF, Lu JH, Chen LL, Yuan Q, Chung SK, Huang L, Li XS, Huang JD, Li M (2012) Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol Aging 33(12):2903–2919
Ennaceur A, Delacour J (1988) A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav Brain Res 31(1):47–59
Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG (2015) Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci 9:191
Ghasemi R, Moosavi M, Zarifkar A, Rastegar K, Maghsoudi N (2015) The interplay of Akt and ERK in Aβ toxicity and insulin-mediated protection in primary hippocampal cell culture. J Mol Neurosci 57(3):325–334
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434):674–678
Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J (2010) GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol 223(2):322–325
Huang HC, Tang D, Xu K, Jiang ZF (2014) Curcumin attenuates amyloid-β-induced tau hyperphosphorylation in human neuroblastoma SH-SY5Y cells involving PTEN/Akt/GSK-3β signaling pathway. J Recept Signal Transduct Res 34(1):26–37
Javadi-Paydar M, Rayatnia F, Fakhraei N, Zakeri M, Mirazi N, Norouzi A, Dehpour AR (2011) Atorvastatin improved scopolamine-induced impairment in memory acquisition in mice: involvement of nitric oxide. Brain Res 1386:89–99
Kim HG, Park G, Lim S, Park H, Choi JG, Jeong HU, Kang MS, Lee MK, Oh MS (2015) Mori Fructus improves cognitive and neuronal dysfunction induced by beta-amyloid toxicity through the GSK-3β pathway in vitro and in vivo. J Ethnopharmacol 171:196–204
Kumar P, Jha NK, Jha SK, Ramani K, Ambasta RK (2015) Tau phosphorylation, molecular chaperones, and ubiquitin E3 ligase: clinical relevance in Alzheimer’s disease. J Alzheimers Dis 43(2):341–361
Kurata T, Miyazaki K, Kozuki M, Panin VL, Morimoto N, Ohta Y, Nagai M, Ikeda Y, Matsuura T, Abe K (2011) Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res 1371:161–170
Kurinami H, Sato N, Shinohara M, Takeuchi D, Takeda S, Shimamura M, Ogihara T, Morishita R (2008) Prevention of amyloid beta-induced memory impairment by fluvastatin, associated with the decrease in amyloid beta accumulation and oxidative stress in amyloid beta injection mouse model. Int J Mol Med 21(5):531–537
Lappegård KT, Pop-Purceleanu M, van Heerde W, Sexton J, Tendolkar I, Pop G (2013) Improved neurocognitive functions correlate with reduced inflammatory burden in atrial fibrillation patients treated with intensive cholesterol lowering therapy. J Neuroinflammation 10:78
Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG (2010) Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem 285(36):27737–27744
Liu XH, Geng Z, Yan J, Li T, Chen Q, Zhang QY, Chen ZY (2015) Blocking GSK3β-mediated dynamin1 phosphorylation enhances BDNF-dependent TrkB endocytosis and the protective effects of BDNF in neuronal and mouse models of Alzheimer’s disease. Neurobiol Dis 74:377–3791
Lu D, Goussev A, Chen J, Pannu P, Li Y, Mahmood A, Chopp M (2004) Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma 21(1):21–32
Lu F, Li X, Suo AQ, Zhang JW (2010) Inhibition of tau hyperphosphorylation and beta amyloid production in rat brain by oral administration of atorvastatin. Chin Med J (Engl) 123(14):1864–1870
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W (2013) Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 123(1):224–235
McGuinness B, Passmore P (2010) Can statins prevent or help treat Alzheimer’s disease? J Alzheimers Dis 20(3):925–933
McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A (2008) Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res 1207:225–236
Métais C, Brennan K, Mably AJ, Scott M, Walsh DM, Herron CE (2014) Simvastatin treatment preserves synaptic plasticity in AβPPswe/PS1dE9 mice. J Alzheimers Dis 39(2):315–329
Musiek ES, Holtzman DM (2015) Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci 18(6):800–806
Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, Tuomilehto J, Nissinen A (1998) Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 17(1):14–20
Ott S, Henkel AW, Henkel MK, Redzic ZB, Kornhuber J, Wiltfang J (2011) Pre-aggregated Aβ1-42 peptide increases tau aggregation and hyperphosphorylation after short-term application. Mol Cell Biochem 349(1–2):169–177
Piermartiri TC, Vandresen-Filho S, de Araújo HB, Martins WC, Dal’agnolo D, Stroeh E, Carqueja CL, Boeck CR, Tasca CI (2009) Atorvastatin prevents hippocampal cell death due to quinolinic acid-induced seizures in mice by increasing Akt phosphorylation and glutamate uptake. Neurotox Res 16(2):106–115
Reiserer RS, Harrison FE, Syverud DC, McDonald MP (2007) Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav 6(1):54–65
Reiss AB, Wirkowski E (2007) Role of HMG-CoA reductase inhibitors in neurological disorders: progress to date. Drugs 67(15):2111–2120
Reitz C, Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 88(4):640–651
Roy A, Jana M, Kundu M, Corbett GT, Rangaswamy SB, Mishra RK, Luan CH, Gonzalez FJ, Pahan K (2015) HMG-CoA reductase inhibitors bind to PPARα to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metab 22(2):253–265
Ryder J, Su Y, Ni B (2004) Akt/GSK3beta serine/threonine kinases: evidence for a signalling pathway mediated by familial Alzheimer’s disease mutations. Cell Signal 16(2):187–200
Selkoe DJ (2001) Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis 3(1):75–80
Selley ML (2005) Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res 1037(1–2):1–6
Shinohara M, Sato N, Kurinami H, Takeuchi D, Takeda S, Shimamura M, Yamashita T, Uchiyama Y, Rakugi H, Morishita R (2010) Reduction of brain beta-amyloid (Abeta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Abeta clearance. J Biol Chem 285(29):22091–22102
Sparks DL, Sabbagh M, Connor D, Soares H, Lopez J, Stankovic G, Johnson-Traver S, Ziolkowski C, Browne P (2006) Statin therapy in Alzheimer’s disease. Acta Neurol Scand Suppl 185:78–86
Stancu IC, Vasconcelos B, Terwel D, Dewachter I (2014) Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener 9:51
Sui HJ, Zhang LL, Liu Z, Jin Y (2015) Atorvastatin prevents Aβ oligomer-induced neurotoxicity in cultured rat hippocampal neurons by inhibiting Tau cleavage. Acta Pharmacol Sin 36(5):553–564
Tendolkar I, Enajat M, Zwiers MP, van Wingen G, de Leeuw FE, van Kuilenburg J, Bouwels L, Pop G, Pop-Purceleanu M (2012) One-year cholesterol lowering treatment reduces medial temporal lobe atrophy and memory decline in stroke-free elderly with atrial fibrillation: evidence from a parallel group randomized trial. Int J Geriatr Psychiatry 27(1):49–58
Tiwari SK, Seth B, Agarwal S, Yadav A, Karmakar M, Gupta SK, Choubey V, Sharma A, Chaturvedi RK (2015) Ethosuximide induces hippocampal neurogenesis and reverses cognitive deficits in amyloid-β toxin induced Alzheimer’s rat model via PI3K/Akt/Wnt/β-catenin pathway. J Biol Chem 290(47):28540–28558
Tschäpe JA, Hartmann T (2006) Therapeutic perspectives in Alzheimer’s disease. Recent Pat CNS Drug Discov 1(1):119–127
Vassar R (2014) BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther 6(9):89
Waters DD (2005) Safety of high-dose atorvastatin therapy. Am J Cardiol 96(5A):69F–75F
Zeng Y, Zhang J, Zhu Y, Zhang J, Shen H, Lu J, Pan X, Lin N, Dai X, Zhou M, Chen X (2015) Tripchlorolide improves cognitive deficits by reducing amyloid β and upregulating synapse-related proteins in a transgenic model of Alzheimer’s Disease. J Neurochem 133(1):38–52
Zhang YY, Fan YC, Wang M, Wang D, Li XH (2013) Atorvastatin attenuates the production of IL-1β, IL-6, and TNF-α in the hippocampus of an amyloid β1-42-induced rat model of Alzheimer’s disease. Clin Interv Aging 8:103–110
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 81201050; No.81271209; No.81371224; No.81541087); Natural Science Foundation of Zhejiang province (No.LQ12H09001); Natural Science Foundation of Ningbo (No. 2012A610249); The Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University (to Junfang Zhang) and Zhejiang “Climbing Program” (PD2013104). This project is also sponsored by K.C. Wong Magna funded at Ningbo University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dongsheng Zhou, Huaxia Liu, Chenli Li and Fangyan Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhou, D., Liu, H., Li, C. et al. Atorvastatin ameliorates cognitive impairment, Aβ1-42 production and Tau hyperphosphorylation in APP/PS1 transgenic mice. Metab Brain Dis 31, 693–703 (2016). https://doi.org/10.1007/s11011-016-9803-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-016-9803-4